

Abstract
Introduction
We have previously identified a rare subpopulation of variant human mammary epithelial cells (vHMEC) with repressed p16INK4A that exist in disease-free women yet display premalignant properties, suggesting that they have engaged the process of malignant transformation. In order to gain insight into the molecular alterations required for vHMEC to progress to malignancy, and to characterize the epigenetic events associated with early progression, we examined the effect of oncogenic stress on the behavior of these cells.
Methods
HMEC that express p16INK4A and vHMEC that do not, were transduced with constitutively active Ha-rasV12 and subsequently exposed to serum to determine whether signals from the cellular microenvironment could cooperate with ras to promote the malignant transformation of vHMEC. Epigenetic alterations were assessed using methylation-specific polymerase chain reaction (PCR).
Results
vHMEC expressing Ha-rasV12 (vHMEC-ras) bypassed the classic proliferative arrest that has been previously documented in normal fibroblasts following oncogenic stress, and that we also observe here in normal HMEC. Moreover, vHMEC-ras cells exhibited many additional alterations that are observed during progression to malignancy such as the generation of chromosomal abnormalities, upregulation of telomerase activity, immortalization following exposure to serum, and anchorage-independent growth, but they did not form tumors following orthotopic injection in vivo. Associated with their early progression to malignancy was an increase in the number of genes methylated, two of which (RASSF1A and SFRP1) were also methylated in other immortalized mammary cell lines as well as in breast cancer cells and tissues.
Conclusions
We have characterized a mammary progression model that recapitulates molecular and methylation alterations observed in many breast cancers. Our data suggest that concomitant methylation of RASSF1A and SFRP1 marks an early event in mammary transformation and may thus have prognostic potential.
Introduction
Oncogenic transformation arises from the accumulation of both genetic and epigenetic alterations that result in the activation of oncogenes and inactivation of tumor suppressor genes. Of the many oncogenes activated in human cancers, ras is one of the genes that has been the most extensively studied. Although mutation of ras genes is rare in human breast cancers [1], over 50% of human breast carcinomas express elevated levels of normal Ha-ras protein [2-4]. In addition, higher levels of ras protein have been observed in hyperplasias from patients who subsequently develop cancer, than in hyperplasias from patients who do not [5]. This suggests that alterations in ras expression can occur early in the transformation process, and thus contribute to the initiation of tumorigenesis. Likewise, epigenetic alterations, including DNA methylation and chromatin structure changes, are among the earliest molecular abnormalities to occur during tumorigenesis. Included among the genes epigenetically silenced in breast cancer are genes involved in cell cycle regulation (p16INK4A, CCND2, RASSF1A), cell signaling (SFRP1, SFRP5), differentiation (HOXA9), immortalization (p57), and DNA repair (MGMT, BRCA1) [6-12]. A recent survey of CpG island methylation using an array-based mapping technique revealed that one-third of CpG islands methylated in premalignant lesions are associated with members of various homeobox gene superfamilies, suggesting that methylation of homeobox genes is a frequent and early event in breast cancer [13].
Consistent with this, we have previously identified a rare subpopulation of variant human mammary epithelial cells (vHMEC) that exhibit p16INK4A and HOXA9 promoter hypermethylation, centrosome dysfunction, genomic instability, and COX-2 overexpression [14-17]. We found evidence that cells with these characteristics exist in morphologically normal tissue of disease-free women [18], as well as in ductal carcinoma in situ (DCIS) lesions [19], suggesting that these cells may be precursors to cancer.
In order to gain insight into the molecular alterations required for vHMEC to progress to malignancy, and the epigenetic events associated with that progression, we examined the effect of oncogenic stress on the behavior of HMEC that express p16INK4A, and vHMEC that do not, by expressing constitutively active Ha-rasV12 in these cells. Since vHMEC display some characteristics of tumor cells, suggesting that the process of malignant transformation is initiated in these cells, we hypothesized that vHMEC would be resistant to ras-induced growth arrest, but that HMEC, like normal fibroblasts which have been shown to senesce in response to oncogenic ras [20], would not. Indeed, as expected, vHMEC continued to proliferate following ras expression while HMEC arrested. Moreover, when cultured in a serum-containing environment, vHMEC expressing oncogenic ras spontaneously immortalized, acquired the capacity for anchorage-independent growth, and exhibited de novo DNA methylation at several gene loci frequently methylated in breast cancer. Among the genes methylated, we identified a panel of four genes, two of which (RASSF1A and SFRP1), were also methylated in other immortalized mammary cell lines, suggesting that programs of epigenetic alterations mark early steps in the transformation process. Thus, these early epigenetic alterations may prove useful as biomarkers for early detection of breast cancer.
Materials and methods
Cell culture and breast tissue specimens
HMEC were isolated from reduction mammoplasty tissue as previously described [21]. HMEC were grown in MEGM (Lonza, Walkersville, MD, USA) supplemented with or without 0.5% fetal bovine serum. Initial experiments examining the effect of oncogenic ras on HMEC and vHMEC proliferation were conducted in cells isolated from five different individuals: RM48 (kindly provided by Martha Stampfer, Lawrence Berkeley National Laboratory, Berkeley, CA), RM13, RM15, RM18, and RM21 (cells derived in our laboratory). All subsequent experiments in which cells were exposed to serum were conducted with RM48. All cell cultures were maintained as previously described [21]. vHMEC clones were isolated using standard ring cloning procedures.
Breast specimens originated from biopsies, mastectomies, and reduction mammoplasties. Donors included both breast cancer patients and women with no breast cancer history. Specimens were acquired as either fresh tissues, frozen tissues, which had been stored at -80°C, or as DNA that had already been extracted from frozen tissues archived in the British Columbia Cancer Agency frozen breast tumor bank and Cooperative Human Tissue Network. Protocols for the acquisition of human specimens were approved by the institutional review boards of the institutions providing the specimens (University of British Columbia, British Columbia Cancer Agency Research Ethics Board, Canada, and the Committee for the Protection of Human Subjects, University of California, Berkeley, CA, USA).
Retroviral gene transfer
The pLXSP3 and pLXSP3-Ha-rasV12 retroviral constructs were a gift from Dr. Frank McCormick (UCSF Comprehensive Cancer Center, University of California San Francisco, San Francisco, CA, USA). Amphotropic retrovirus was produced by transfecting Phoenix-A packaging cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Forty-eight to 72 hours post-transfection, virus-containing culture medium was collected and filtered through 0.45-μm syringe filters. Cells were infected by exposing them to virus-containing medium in the presence of 4 μg/mL Polybrene (Sigma-Aldrich, St. Louis, MO, USA). Forty-eight hours following infection, transduced cells were selected for four to six days in medium containing 2 μg/mL puromycin. Experiments were conducted four to six days following exposure to puromycin to allow selection of cells that exhibit puromycin resistance.
Immunoblot analysis
Cells were washed twice with ice-cold phosphate buffered saline (PBS) and lysed with 50 mM Tris, 150 mM NaCl buffer containing 1% Nonidet P-40, 0.25% deoxycholate, 1 mM EDTA, 20 mM sodium fluoride, 1 mM sodium orthovanadate, and 1× cocktail of Complete protease inhibitors (Roche Applied Science, Indianapolis, IN, USA). Protein content was quantitated utilizing the BCA protein assay reagent (Pierce, Rockford, IL, USA). Protein extracts were separated by SDS-PAGE using 4-20% gradient polyacrylamide gels (Cambrex, East Rutherford, NJ, USA), and transferred onto Hybond-P membranes (GE Healthcare Bio, Piscataway, NJ, USA) at 100 volts for two hours. Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing Tween (TBS-T) (20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Tween 20 (v/v)) for one hour at room temperature and incubated with the primary antibodies diluted in blocking buffer overnight at 4°C. The rabbit polyclonal antibody against Ha-ras (#SC520) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The mouse monoclonal antibody against β-actin (#5441) was obtained from Sigma-Aldrich (St. Louis, MO, USA). Following incubation with primary antibodies, the membranes were washed three times for 10 minutes with TBS-T, incubated with horseradish peroxidase-conjugated secondary antibodies for one hour at room temperature, and rewashed three times for 10 minutes with TBS-T. Immunoreactive bands were visualized by chemiluminescence using the SuperSignal West Pico Chemiluminescent Substrate System (Pierce, Rockford, IL, USA).
