

Abstract
The bifunctional enzyme UDP-GlcNAc 2-epimerase/ ManNAc kinase (GNE/MNK), encoded by the GNE gene, catalyzes the first two committed, rate-limiting steps in the biosynthesis of N-acetylneuraminic acid (sialic acid). GNE/MNK is feedback inhibited by binding of the downstream product, CMP-sialic acid in its allosteric site. GNE mutations can result in two human disorders, hereditary inclusion body myopathy (HIBM) or sialuria. So far, no active site geometry predictions or conformational transitions involved with function are available for mammalian GNE/MNK. The N-terminal GNE domain is homologous to various prokaryotic 2-epimerases, some of which have solved crystallographic structures. The C-terminal MNK domain belongs to the sugar kinases superfamily; its crystallographic structure is solved at 2.84 Å and three-dimensional structures have also been reported for several other kinases. In this work, we employed available structural data of GNE/MNK homologs to model the active sites of human GNE/MNK and identify critical amino acid residues responsible for interactions with substrates. In addition, we modeled effects of GNE/MNK missense mutations associated with HIBM or sialuria on helix arrangement, substrate binding, and enzyme action. We found that all reported mutations are associated with the active sites or secondary structure interfaces of GNE/MNK. The Persian-Jewish HIBM founder mutation p.M712T is located at the interface α4α10 and likely affects GlcNAc, Mg2+, and ATP binding. This work contributes to further understanding of GNE/MNK function and ligand binding, which may assist future studies for therapeutic options that target misfolded GNE/MNK in HIBM and/or sialuria.
Keywords: GNE, MNK, homology model, missense mutation, protein structure, sialic acid
Introduction
Uridine diphosphate (UDP)-N-acetylglucosamine (GlcNAc) 2-epimerase/N-acetylmannosamine (ManNAc) kinase (GNE/ MNK) is a bifunctional enzyme of sialic acid biosynthesis encoded by the GNE gene (Hinderlich et al. 1997; Eisenberg et al. 2001). One domain (GNE) carries out epimerase function; the other domain (MNK) is responsible for kinase activity. GNE mutations can result in two human disorders, hereditary inclusion body myopathy (HIBM) or sialuria (see Table I) (Huizing 2005).
Table I.
GNE mutations (GenBank NM_005476) resulting in HIBM (white background) or sialuria (gray background)
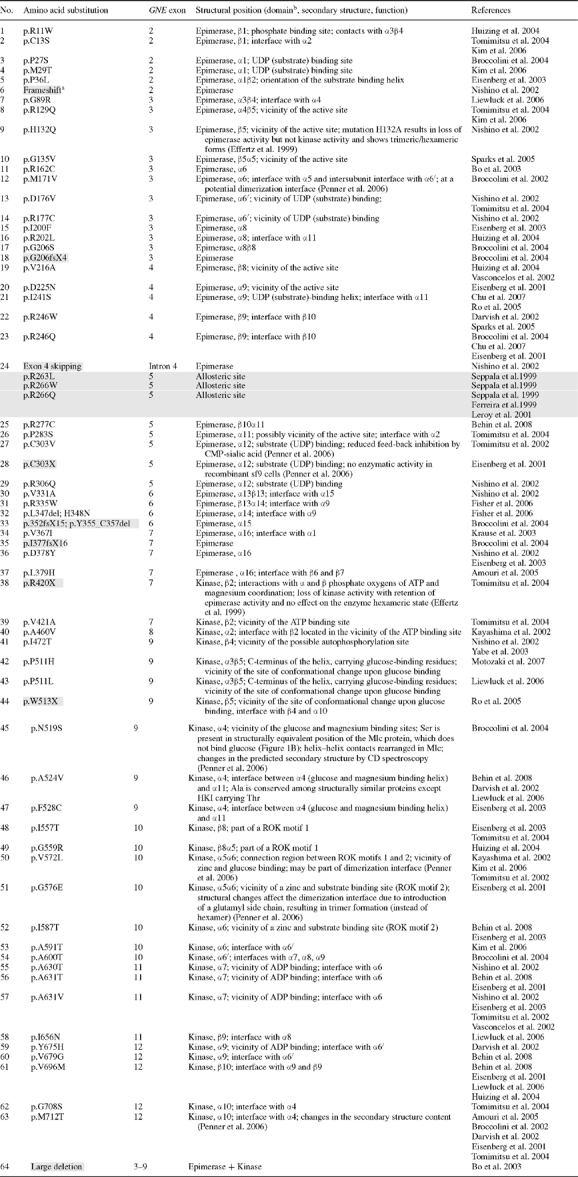 |
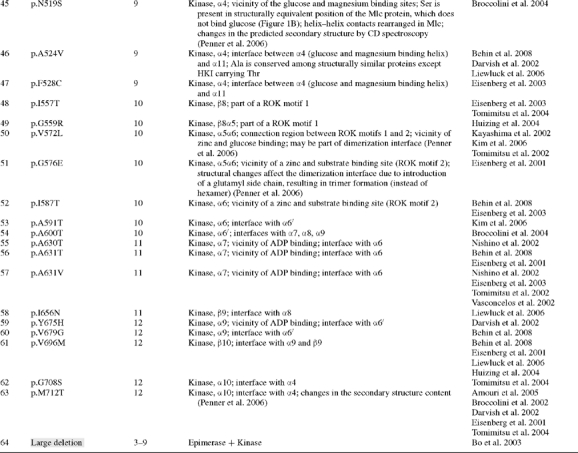 |