Cell cycle analysis
Cells were metabolically labeled with 10 μmol/L bromodeoxyuridine for four hours, trypsinized, and fixed with ice-cold 70% ethanol. Nuclei were isolated and stained with propidium iodide and FITC-conjugated anti-bromodeoxyuridine antibodies (Becton Dickinson, San Jose, CA, USA) and analyzed by flow cytometry using a FACS-Sorter (Becton Dickinson, San Jose, CA, USA) and the CellQuest software (Becton Dickinson, San Jose, CA, USA).
Chromosomal analysis
Metaphase spreads were prepared from cells treated with 100 ng/ml Colcemid (KaryoMAX, GibcoBRL) for six hours. Standard G-banding karyotypic analysis was performed on at least 50 metaphase spreads for each cell population.
Centrosome analysis
Centrosomes were stained and analyzed as previously described [17]. Briefly, cells were grown on coverslips, fixed in methanol at -20°C for five minutes and stained using a standard immunocytochemistry protocol with a primary monoclonal antibody that recognizes γ-tubulin (1 μg/mL; clone GTU-88, Sigma) and a secondary fluorescein isothiocyanate (FITC) conjugated sheep F(AB')2 fragment to mouse IgG (whole molecule) (ICN Pharmaceuticals, Inc., Costa Mesa, CA, USA). Samples were analyzed on a Zeiss 510 LSM Confocal Microscope (Carl Zeiss AG, Oberkochen, Germany). Statistical significance was determined by the two-sided Fisher exact test (95% confidence interval).
Telomerase activity assay
Telomerase activity was measured using the Quantitative Telomerase Detection Kit from Allied Biotech (Vallejo, CA, USA) following the manufacturer's directions. Briefly, 1 × 106 cells were harvested, snap frozen in liquid nitrogen, and subsequently lysed using the provided lysis buffer. Protein content in the lysates was determined utilizing the BCA protein assay reagent (Pierce, Rockford, IL, USA). The amount of telomerase activity in 1 μg of lysate was compared to the standard provided in the kit. Each sample was analyzed in triplicate. A no template control, heat inactivated sample, and cell lysates from telomerase positive (MDA-MB-231) and telomerase negative (U2OS) cells were included in each experiment.
Soft agar colony assay
Cells were harvested, counted, and resuspended in media containing 0.6% agarose, 5% serum and 10 mM Hepes. Cell suspensions were then plated on top of a bottom layer of agarose in 35 mm dishes at a concentration of 50,000 cells per dish, in triplicate. After 14 days, colonies were counted manually in eight different fields. The data are presented as the average of the sum of eight different fields counted.
In vivo tumor studies
An IRES-based bicistronic lentiviral vector encoding green fluorescent protein (GFP) and firefly luciferase was obtained from Dr. Sanjiv Sam Gambhir (Stanford University, Palo Alto, CA, USA) and utilized to generate the cells used for the in vivo tumor studies. Briefly, virus was produced by co-transfecting 293T cells with the CSCMV-FLuc-EGFP vector along with a defective packaging vector encoding the HIV gal, pol, rev, and tat genes, and a plasmid coding for the VSVG envelope protein. Clones isolated from a pool of immortalized vHMEC-ras0.5 cells were transduced with lentiviral particles. Twenty-four hours following transduction, the cells were split, allowed to expand for one week, and then FACS sorted to collect GFP-expressing cells. Expression of GFP-luciferase did not alter the growth characteristics of these cells (see Additional file 1). One, four, or ten million cells expressing GFP-luciferase were injected directly into the surgically exposed #4 mammary fat pads of six to eight week old female SCID-Beige mice. Cell growth and survival were monitored weekly by bioluminescence imaging utilizing the Xenogen IVIS imaging system (Caliper Life Sciences, Hopkinton, MA, USA). Quantitation of photon emission from the bioluminescent signal was performed utilizing the acquisition and analysis software Living Image (Xenogen). All experiments involving animals were conducted in compliance with the Institutional Animal Care and Use Committee guidelines.
Methylation-specific PCR
Genomic DNA was isolated from cells using the Wizard Genomic DNA Isolation kit (Promega, Madison, WI, USA). Approximately 750 ng of DNA was bisulfite-treated with the EZ DNA Methylation Gold kit according to the manufacturer's protocol (Zymo Research, Orange, CA, USA). Methylation-specific PCR (MSP) was performed on bisulfite-modified DNA using previously described primer pairs and PCR cycle conditions for p16INK4A [22], HOXA9 [16], CCND2 [12], RASSF1A [23], SFRP1 [24], p57, MGMT [25], and THBS1 [26]. Control templates from human genomic lymphocyte DNA either treated with SssI methylase (methylated control) or untreated (unmethylated control) and a no template (water) control were included in each experiment. PCR products were electrophoresed on 3% agarose gels, stained with ethidium bromide, and visualized under UV illumination.
Real-time quantitative PCR
Total RNA was isolated from cells and cDNA synthesized using standard methods. Quantitative PCR (Taqman, Applied Biosystems, Inc., Foster City, CA, USA) was performed on cDNA using the standard curve method with primer/probe sets (Applied Biosystems, Inc.) for SFRP1 (Hs00610060_m1), RASSF1A (Hs00200394_m1), and MGMT (Hs01037698_m1). The expression of GUSB (IDT), an external control, was used to normalize for variances in input cDNA. The forward and reverse primer sequences for GUSB were: 5'-CTCATTTGGAATTTTGCCGATT-3', 5'-CCGAGGAAGATCCCCTTTTTA-3', 5' FAM-TGAACAGTCACCGACGAGAGTGCTGGTA-TAM 3', respectively. Each experiment was performed at least in triplicate. Error bars represent the standard deviation in a representative experiment.
Results
vHMEC are resistant to Ha-ras-induced proliferative arrest and display chromosomal abnormalities
To test the effect of oncogenic stress on normal HMEC and vHMEC with repressed p16INK4A, cells were retrovirally transduced with constitutively active Ha-rasV12. Expression of Ha-rasV12 was confirmed by immunoblot analysis (Figure 1a). Despite oncogenic ras expression, vHMEC failed to undergo the classic proliferative arrest that has been previously documented in normal fibroblasts [20], and that we also observe here in normal HMEC (Figure 1b). Instead, they continued to proliferate at the same rate as vHMEC expressing the control vector (vHMEC-vector). The percentage of cells in S phase was not affected by expression of oncogenic ras in vHMEC, but was significantly reduced in HMEC (Figure 1c). These data demonstrate that vHMEC are resistant to Ha-rasV12-induced proliferative arrest.
Figure 1.

vHMEC expressing Ha-ras are resistant to proliferative arrest and display increased numbers of chromosomal abnormalities. (a) Immunoblot analysis demonstrating Ha-rasV12 expression in HMEC and vHMEC following retroviral transduction with pLXSP3-Ha-rasV12 (r) or the control pLXSP3 vector (v). Constructs were expressed in HMEC and vHMEC derived from five different individuals. A representative blot is shown along with actin as a loading control. (b) Growth curves demonstrating that HMEC underwent a proliferative arrest in response to oncogenic ras (orange line, left graph), while vHMEC continued to proliferate (orange line, right graph). (c) Cell cycle analysis of HMEC and vHMEC expressing Ha-rasV12 or control vector demonstrating that the number of cells in S-phase dropped from 33.8% to 8.8% following Ha-rasV12 expression in HMEC, but remained the same in vHMEC (37.9% and 37.6%, respectively). (d) Chromosomal analysis of vHMEC-vector and vHMEC-ras cells. Control vHMEC (vector) and vHMEC expressing oncogenic Ha-RasV12 (ras) were harvested at different passages (P+1, P+5, and P+8), as indicated, and processed for metaphase analysis. Standard G-banding karyotypic analysis was performed on at least fifty metaphase spreads for each cell population. Aneuploidy refers to additions or deletions of whole chromosomes. Structural abnormalities include all deletions, duplications, rings, marker chromosomes, chromatid exchanges and translocations. The total number of abnormalities includes all structural abnormalities and telomeric associations, not including numerical abnormalities.
Progression to malignancy is often associated with an increase in genomic instability. Since vHMEC expressing oncogenic ras (vHMEC-ras) were capable of bypassing the proliferative barrier normally induced by oncogenic stress, we examined whether these cells displayed any alterations in genomic integrity beyond those previously detected in vHMEC [15,17]. This analysis could not be performed in HMEC expressing oncogenic ras since the cells arrested. In vHMEC, no increases in the frequency of centrosome abnormalities were detected between the vHMEC vector control and vHMEC ras-expressing cell populations (data not shown). However, karyotypic analysis revealed a number of chromosomal abnormalities, including structural abnormalities, telomeric associations, and alterations in ploidy (Figure 1d). Thus, upon continued propagation, vHMEC-ras cells become increasingly genomically unstable, manifesting genomic changes at an earlier passage and an increased frequency than detected in cultured vHMEC.
Signals from the extracellular environment can cooperate with oncogenic ras to immortalize vHMEC and upregulate telomerase activity
While oncogenic ras can cooperate with a number of viral and cellular genes to transform cells [27,28], whether it can do so in concert with signals from the extracellular milieu has not been examined. Since signals from the microenvironment can promote mammary tumorigenesis [29], we examined whether changes in the cellular environment could influence the effect of ras in vHMEC, and facilitate their progression to malignancy. Studies have shown that the gene expression pattern of cultured primary fibroblasts in response to serum exposure resembles that of a wounding response, and that this wound-response signature is strongly predictive of metastasis and progression for a variety of carcinomas [30]. Therefore, we exposed both vHMEC-vector and vHMEC-ras cells at several points in their growth curve to media containing 0.5% serum. Addition of serum while the cells were still in the exponential growth phase induced a proliferative arrest in both cell populations (data not shown). In contrast, addition of serum while the cells were in a highly mutagenic phase (agonescence) was sufficient to cause immortalization of vHMEC-ras (vHMEC-ras0.5) but not control vHMEC (Figure 2A). This suggests that an additional event, likely conferred by the genomic instability that is exhibited at agonescence (growth plateau) [15], is required for serum to cooperate with ras in immortalizing vHMEC.
Figure 2.
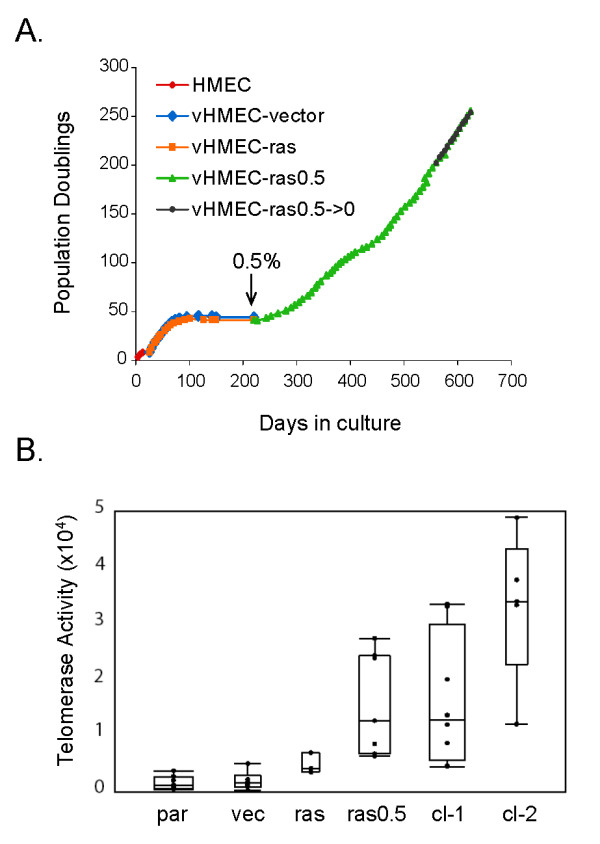
Signals from the extracellular environment cooperate with ras to immortalize vHMEC and upregulate telomerase activity. (a) Growth curves of HMEC (red line), vHMEC expressing control vector (blue line), and vHMEC expressing Ha-rasV12 (orange line). Arrow indicates time at which vHMEC-ras cells were exposed to serum (day 220). These cells demonstrated increased population doublings within two passages (day 243) in serum-containing media, and are referred to as vHMEC-ras0.5 cells. Their growth curve is depicted in green. After 560 days, the vHMEC-ras0.5 cells were cultured in the absence of serum and continued to proliferate. These cells are referred to as vHMEC-ras0.5- >0 and their growth curve is depicted in black. (b) Telomerase activity assay depicting the amount of telomerase activity in lysates prepared from parental vHMEC (par), vHMEC-vector (vec), vHMEC-ras (ras), and vHMEC-ras0.5 (ras0.5) cells, as well as two clones isolated from the ras0.5 cell population (cl-1 and cl-2).
To address whether constitutive extracellular stimulation resulting from the exposure to serum was required for the continued proliferation, vHMEC-ras0.5 cells were cultured in mammary epithelial growth medium without serum (vHMEC-ras0.5->0). We found that the cells were capable of continued proliferation in the absence of serum, indicating that once immortalization is initiated by extracellular serum stimulation, proliferation is no longer dependent on the initial signals provided by the serum (Figure 2a). Consistent with this, immortalized vHMEC-ras0.5 cells displayed an increase in telomerase activity relative to the parental, vector control, and vHMEC-ras cells (Figure 2b). In addition, two clones (cl-1 and cl-2) isolated from the vHMEC-ras0.5 cell population also displayed an increase in telomerase activity (Figure 2b).
Immortalized vHMEC expressing oncogenic ras are capable of anchorage-independent growth but are not tumorigenic in vivo
Since vHMEC expressing oncogenic ras exhibited many characteristics observed during the progression to malignancy, including increased genomic instability, upregulation of telomerase activity, and unlimited proliferative capacity, we then asked if the accumulation of these molecular alterations had rendered these cells tumorigenic. Since anchorage-independent growth is a requirement for tumorigenicity, we first examined whether vHMEC expressing oncogenic ras could grow in soft agar. As shown in Figure 3a, the parental, vector control, and non-immortalized vHMEC-ras cells failed to grow in soft agar. In contrast, the immortalized vHMEC-ras0.5 as well as two clones isolated from vHMEC-ras0.5 cells (cl-1 and cl-2), displayed some, albeit weak, capacity for anchorage-independent growth, suggesting that they may have some tumorigenic potential.
Figure 3.

Immortalized variant human mammary epithelialcells expressing oncogenic ras are capable of anchorage-independent growth but are not tumorigenic in vivo. (a) Soft agar colony assay. Parental vHMEC (par), vHMEC-vector (vec), vHMEC-ras (ras), vHMEC-ras0.5 (ras0.5), clone 1 (cl-1), and clone 2 (cl-2), which were isolated from the vHMEC-ras0.5 cells, were plated in 35 mm dishes at a concentration of 50,000 cells per dish, in triplicate. After 14 days, colonies were counted manually in eight different fields. The data are presented as the average of the sum of eight different fields counted. (b) Bioluminescence imaging of SCID-Beige mice following orthotopic injection of 1 × 106 (top panel), 4 × 106 (middle panel), or 10 × 106 (bottom panel) vHMEC-ras clone 1 cells expressing GFP-luciferase into the left and right #4 mammary fat pad. Cell growth and survival was monitored weekly by bioluminescence imaging utilizing the Xenogen IVIS imaging system. Representative images of each experiment are shown.
Since clone 1 exhibited the highest capacity for anchorage-independent growth in the soft agar assay, we decided to examine whether this clone could grow following orthotopic injection into the mammary glands of immunocompromised mice. In order to facilitate the monitoring of its growth in vivo, clone 1 was engineered to express firefly luciferase. The light emitted by the clone following administration of the luciferin substrate allowed us to directly visualize the cells in live animals by bioluminescence imaging.
For these experiments, we injected either 1 × 106 (n = 6), 4 × 106 (n = 12) or 10 × 106 (n = 11) clone 1 cells expressing luciferase directly into the surgically exposed #4 mammary fat pads of six to eight week old female Scid/beige mice. In all the glands injected with either 1 × 106 or 4 × 106 clone 1 cells, the luminescent signal faded within three weeks following injection, suggesting that the majority of cells were not surviving in vivo (Figure 3b). Likewise, in the majority of the glands injected with 10 × 106 clone 1 cells, the luminescent signal faded within three to four weeks, while in one gland, the bioluminescent signal persisted up to eight weeks (Figure 3b and data not shown). No palpable tumors ever formed in any of these mice. Histological and whole mount analyses of their mammary glands did not reveal any signs of tumor growth or hyperplasia (data not shown). Thus, under the experimental conditions tested, these cells were not tumorigenic, suggesting that additional alterations/mutations are required for complete transformation.