aGray shaded; severe mutations, likely resulting in nonsense mediated RNA decay and limited GNE/MNK protein expression.
bGNE/MNK amino acid residues 1–378 are suggested to regulate epimerase enzymatic activity, and residues 410–722 regulate kinase enzymatic activity (Effertz et al. 1999).
In mammalian GNE/MNK, the end product of sialic acid synthesis, CMP-sialic acid, feedback-inhibits GNE-epimerase activity by binding in the allosteric site of GNE/MNK. This feedback-inhibition mechanism is significantly compromised in sialuria patients, resulting in cytoplasmic accumulation and urinary excretion of large quantities of free sialic acid (Seppala et al. 1991; Leroy et al. 2001). Sialuria is an autosomal dominant disorder; all known sialuria patients are heterozygous mutated for a missense mutation in one of two amino acids, arginine at position 263 (R263L) or arginine at position 266 (R266Q; R266W) (see Table I) (Enns et al. 2001; Ferreira et al. 1999; Seppala et al. 1999; Leroy et al. 2001). The clustering of these mutations in the region of codons 263–266 supports the hypothesis that this region is part of the allosteric site for CMP-sialic acid binding. However, the exact dimensions of the allosteric site remain to be determined. Additional reports suggested experimental ‘sialuria’ mutations; the C303V mutation showed reduced feedback inhibition by CMP-sialic acid (Penner et al. 2006), and amino acids D255, E260, K268, and N275 were found to be mutated in an experimental mutational screen for cells with high levels of sialic acid (Yarema et al. 2001).
In contrast, HIBM is an autosomal recessive disorder; patients harbor two GNE mutations outside the enzyme's allosteric site, in either the GNE and/or MNK coding domains (Eisenberg et al. 2003). HIBM mutations lead to decreased GNE and MNK enzymatic activities (Effertz et al. 1999; Sparks et al. 2005, Penner et al. 2006) and, in some patients, decreased sialylation of glycoproteins (Huizing et al. 2004; Tajima et al. 2005). To date, more than 60 HIBM-associated GNE mutations (predominantly missense, see Table I) are identified. Mutations in one enzymatic domain also affect the activity of the other domain (Sparks et al. 2005; Penner et al. 2006). GNE/MNK exists in two major oligomeric states, tetramers and dimers, which are in a dynamic interplay with monomers and higher aggregates. The fully functional tetrameric state of GNE/MNK is stabilized by ligands of the GNE domain, UDP-N-acetylglucosamine and CMP-N-acetylneuraminic acid (Ghaderi et al. 2007).