Progression to malignancy is associated with DNA methylation at several gene loci
In order to determine whether the accumulation of alterations observed in our progression model was also accompanied by additional epigenetic alterations, we used methylation-specific PCR (MSP) to determine whether genes that are commonly epigenetically silenced in breast cancer, were also silenced in our cell model. A list of the MSP primers used, along with their location relative to the transcription start site of each gene, is presented in Table 1. Coincident with the p16INK4A and HOXA9 promoter hypermethylation we have previously documented in vHMEC [16], we observed methylation of the CCND2 promoter in these same cells (Figure 4, Table 2, Additional file 2). As the cells progressed further towards malignancy and became immortalized, DNA methylation was observed at several additional gene loci including, RASSF1A, SFRP1, p57, and MGMT. In order to determine whether this panel of four genes represented a program of epigenetic alterations important for the immortalization of mammary epithelial cells, we examined whether these genes were methylated in the immortalized, but non-tumorigenic 184-A1 and MCF-10A mammary cells, as well as in the MCF-7 and MDA-231 breast cancer cell lines. DNA methylation of the RASSF1A gene, and of one of the CpG islands of the SFRP1 gene (SFRP1-exon1), was observed in all four of the cell lines examined, while methylation of the p57 gene was observed in two of the four cell lines (MCF-10A and MCF-7), and methylation of the MGMT gene was observed only in the MDA-231 cell line (Figure 4, Table 2). The gene encoding thrombospondin (THBS1), which is also frequently methylated in breast cancer, remained unmethylated in all the cells. These data suggest that concomitant DNA methylation of the RASSF1A and SFRP1 (exon 1) genes may play a role in the early steps of mammary transformation.
Table 1.
MSP primers
| Gene Name | Forward Primer (5'-3') | Reverse Primer (5'-3') | Product size (bp) | Primer location relative to TSS (bp) | Ref |
|---|---|---|---|---|---|
| p16INK4A-U | TTA TTA GAG GGT GGG GTG GAT TGT | CAA CCC CAA ACC ACA ACC ATA A | 151 | +132 to 283 | [22] |
| p16INK4A-M | TTA TTA GAG GGT GGG GCG GAT CGC | GAC CCC GAA CCG CGA CCG TAA | 150 | +132 to 282 | |
| HOXA9-U | TTG GGG TTA GAT AGG GAG TTG GGA | AAA AAT AAA AAC AAA AAA CAA ACA AA | 167 | -4453 to -4286 | [16] |
| HOXA9-M | TCG GGG TTA GAT AGG GAG TCG GGA | AAA ATA AAA ACG AAA AAC AAA CGA A | 166 | -4453 to -4287 | |
| CCND2-U | AGA GTA TGT GTT AGG GTT GAT T | ACA TCC TCA CCA ACC CTC CA | 108 | -1167 to -1059 | [12] |
| CCND2-M | CGG CGA TTT TAT CGT AGT CG | CTC CAC GCT CGA TCC TTC G | 101 | -1138 to -1037 | |
| RASSF1A-U | TTT GGT TGG AGT GTG TTA ATG TG | CAA ACC CCA CAA ACT AAA AAC AA | 108 | +201 to +309 | [23] |
| RASSF1A-M | GTG TTA ACG CGT TGC GTA TC | AAC CCC GCG AAC TAA AAA CGA | 94 | +213 to +307 | |
| SFRP1-U* | GTT TTG TAG TTT TTG GAG TTA GTG TTG TGT | CTC AAC CTA CAA TCA AAA ACA ACA CAA ACA | 135 | -2 to +133 | [24] |
| SFRP1-M | TGT AGT TTT CGG AGT TAG TGT CGC GC | CCT ACG ATC GAA AAC GAC GCG AAC G | 126 | +2 to +128 | |
| SFRP1-U** | TTT TAG TAA ATT GAA TTT GTT TGT GA | TAA AAT ACA CAA AAC TGG TAG AAC | 149 | -151 to -2 | [40] |
| SFRP1-M | TTT AGT AAA TCG AAT TCG AAT TCG TTC GC | TAA AAT ACG CGA AAC TCC TAC G | 148 | -150 to -2 | |
| p57-U | GTT GTT TGT GTT TGT GTA GTT TT | AAA AAT CCC ACA AAC AAC AAA ACA | 91 | +248 to +339 | |
| p57-M | TTG TTC GCG TTT GCG TAG TTT C | AAA TCC CAC GAA CGA CAA AAC G | 88 | +249 to +337 | |
| MGMT-U | TTT GTG TTT TGA TGT TTG TAG GTT TTT GT | AAC TCC ACA CTC TTC CAA AAA CAA AAC A | 93 | +30 to +123 | [25] |
| MGMT-M | TTT CGA CGT TCG TAG GTT TTC GC | GCA CTC TTC CGA AAA CGA AAC G | 81 | +36 to +117 | |
| THBS1-U | TTG AGT TTG TGT GGT GTA AGA GTA T | CCC CAC TAC CTA ACA CAC AAC T | 156 | -280 to -124 | [26] |
| THBS1-M | GTT CGC GTG GCG TAA GAG TAC | CGC TAC CTA ACG CGC AAC T | 149 | -276 to -127 |
*exon 1
**promoter
Figure 4.

Progression to malignancy is associated with DNA hypermethylation at several gene loci. MSP analysis of CCND2, RASSF1A, SFRP1, p57, MGMT, and THBS1 in the cells indicated using primer sets listed in Table 1 that specifically amplify either methylated (m) or unmethylated (u) DNA. Positive (+) and (-) controls for the methylated product, as well as a H2O negative control, were included in all experiments. All MSP experiments were performed on at least two independent cell populations. The data presented for the SFRP1-exon1 MSP are from two different experiments. Analysis of the primary cells (HMEC, vHMEC, vector, ras, and ras0.5) and the cell lines (184A1, MCF10A, MCF7, and MDA231) was done separately and merged. The data presented for all the other genes were obtained in the same experiment. Replicate experiments are available online in Additional file 2.
Table 2.
Methylation-specific PCR analysis of genes methylated in the human mammary epithelial cell progression model and in mammary cell lines
| HMEC | vHMEC | vector | ras | ras0.5 | 184-A1 | MCF-10A | MCF-7 | MDA-231 | |
|---|---|---|---|---|---|---|---|---|---|
| p16 INK4A | U | M | M | M | M | M | M | M | M |
| HOXA9 | U | M | M | M | M | M | M | M | M |
| CCND2 | U | M | M | M | M | M | M | M | M |
| RASSF1A | U | U | U | U | U/M | M | M | M | M |
| SFRP1* | U | U | U | U | M | U/M | U/M | M | M |
| p57 | U | U | U | U | M | U | M | M | U |
| MGMT | U | U | U | U | U/M | U | U | U | U/M |
| THBS1 | U | U | U | U | U | U | U | U | U |
SFRP1* Methylation status of the CpG island located in exon 1 is shown.
In order to address this question further, we examined whether methylation of the RASSF1A and SFRP1 genes led to their silencing. As shown in Figure 5, qPCR analysis revealed that the expression of RASSF1A was reduced in all the cells that exhibited DNA methylation at the RASSF1A gene locus. Similarly, SFRP1 expression also correlated with DNA methylation in all the cell lines except the MCF-10A cells, where methylation was observed in the CpG island located in the 3' promoter region that extends into the first exon, but not in the CpG island located in the 5' promoter region (Figure 4), suggesting that methylation in the 5' promoter region may be required for silencing. Upon careful examination, we observed that the degree to which RASSF1A and SFRP1 expression was repressed, corresponded, for the most part, to the extent of DNA methylation, that is, in cells where complete methylation was observed (labeled as "M"), mRNA expression was significantly repressed (except in 184-A1 cells where approximately 50% of RASSF1A expression was retained). In contrast, in cells where both methylated and unmethylated products were observed (labeled as "U/M"), mRNA expression was reduced, but still present. This was also true for MGMT expression, which we examined for comparison (Figure 5c).
Figure 5.
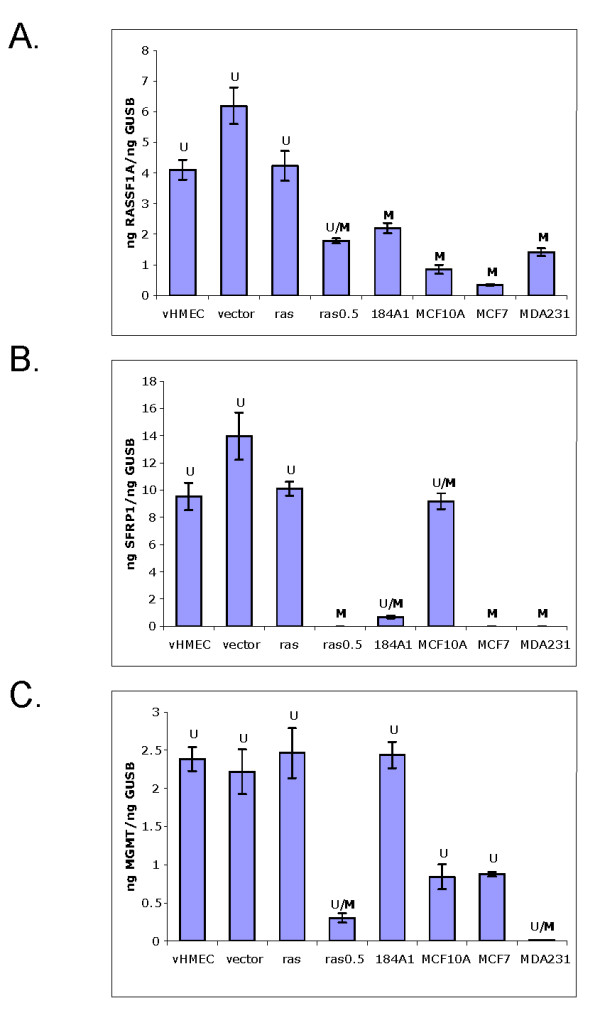
DNA methylation correlates with reduced gene expression. Quantitative RT-PCR for (a) RASSF1A; (b) SFRP1; and (c) MGMT. Methylation status as evaluated by MSP is indicated as follows: Unmethylated (U), partially methylated (U/M), or methylated (M). For SFRP1, the U/M labels refer to the methylation status of the CpG island that extends into exon 1.
Concomitant DNA methylation of RASSF1A and SFRP1 occurs in malignant and premalignant breast lesions
If concomitant DNA methylation of the RASSF1A and SFRP1 genes observed in vitro is biologically relevant in vivo, we reasoned that DNA methylation of these genes would also be present in breast cancer tissues, but not in normal breast cells or tissues. Therefore, we examined the methylation status of RASSF1A and SFRP1 in DNA isolated from 12 invasive ductal breast carcinomas (IDC), seven normal breast tissues, and three vHMEC populations isolated from disease-free reduction mammoplasties. As illustrated in Figure 6a and 6b, and summarized in Table 3, this analysis revealed that the RASSF1A locus was methylated in 10 out of 12 IDC, none of the vHMECs, and only one of the seven normal tissues (N3, where methylation was very faint). Similarly, the SFRP1 locus was methylated in all 12 IDC, none of the vHMECs, and only one of the seven normal tissues. Notably, the normal tissue in which DNA methylation was detected at the SFRP1 locus, was also the one in which faint DNA methylation at the RASSF1A locus was detected (N3), suggesting that the transformation process may already be initiated in these cells, despite their normal histological appearance. Concomitant methylation of RASSF1A and SFRP1 appears to be targeted and specific as the p57 and MGMT gene loci were methylated in none or only one of the IDC analyzed, respectively, and in none of the vHMECs or normal tissues (Figure 6a). These data demonstrate that the concomitant DNA methylation of the RASSF1A and SFRP1 genes observed upon immortalization of mammary epithelial cells in vitro, is biologically relevant in that it can also be detected in both breast cancer cell lines and in breast cancer tissues in vivo.
Figure 6.

Concomitant methylation of RASSF1A and SFRP1 in malignant and premalignant breast tissues. (a) MSP analysis of RASSF1A, SFRP1-exon1, p57, and MGMT in normal and malignant (invasive ductal carcinoma) breast tissues using primer sets that specifically amplify either methylated (m) or unmethylated (u) DNA, as in Figure 4 B and C. MSP analysis of RASSF1A and SFRP1 in normal, hyperplasia without atypia (H1 and H2), atypical ductal hyperplasia (ADH, H3), invasive ductal carcinoma (b), and ductal carcinomas in situ (c) tissues. Additional diagnostic information about these samples is available in Table 3.
Table 3.
Description of normal, premalignant, and malignant breast tissues utilized for MSP analysis along with methylation status of RASSF1A and SFRP1 in each tissue
| Sample | Description | RASSF1A | SFRP1 |
|---|---|---|---|
| N1 | Normal reduction mammoplasty tissue | U | U |
| N2 | Normal reduction mammoplasty tissue | U | U |
| N3 | Normal reduction mammoplasty tissue | M | M |
| N4 | Normal reduction mammoplasty tissue | U | U |
| N5 | Normal reduction mammoplasty tissue | U | U |
| N6 | Normal reduction mammoplasty tissue | U | U |
| N7 | Normal reduction mammoplasty tissue | U | U |
|
Total Normal Methylated |
1/7 | 1/7 | |
| H1 | Lobular hyperplasia (85% of mammary cells), blunt duct hyperplasia (15% of mammary cells) | U | U |
| H2 | Normal (80%), intraductal hyperplasia (10%), cystic hyperplasia (10%) | U | U |
|
Total Hyperplasia Methylated |
0/2 | 0/2 | |
| H3 | Focal atypical terminal duct hyperplasia (single focus); fibrocystic disease | M | M |
|
Total ADH Methylated |
1/1 | 1/1 | |
| D1 | CCL, DCIS | M | M |
| D2 | DCIS | M | M |
| D3 | DCIS, cysts, normal breast tissue | M | M |
| D4 | DCIS and normal tissue | U | M |
| D5 | Extensive comedo DCIS, calcifications | M | M |
| D6 | Extensive DCIS | M | M |
| D7 | Extensive DCIS and normal tissue | M | M |
| D8 | DCIS, calcifications | U | M |
| D9 | Extensive DCIS | M | M |
| D10 | DCIS with calcifications | M | M |
| D11 | Extensive comedo DCIS | M | M |
|
Total DCIS Methylated |
9/11 | 11/11 | |
| C1 | IDC, grade 2, node-, ER+ | M | M |
| C2 | IDC, grade 3, node+, ER- | M | M |
| C3 | IDC, grade 3, node+, ER- | U | M |
| C4 | IDC, grade 3, node-, ER- | M | M |
| C5 | IDC, grade 3, node+, ER+ | M | M |
| C6 | IDC, grade 1, node+, ER+ | M | M |
| C7 | IDC, grade 3, node+, ER- | M | M |
| C8 | IDC, DCIS, CCL, normal breast | M | M |
| C9 | IDC, DCIS | M | M |
| C10 | IDC, DCIS, normal breast | U | M |
| C11 | IDC and extensive DCIS | M | M |
| C12 | IDC and extensive comedo DCIS with calcifications | M | M |
|
Total IDC Methylated |
10/12 | 12/12 |
ADH = atypical ductal hyperplasia; CCL = columnar cell lesion; DCIS = ductal carcinoma in situ; IDC = invasive ductal carcinoma; ER = estrogen receptor; U = unmethylated; M = methylated. For SFRP1, methylation status of the CpG island located in exon 1 is shown.