The GNE/MNK allosteric site appears to exist only in the mammalian enzyme; prokaryotic 2-epimerases have no allosteric feedback inhibition. In prokaryotes, epimerase and kinase functions are carried out by two separate enzymes. In mammals, a bifunctional enzyme may have evolved by gene fusion of the two independent enzymes responsible for epimerase and kinase activity. Similarities between mammalian GNE/MNK N-terminal regions with prokaryotic UDP-GlcNAc 2-epimerases and mammalian C-terminal regions with members of the sugar kinase superfamily assisted in identifying several characteristic motifs of the GNE and MNK enzymatic domains (Effertz et al. 1999).
Assessment of the structural changes introduced by a selected group of HIBM mutations was previously performed using CD spectroscopy and gel filtration analysis together with three-dimensional modeling based on Escherichia coli GNE and E. coli glucokinase three-dimensional structures (Penner et al. 2006). In the current study, we build on the previous study by employing all available structural data of GNE/MNK homologs together with interhelical angle calculations to model the structure and active sites of the GNE/MNK enzyme. In addition, we modeled effects of all reported missense mutations to date associated with HIBM or sialuria on interhelical arrangements, substrate binding, and enzyme action. These data contribute to predicting the pathogenicity of newly identified GNE/MNK mutations and are invaluable for further understanding of GNE/MNK ligand binding and function, which may assist in designing future studies for therapeutic options targeting mutated GNE/MNK in HIBM and/or sialuria.
Results
Three-dimensional model of the GNE domain
A three-dimensional structure of the GNE domain of the GNE/MNK enzyme was assembled using different tools. First, amino acid similarities between the Homo sapiens GNE/MNK enzyme and Vibrio cholera (27% homology), E. coli (20% homology), Bacillus subtilis (25% homology), Bacillus anthracis (18% homology), and Thermus thermophilus (21% homology) 2-epimerases (Figure 1A) were considered. Second, the crystallographic structures of the E. coli GNE enzyme unbound and in complex with UDP-N-acetylglucosamine (pdb code 1f6d, 1vgv), the V. cholera enzyme (pdb code 1dzc), B. subtilis enzyme (pdb code 1o6c), and the B. anthracis enzyme in complex with reaction intermediate UDP and with allosteric activator UDP-N-acetylglucosamine (pdb code 3beo) were used to identify residues involved in ligand binding in the H. sapiens GNE domain. Third, similarity of the E. coli GNE enzyme to phosphoglycosyl transferases was employed, including glycogen phosphorylase (GP), phage T4 glycosyltransferase (BGT), and bacterial wall peptidoglycan hydrolase MurG (Campbell et al. 2000). Tertiary structures of these enzymes contain the same fold while amino acid sequences differ strongly. All secondary structure elements except α2 and α8 can be superimposed in spite of less than 10% sequence identity between BGT or GP and GNE. And fourth, amino acids at helix–helix interfaces were considered to predict possible changes in the arrangement of helices in the human GNE domain of the GNE/MNK enzyme compared to homologous and distantly related structures.
Fig. 1.

Sequence alignments of GNE/MNK. (A) Sequence alignments of mammalian and prokaryotic 2-epimerases (similar to the mammalian GNE/MNK N-terminal domain) and hexokinases (similar to the GNE/MNK C-terminal domain). Amino acids mutated in HIBM (G135, V216, R246, A631, M712, etc.), histidines, mutations of which result in loss of epimerase activity, and absolutely conserved in pro- and eukaryotic organisms ligand binding amino acids are in bold. Secondary structure assignments are shown (α-helices green and β-strands blue). For the epimerase domain, amino acids involved in allosteric regulation (#) in the B. anthracis enzyme and forming an active site (°) in the E. coli enzyme are shown in the upper line. For the kinase domain, amino acids of β1, β3α1, β4α3, β5α4, β8α5 loops involved in glucose binding (*), ADP binding (+), N-acetyl binding (^), and Mg2+ coordination (!) in S. tokodaii hexokinase /2e2o/ are shown in the upper line. Predicted allosteric site residues of the human enzyme and two phosphate binding motifs are underlined. The ATP phosphate binding motif (dark gray) and two characteristic ROK Zn2+ binding motifs (light gray) are shaded. (B) Sequence homology between parts of human GNE/MNK (upper line)/3eo3/ and the glucose binding helices of Arthrobacter sp. glucomannokinase (GMK)/1woq/, S. tokodaii hexokinase /2e2o/, and human hexokinase I (HKI)/1dgk/. Identical residues are in bold. Glucose binding residues are underlined.