In order to determine whether concomitant DNA methylation of the RASSF1A and SFRP1 genes marks an early event in mammary transformation, we analyzed the methylation status of these two genes in DNA obtained from early lesions. This analysis was performed on two cases of hyperplasia without atypia (H1 and H2), one case of hyperplasia with atypia (H3), and 11 cases of DCIS obtained from women with IDC (see Table 3 for a more detailed description of the samples analyzed). As shown in Figure 6b, DNA methylation was not detected in the hyperplasias without atypia at either gene locus (H1 and H2). In contrast, concomitant DNA methylation of RASSF1A and SFRP1 was detected in the atypical ductal hyperplasia (H3), which unlike the hyperplasias without atypia, is a premalignant lesion associated with risk for progression to cancer. In the DCIS samples, DNA methylation at the RASSF1A locus was observed in 9 of the 11 cases, and methylation of SFRP1 was observed in all 11 cases (Figure 6c and Table 3). These data demonstrate that concomitant DNA methylation of RASSF1A and SFRP1 occurs in premalignant lesions and may mark an early event in mammary transformation.
Discussion
Malignant progression involves an accumulation of multiple alterations in cellular growth control over an extended period of time. Many studies have shown that oncogenic ras can cooperate with other oncogenic alterations as well as viral oncoproteins to transform primary human cells [28,31]. However, whether oncogenic ras can cooperate with alterations in the extracellular environment to transform primary cells is unknown. Having observed that, unlike HMEC, vHMEC were resistant to the proliferative arrest induced by oncogenic ras and displayed an increase in genomic instability, we sought to determine whether signaling induced by the microenvironment could cooperate with oncogenic stress to transform vHMEC.
In cells isolated from one out of three individuals tested, extracellular stimulation with 0.5% serum at agonescence (which is a highly mutagenic growth plateau), led to the immortalization of vHMEC expressing oncogenic ras. In contrast, stimulation with serum during the exponential phase of growth (prior to agonescence) led to proliferative arrest in these cells. This suggests that an additional mutation, likely conferred by the genomic instability that is exhibited at agonescence [15], is required for serum to cooperate with ras in immortalizing vHMEC.
Altering the cellular microenvironment by simply changing culture conditions can have profound effects on cell behavior. Consistent with this, Shay and colleagues have demonstrated that HMEC expressing the catalytic subunit of telomerase can achieve immortality when grown on fibroblast feeder layers in the presence of 1% serum [32]. In addition, extended cultivation of mammary cells isolated from fibrocystic mammary tissue in growth media containing low concentrations of calcium is what led to the immortalization of the widely used non-tumorigenic MCF-10A mammary epithelial cell line [33]. These data highlight the importance of signals from the microenvironment in modulating cellular behaviors that can influence tumorigenesis.
Interestingly, transient alterations in the cellular environment can permanently alter cell behavior. For example, studies have shown that the non-tumorigenic BPH-1 prostatic epithelial cells can be induced to form tumors in nude mice when recombined with carcinoma-associated fibroblasts or when exposed to carcinogenic doses of steroid hormones. That newly acquired tumorigenicity is maintained in subsequent re-grafting of cells even in the absence of carcinoma-associated fibroblasts or steroid hormones [34]. Likewise, once immortalized, our vHMEC-ras0.5 cells no longer required sustained serum stimulation for continued proliferation, suggesting that signaling induced by serum led to additional alterations sufficient to maintain growth.
Among the alterations we observed in the immortalized vHMEC-ras0.5 cells were an upregulation in telomerase activity as well as DNA methylation at several gene loci, including the RASSF1A, SFRP1, p57, and MGMT gene loci. Two of these four genes, RASSF1A and SFRP1, were also methylated in four other immortalized mammary cell lines we examined, two of which were non-tumorigenic (184-A1 and MCF-10A), and two of which were tumorigenic (MCF-7 and MDA-231). In addition, concomitant DNA methylation of both genes was also observed in malignant (10 out of 12 IDC) and premalignant (1 ADH and 9 out of 11 DCIS) breast lesions. Interestingly, in the reduction mammoplasty tissues in which some hyperplasia without atypia was diagnosed (H1 and H2), no DNA methylation was detected. In contrast, in the reduction mammoplasty tissue with documented atypical ductal hyperplasia (H3), we observed concomitant methylation of RASSF1A and SFRP1 (Figure 6 and Table 3). While hyperplasias without atypia are not associated with any increased risk for developing breast cancer, atypical ductal hyperplasia (ADH) has been associated with a four- to five-fold increased risk of developing cancer [35], and is thus considered a premalignant lesion. Hence, we have analyzed 12 premalignant lesions (1 ADH and 11 DCIS) and found concomitant methylation of RASSF1A and SFRP1 in 10 of these 12 samples. Although these data need to be confirmed in a larger sample set, they strongly suggest that concomitant methylation of RASSF1A and SFRP1 marks an early event in mammary transformation and may thus have diagnostic and/or prognostic potential.
The concomitant methylation of RASSF1A and SFRP1 also provides possible insights into the biology of early transformation. The RASSF1A gene encodes ras association domain family member 1, and the SFRP1 gene encodes the Wnt signaling antagonist, secreted frizzled-related protein 1, both of which play an important role in cell proliferation [24,36,37]. Consistent with its role in regulating cell proliferation, RASSF1A has been reported to be the most frequently methylated gene in SV40- and hTERT-immortalized prostate epithelial cells [38]. Similarly, we have observed that RASSF1A is methylated in the SV40-immortalized human skin fibroblast cell line, GM847 (data not shown). These data suggest that epigenetic alterations in RASSF1A occur early in several cell types, not just mammary cells. This is further supported by the fact that RASSF1A-deficient mice display increased susceptibility to spontaneous tumor development in multiple organs, including breast, lung, and skin [39]. Compelling evidence suggests that activation of Wnt signaling plays an important role in breast cancer, and that loss of SFRP1 function is a key mechanism by which Wnt signaling is activated under such circumstances [24]. Like RASSF1A, SFRP1 has also been shown to be silenced through promoter methylation in breast cancer [12,24,40]. We now observe concomitant methylation of RASSF1A and SFRP1 in three immortalized but non-tumorigenic mammary cell lines as well as in premalignant breast lesions (ADH and DCIS), suggesting that these epigenetic alterations occur early in the transformation process.
Interestingly, we observed a correlation between SFRP1 DNA methylation and repression of its expression in all the cell lines we examined except the MCF-10A cells where both methylated and unmethylated DNA products were detected by MSP. While moderate levels of gene expression are expected under circumstances where only partial methylation is observed at a gene locus, the robust expression of SFRP1 in MCF-10A cells was not anticipated. Although we detected DNA methylation in the first exon of SFRP1 in MCF-10A cells, previous studies by Lo et al., have demonstrated that the promoter region of SFRP1 is unmethylated in these cells [40]. We therefore performed additional experiments with MSP primers designed to survey the methylation status of CpG islands located in the promoter region of SFRP1, as described in Lo et al., and confirmed the unmethylated status in that specific region (Figure 4). This supports the well established concept that the location of the CpG dinucleotides subjected to methylation plays an important role in determining whether genes are silenced or not. Consistent with this, CST6, which encodes the cysteine protease inhibitor, cystatin M, has been shown to be silenced following DNA methylation of CpG islands located within the promoter region, but not within exon 1 [41]. Of particular note, in MCF-7 and MDA-231 cells where SFRP1 expression is completely abrogated, we observed complete methylation in CpG islands located in the first exon and in the promoter region of SFRP1 in these cells. Interestingly, the frequency of DNA methylation in the promoter region and first exon seems to differ. While we observed DNA methylation in exon 1 of the SFRP1 gene in 100% of the DCIS and IDC samples we examined, methylation in the promoter region of SFRP1 has been reported to occur in approximately 68% of high-grade DCIS and invasive breast cancers [7,40]. These data suggest that there may be different functional consequences to DNA methylation at different CpG islands and that methylation events within a single gene can occur at different frequencies.
While DNA methylation is often associated with gene silencing, methylation marks that are not associated with gene silencing are nonetheless significant. For example, promoter methylation of TWIST1, an anti-apoptotic and pro-metastatic transcription factor, is significantly more prevalent in malignant breast tissue than in healthy breast tissue [42], and even more prevalent in metastatic lesions relative to matched primary cancers [43]. However, there is no correlation between TWIST1 promoter methylation and TWIST1 mRNA or protein expression [42]. Methylation of the TWIST1 promoter may lead to the recruitment of chromatin remodeling proteins that can alter the function and/or expression of neighboring genes. Alternatively, TWIST1 expression may be regulated by a balance between promoter and intragenic CpG methylation as there is evidence that intragenic CpG methylation can promote gene expression [44]. Thus, even though TWIST1 expression is not repressed by methylation, the compelling association between TWIST1 CpG methylation and malignancy makes it a useful biomarker for breast cancer diagnosis and prognosis. Likewise, SFRP1 methylation in the first exon may be of significance despite the lack of methylation in the 5' promoter region, and the absence of gene silencing. In support of this, recent studies have demonstrated that more than half of the methylated CpG islands in normal genomes fall within the body of the genes or in downstream regions [45]. In addition, numerous CpG islands mapping to intragenic or downstream regions of genes have been reported to be heavily methylated in DCIS, highlighting the significance of intragenic CpG island methylation in the early stages of breast cancer [13].
Conclusions
Cells without p16 function are resistant to arrest induced by ras-associated oncogenic stress. The accelerated accumulation of genetic and epigenetic events dictate their ability to bypass additional arrest signals allowing rare emergent subpopulations to immortalize, and grow in soft agar. We have identified a multigene methylation pattern acquired during this in vitro progression to malignancy that is detected in vivo in both premalignant and malignant lesions. This model will thus allow further study of the mechanisms underlying the accumulation of epigenetic alterations that occur during progression to malignancy. By characterizing the methylation profiles that manifest at different stages of transformation, biomarkers with diagnostic and/or prognostic value could eventually be identified.
Abbreviations
ADH: atypical ductal hyperplasia; BCA: bicinchoninic acid; CCL: columnar cell lesion; DCIS: ductal carcinoma in situ; ER: estrogen receptor; FITC: fluorescein isothiocyanate; GFP: green fluorescent protein; HMEC: human mammary epithelial cells; IDC: invasive ductal carcinoma; MSP: methylation-specific PCR; PBS: phosphate-buffered saline; TBS-T: tris-buffered saline containing tween; vHMEC: variant HMEC with repressed p16INK4A.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ND participated in the design of the studies, performed the immunoblot analysis, the serum-free cell culture experiments, the soft agar assays, the in vivo tumor studies, the bioluminescence imaging analysis, part of the MSP experiments, and wrote the manuscript. YGC generated the vHMEC cells expressing oncogenic ras and exposed them to serum. MBW initiated the MSP experiments, which were repeated and completed by MS and ND. SN performed the qPCR experiments, mapped all the MSP primers, and generated Table 1. GB, GT, and SA provided the premalignant and malignant tissue DNA samples. MLG generated the HMEC expressing oncogenic ras and performed the BrDU analysis. CAF performed the telomerase assay. KMM performed the centrosome analysis. TDT contributed to the conception and design of the experiments, and the critical revision of the manuscript. All authors read and approved the final manuscript.
Supplementary Material
A PowerPoint file containing a figure illustrating the growth curves of vHMEC-ras0.5 clone 1 cells and vHMEC-ras0.5 clone 1 cells expressing GFP and luciferase (clone 1 GFP-Lux), which indicates that expression of GFP and luciferase does not alter the growth characteristics of these cells.
A PowerPoint file containing a figure illustrating MSP analysis of RASSF1A and SFRP1 in vHMEC, ras0.5, and two different preparations of 184A1 cells, which demonstrates that both genes are methylated in ras0.5 and 184A1 cells, but unmethylated in vHMEC. The experiments were conducted using primer sets listed in Table 1 that specifically amplify either methylated (m) or unmethylated (u) DNA. Positive (+) and (-) controls for the methylated product, as well as a H2O negative control are shown.
Contributor Information
Nancy Dumont, Email: nancy.dumont@ucsf.edu.
Yongping G Crawford, Email: crawford.yongping@gene.com.
Mahvash Sigaroudinia, Email: mahvash.sigaroudinia@ucsf.edu.
Shefali S Nagrani, Email: shefali.nagrani@ucsf.edu.
Matthew B Wilson, Email: matthew.b.wilson@gmail.com.
Gertrude C Buehring, Email: buehring@calmail.berkeley.edu.
Gulisa Turashvili, Email: gturashv@bccrc.ca.
Samuel Aparicio, Email: saparicio@bccrc.ca.
Mona L Gauthier, Email: mona.gauthier@gmail.com.
Colleen A Fordyce, Email: Colleen.Fordyce@ucsf.edu.
Kimberly M McDermott, Email: kimberlymariemcdermott@gmail.com.
Thea D Tlsty, Email: thea.tlsty@ucsf.edu.
Acknowledgements
We would like to thank Krystyna Kozakiewicz for performing the chromosomal analysis, Chira Chen-Tanyolac for assistance with tumor DNA isolation, Dr. Sanjiv Sam Gambhir (Stanford University, Palo Alto, CA) for providing us with the GFP-luciferase lentiviral construct, and Dr. Philippe Gascard for critical discussions, review of the manuscript, and assistance with MSP primer mapping. This research was supported by the Susan G. Komen Foundation, postdoctoral award PDF124906 (ND), the NCI Institutional Training Grant T32 CA009043 (MBW), the Department of Defense Breast Cancer Research Program Concept Award BC023982 and 14OB-0165 (TDT), the NIH/NCI CA097214 and CA122024 grants (TDT).
References
- Theillet C, Lidereau R, Escot C, Hutzell P, Brunet M, Gest J, Schlom J, Callahan R. Loss of a c-H-ras-1 allele and aggressive human primary breast carcinomas. Cancer Res. 1986;46:4776–4781. [PubMed] [Google Scholar]
- Spandidos DA, Agnantis NJ. Human malignant tumours of the breast, as compared to their respective normal tissue, have elevated expression of the Harvey ras oncogene. Anticancer Res. 1984;4:269–272. [PubMed] [Google Scholar]
- Hand PH, Vilasi V, Thor A, Ohuchi N, Schlom J. Quantitation of Harvey ras p21 enhanced expression in human breast and colon carcinomas. J Natl Cancer Inst. 1987;79:59–65. [PubMed] [Google Scholar]
- De Biasi F, Del Sal G, Hand PH. Evidence of enhancement of the ras oncogene protein product (p21) in a spectrum of human tumors. Int J Cancer. 1989;43:431–435. doi: 10.1002/ijc.2910430315. [DOI] [PubMed] [Google Scholar]
- Ohuchi N, Thor A, Page DL, Hand PH, Halter SA, Schlom J. Expression of the 21,000 molecular weight ras protein in a spectrum of benign and malignant human mammary tissues. Cancer Res. 1986;46:2511–2519. [PubMed] [Google Scholar]
- Cheng X. Silent assassin: oncogenic ras directs epigenetic inactivation of target genes. Sci Signal. 2008;1:pe14. doi: 10.1126/stke.113pe14. [DOI] [PubMed] [Google Scholar]
- Veeck J, Niederacher D, An H, Klopocki E, Wiesmann F, Betz B, Galm O, Camara O, Durst M, Kristiansen G, Huszka C, Knüchel R, Dahl E. Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene. 2006;25:3479–3488. doi: 10.1038/sj.onc.1209386. [DOI] [PubMed] [Google Scholar]
- Munot K, Bell SM, Lane S, Horgan K, Hanby AM, Speirs V. Pattern of expression of genes linked to epigenetic silencing in human breast cancer. Hum Pathol. 2006;37:989–999. doi: 10.1016/j.humpath.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Ordway JM, Budiman MA, Korshunova Y, Maloney RK, Bedell JA, Citek RW, Bacher B, Peterson S, Rohlfing T, Hall J, Brown R, Lakey N, Doerge RW, Martienssen RA, Leon J, McPherson JD, Jeddeloh JA. Identification of novel high-frequency DNA methylation changes in breast cancer. PLoS ONE. 2007;2:e1314. doi: 10.1371/journal.pone.0001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeck J, Geisler C, Noetzel E, Alkaya S, Hartmann A, Knuchel R, Dahl E. Epigenetic inactivation of the Secreted frizzled-related protein-5 (SFRP5) gene in human breast cancer is associated with unfavorable prognosis. Carcinogenesis. 2008;29:991–998. doi: 10.1093/carcin/bgn076. [DOI] [PubMed] [Google Scholar]