Similarities between H. sapiens GNE, bacterial 2-epimerases, and phosphoglycosyl transferases (Figure 1A) and the presence of common ligands suggest that the GNE domain of the human GNE/MNK enzyme possess a similar structure, which contains two α/β domains that form a cleft at the domain interface harboring the active site. The topology of both domains is similar to the Rossmann dinucleotide binding fold (Rao and Rossmann 1973). The N-terminal part of the GNE domain has a 7-stranded parallel β-sheet β1–β7 sandwiched between a total of seven α-helices (Figure 1A, Table II). The C-terminal part of the GNE domain contains a 6-stranded β-sheet, β8–β13, surrounded by a total of seven α-helices α8–α15 (Figure 1A, Table II). Interfaces between secondary structure elements of 2-epimerases consist of α–α (between a pair of α-helices) and α–β (between α-helix and β-sheet) types. Modeling of interfaces between α-helices shows that in spite of low overall homology (18–27%) between H. sapiens 2-epimerase and bacterial 2-epimerases, homology at helix–helix interfaces is higher (42%) indicating that these proteins have similar folds. For example, interactions between α-helices α3 and α4 show homology among all aligned structures with interhelical angles being between −53 and −43 degrees (see Material and methods; Modeling of GNE/MNK interhelical angles). This α3α4 interface also belongs to the same group of interfaces as those found in TIM-barrel proteins Mycobacterium tuberculosis 2-isopropyl-malate synthase and Aeropyrum pernix deoxyribose phosphate aldolase (Kurochkina 2007). A detailed structure of interfaces that are not conserved has yet to be determined.
Table II.
Secondary structure assignments and topology of the β-structures in the GNE/MNK enzyme
| GNE (predicted model) | MNK (crystallographic structure 3eo3) | ||||||
|---|---|---|---|---|---|---|---|
| Domain I | Domain II | Domain I (small) | Domain II (large) | ||||
| ↑ ↑ ↑ ↑ ↑ ↑ ↓ | ↑ ↑ ↑ ↑ ↑ ↑ | ↑ ↓ ↑ ↑ ↑ | ↑ ↓ ↑ ↑ ↑ | ||||
| β3 β2 β1 β4 β5 β6 β7 | β10 β9 β8 β11 β12 β13 | β3 β2 β1 β4 β5 | β8 β7 β6 β9 β10 | ||||
| Amino acid # | Amino acid # | Amino acid # | Amino acid # | ||||
| α8 | H199-M203 | β1 | S408-L414 | β6 | F537-T542 | ||
| β1 | L10-T16 | β8 | K212-L218 | β2 | N418-S425 | β7 | I546-H552 |
| α1 | N18-E35 | α9 | P220-S242 | β3 | I430-F437 | β8 | E555-L556 |
| β2 | E40-L46 | β9 | K245-L248 | α1α2 | T441-L463 | α5 | E566-L570 |
| α2 | H49-E62 | α10 | F249-V260 | α6 | C586-S592 | ||
| β3 | F66-D67 | β10 | H273-F276 | α6′ | S592-E606 | ||
| α3 | D80-K103 | α11 | K280-H293 | α7 | G623-L633 | ||
| β4 | I106-H110 | β11 | C296-I298 | α8 | N635-N661 | ||
| α4 | R113-M126 | α12 | S301-F311 | β4 | I467-S473 | β9 | L664-S668 |
| β5 | I130-I133 | β12 | P314-I316 | α3 | L500-H509 | α9 | L671-Q692 |
| α5 | D143-A154 | α13 | Q322-G328 | β5 | V512-D515 | β10 | D695-V698 |
| β6 | Y156-V158 | β13 | L332-V334 | α4 | N516-F528 | ||
| α6 | T161-M171 | α14 α15 | D338-S358 | α10 | D703-T717 | ||
| α6′ | D174-R177 | ||||||