- Kobatake T, Yano M, Toyooka S, Tsukuda K, Dote H, Kikuchi T, Toyota M, Ouchida M, Aoe M, Date H, Pass HI, Doihara H, Shimizu N. Aberrant methylation of p57KIP2 gene in lung and breast cancers and malignant mesotheliomas. Oncol Rep. 2004;12:1087–1092. [PubMed] [Google Scholar]
- Fackler MJ, McVeigh M, Evron E, Garrett E, Mehrotra J, Polyak K, Sukumar S, Argani P. DNA methylation of RASSF1A, HIN-1, RAR-beta, Cyclin D2 and Twist in in situ and invasive lobular breast carcinoma. Int J Cancer. 2003;107:970–975. doi: 10.1002/ijc.11508. [DOI] [PubMed] [Google Scholar]
- Tommasi S, Karm DL, Wu X, Yen Y, Pfeifer GP. Methylation of homeobox genes is a frequent and early epigenetic event in breast cancer. Breast Cancer Res. 2009;11:R14. doi: 10.1186/bcr2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford YG, Gauthier ML, Joubel A, Mantei K, Kozakiewicz K, Afshari CA, Tlsty TD. Histologically normal human mammary epithelia with silenced p16(INK4a) overexpress COX-2, promoting a premalignant program. Cancer Cell. 2004;5:263–273. doi: 10.1016/S1535-6108(04)00023-6. [DOI] [PubMed] [Google Scholar]
- Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, Tlsty TD. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature. 2001;409:633–637. doi: 10.1038/35054579. [DOI] [PubMed] [Google Scholar]
- Reynolds PA, Sigaroudinia M, Zardo G, Wilson MB, Benton GM, Miller CJ, Hong C, Fridlyand J, Costello JF, Tlsty TD. Tumor suppressor p16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J Biol Chem. 2006;281:24790–24802. doi: 10.1074/jbc.M604175200. [DOI] [PubMed] [Google Scholar]
- McDermott KM, Zhang J, Holst CR, Kozakiewicz BK, Singla V, Tlsty TD. p16(INK4a) prevents centrosome dysfunction and genomic instability in primary cells. PLoS Biol. 2006;4:e51. doi: 10.1371/journal.pbio.0040051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst CR, Nuovo GJ, Esteller M, Chew K, Baylin SB, Herman JG, Tlsty TD. Methylation of p16(INK4a) promoters occurs in vivo in histologically normal human mammary epithelia. Cancer Res. 2003;63:1596–1601. [PubMed] [Google Scholar]
- Gauthier ML, Pickering CR, Miller CJ, Fordyce CA, Chew KL, Berman HK, Tlsty TD. p38 regulates cyclooxygenase-2 in human mammary epithelial cells and is activated in premalignant tissue. Cancer Res. 2005;65:1792–1799. doi: 10.1158/0008-5472.CAN-04-3507. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Hammond SL, Ham RG, Stampfer MR. Serum-free growth of human mammary epithelial cells: rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc Natl Acad Sci USA. 1984;81:5435–5439. doi: 10.1073/pnas.81.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Guan M, Su B, Liu W, Xu M, Lu Y. Hypermethylation of the RASSF1A gene in gliomas. Clin Chim Acta. 2004;349:173–179. doi: 10.1016/j.cccn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Toyota M, Caraway H, Gabrielson E, Ohmura T, Fujikane T, Nishikawa N, Sogabe Y, Nojima M, Sonoda T, Mori M, Hirata K, Imai K, Shinomura Y, Baylin SB, Tokino T. Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer. 2008;98:1147–1156. doi: 10.1038/sj.bjc.6604259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petko Z, Ghiassi M, Shuber A, Gorham J, Smalley W, Washington MK, Schultenover S, Gautam S, Markowitz SD, Grady WM. Aberrantly methylated CDKN2A, MGMT, and MLH1 in colon polyps and in fecal DNA from patients with colorectal polyps. Clin Cancer Res. 2005;11:1203–1209. [PubMed] [Google Scholar]
- Guerrero D, Guarch R, Ojer A, Casas JM, Ropero S, Mancha A, Pesce C, Lloveras B, Garcia-Bragado F, Puras A. Hypermethylation of the thrombospondin-1 gene is associated with poor prognosis in penile squamous cell carcinoma. BJU Int. 2008;102:747–755. doi: 10.1111/j.1464-410X.2008.07603.x. [DOI] [PubMed] [Google Scholar]
- Dimri G, Band H, Band V. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res. 2005;7:171–179. doi: 10.1186/bcr1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, Rijn M van de, Botstein D, Brown PO. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6:171–183. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Herbert BS, Wright WE, Shay JW. p16(INK4a) inactivation is not required to immortalize human mammary epithelial cells. Oncogene. 2002;21:7897–7900. doi: 10.1038/sj.onc.1205902. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, Sudilovsky D, Cunha GR. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61:8135–8142. [PubMed] [Google Scholar]
- Page DL, Dupont WD, Rogers LW, Rados MS. Atypical hyperplastic lesions of the female breast. A long-term follow-up study. Cancer. 1985;55:2698–2708. doi: 10.1002/1097-0142(19850601)55:11<2698::AID-CNCR2820551127>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Thaler S, Hahnel PS, Schad A, Dammann R, Schuler M. RASSF1A Mediates p21Cip1/Waf1-Dependent Cell Cycle Arrest and Senescence through Modulation of the Raf-MEK-ERK Pathway and Inhibition of Akt. Cancer Res. 2009. [DOI] [PubMed]
- Shivakumar L, Minna J, Sakamaki T, Pestell R, White MA. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol Cell Biol. 2002;22:4309–4318. doi: 10.1128/MCB.22.12.4309-4318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang J, Bates S, Li JJ, Peehl DM, Rhim JS, Pfeifer GP. A methylation profile of in vitro immortalized human cell lines. Int J Oncol. 2005;26:275–285. [PubMed] [Google Scholar]
- Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP. Tumor susceptibility of Rassf1a knockout mice. Cancer Res. 2005;65:92–98. [PubMed] [Google Scholar]
- Lo PK, Mehrotra J, D'Costa A, Fackler MJ, Garrett-Mayer E, Argani P, Sukumar S. Epigenetic suppression of secreted frizzled related protein 1 (SFRP1) expression in human breast cancer. Cancer Biol Ther. 2006;5:281–286. doi: 10.4161/cbt.5.3.2384. [DOI] [PubMed] [Google Scholar]
- Rivenbark AG, Livasy CA, Boyd CE, Keppler D, Coleman WB. Methylation-dependent silencing of CST6 in primary human breast tumors and metastatic lesions. Exp Mol Pathol. 2007;83:188–197. doi: 10.1016/j.yexmp.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gort EH, Suijkerbuijk KP, Roothaan SM, Raman V, Vooijs M, Wall E van der, van Diest PJ. Methylation of the TWIST1 promoter, TWIST1 mRNA levels, and immunohistochemical expression of TWIST1 in breast cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:3325–3330. doi: 10.1158/1055-9965.EPI-08-0472. [DOI] [PubMed] [Google Scholar]
- Mehrotra J, Vali M, McVeigh M, Kominsky SL, Fackler MJ, Lahti-Domenici J, Polyak K, Sacchi N, Garrett-Mayer E, Argani P, Sukumar S. Very high frequency of hypermethylated genes in breast cancer metastasis to the bone, brain, and lung. Clin Cancer Res. 2004;10:3104–3109. doi: 10.1158/1078-0432.CCR-03-0118. [DOI] [PubMed] [Google Scholar]
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, Jackson D, Clee C, Plumb R, Rogers J, Humphray S, Cox T, Langford C, Bird A. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A PowerPoint file containing a figure illustrating the growth curves of vHMEC-ras0.5 clone 1 cells and vHMEC-ras0.5 clone 1 cells expressing GFP and luciferase (clone 1 GFP-Lux), which indicates that expression of GFP and luciferase does not alter the growth characteristics of these cells.
A PowerPoint file containing a figure illustrating MSP analysis of RASSF1A and SFRP1 in vHMEC, ras0.5, and two different preparations of 184A1 cells, which demonstrates that both genes are methylated in ras0.5 and 184A1 cells, but unmethylated in vHMEC. The experiments were conducted using primer sets listed in Table 1 that specifically amplify either methylated (m) or unmethylated (u) DNA. Positive (+) and (-) controls for the methylated product, as well as a H2O negative control are shown.