| β7 | I178-L180 | ||||||
| α16 | A366-L379 | α7 | T184-V193 | ||||
Predicted active site of the GNE domain
The active site of the GNE domains of prokaryotic epimerases contains about 10 conserved residues (Campbell et al. 2000). In the E. coli enzyme, amino acid residues recognized for importance for catalysis can be subdivided into several groups: (1) residues responsible for stabilization of the active site structure (P11, E12, G94, H115, G119, and E132); (2) residues facilitating interdomain rotation located in the interdomain hinge region (G170, D175); (3) residues binding the UDP portion of the substrate (R10, S290, E296); (4) residues forming the vicinity of the active site (K15, D95, E117, E131, R135, P18, V20, E132, G291, T96, Q271, Y273, F276), and (5) possible general catalyst H213 (Campbell et al. 2000).
Since most of these residues are conserved in the H. sapiens 2-epimerase GNE domain, they may play a similar role in the human GNE/MNK enzyme. H. sapiens GNE/MNK residues corresponding to E. coli GNE residues may be assigned similar structural roles and can be subdivided into the same functional groups: (1) residues A20, D21, G111, H132, G136, and D144 (corresponding to residues P11, E12, G94, H115, G119, and E132 of the E. coli enzyme) responsible for stabilization of the active site structure; (2) residues G182 and D187 in the interdomain hinge region that facilitate interdomain rotation (corresponding to E. coli residues G170 and D175); (3) residues R19, S301, and E307 binding the UDP portion of the substrate (R10, S290, and E296 of the E. coli 2-epimerase); (4) residues K24, D112, E134, D143, R147, P27, M29, D144, S302, R113, forming the vicinity of the active site (K15, D95, E117, E131, R135, P18, V20, E132, G291, T96, Q271, Y273, F276 of the E. coli 2-epimerase), (5) possible general catalyst H220 (H213 of the E. coli 2-epimerase), and (6) H45 (H45 of the rat 2-epimerase) necessary for epimerase reaction.
Residues H45 and H132 of the rat GNE (98% homologous to H. sapiens GNE, residues 45 and 132 are also histidines in H. sapiens GNE), which correspond to H39 and H115 of the E. coli enzyme, were found to be important for epimerase activity (Effertz et al. 1999). Mutation H132A resulted in loss of epimerase activity but not of kinase activity nor loss of the formation of trimeric/hexameric forms of the enzyme suggesting its involvement partially in the formation of the enzyme oligomeric state. Mutation H45A resulted in loss of epimerase activity but not of kinase activity nor loss of the formation of hexameric forms of the enzyme indicating its direct involvement in the epimerization process (Effertz et al. 1999).
Based on the above data, the following residues are part of the putative active site of the human 2-epimerase enzyme activity of GNE/MNK: R19, A20, D21, K24, P27, M29, H45 G111, D112, H132, E134, G136, D143, D144, D147, D182, D187, H220, S301, G302, and E307 (also shown in Figures 1A, 3, and Table III).
Fig. 3.

The N-acetylmannosamine kinase (MNK) domain of the human GNE/MNK protein. (A) Mutated residues Met712 at the interface of the α-helices (green) and β-sheets (blue), Asn519, Gly576, Ile587, Ile472, Ser473 and proposed glucose binding residues Asn516, Asp517, Glu566 are shown (oxygen – red, nitrogen – dark blue, carbon – gray/black, and sulfur – yellow). The two ATP binding motifs characteristic for sugar kinases are in magenta; glucose binding helices, C-terminal part of ROK1 motif and N-terminal region of ROK2 motif are gray and cyan ribbons. (B) Amino acid sequence alignment of the two interacting helices α4 and α10 containing GNE mutations M712T and A524V of H. sapiens MNK, S. tokodaii, H. sapiens (HKI) and S. cereviciae (PII) hexokinases, A. sp glucomannokinase, H. sapiens GlcNAc kinase and E. coli, Plasmodium falciparum and Thermococcus kodakarensis glycerol kinases. For selelomethionine, m is used. (C) Helices α4 (dark gray) and α10 (light gray) of H. sapiens MNK, S. tokodaii hexokinase, H. sapiens hexokinase, and E. coli glycerol kinases superimposed.
Table III.
Active sites and allosteric sites of the GNE/MNK enzyme
| Site | Function | Domain | Residues | References |
|---|---|---|---|---|
| 2-Epimerase active site | Stabilization of the active site structure | GNE | D21, G111, H132, G136, D144 | E12, G94, H115, G119, E132 of E.coli 2-epimerase (Campbell et al. 2000) |
| Interdomain hinge region | GNE | G182, D187 | G170, D175 of E.coli 2- epimerase (Campbell et al. 2000) | |
| Binding of the UDP portion of the substrate | GNE | R19, S301, E307 | R10, S290, E296 of E. coli 2-epimerase (Campbell et al. 2000) | |
| Vicinity of the active site | GNE | K24, P27, M29, D112, E134, D143, D144, R147, S302, R113 | K15, P18, V20, D95, E117, E131, E132, R135, G291 (Campbell et al. 2000), T96, Q271, Y273, F276 of E. coli 2-epimerase (Badger et al. 2005) | |
| Possible general catalyst | GNE | H220 | H213 of E. coli 2-epimerase (Campbell et al. 2000) | |
| Required for 2-epimerase activity | GNE | H45, H132 | H45, H132 of rat 2-epimerase (Effertz et al. 1999) | |
| Kinase active site | ATP binding motifs DXGGT and GTG | MNK | G416, G545 | G11, G117 of S. tokodaii hexokinase (Nishimazu et al. 2007) |
| Interaction with ATP-complexed Mg2+ | MNK | D413 | (Effertz et al. 1999) | |
| Interaction with α and β phosphate oxygens | MNK | R420 | ||
| Glucose binding | MNK | N516, E566, E588 | N122, E168, E180 of Arthrobacter sp. Glucomannokinase (Mukai et al. 2004) | |
| Abstraction of a proton from the 6-hydroxyl group of glucose as a catalytic base | MNK | D517 | D95 of Sulfolobas tokodaii hexokinase (Nishimazu et al. 2007) | |
| Zn2+ binding site | MNK | H569, C579, C581, C586 | (Tong et al. 2009) | |
| Allosteric site | Allosteric regulation by CMP-sialic acid | GNE | D255, E260, R263, R266, K268, N275 | (Ferreira et al. 1999; Seppala et al. 1999; Leroy et al. 2001; Enns et al. 2001; Yarema et al. 2001; Penner et al. 2006) |
Predicted active site of the MNK domain
The active site of the kinase domain (MNK) of human GNE/MNK was predicted using sequence alignment and structural alignment tools with previously studied hexokinases and glucokinases. In addition, we used the crystallographic structure of the MNK domain, which was determined in the ligand-free state (Tong et al. 2009). The three-dimensional structure of Sulfolobus tokodaii hexokinase was determined in the apo form and in complex with glucose, ADP, and xylose/Mg2+/ADP (Nishimazu et al. 2007). This hexokinase enzyme is known to phosphorylate glucose, GlcNAc, mannose, and glucosamine. It binds glucose via the β3α1 and β4α3 loops of the small kinase domain and the β5α4 and β8α5 loops of the large kinase domain. Glucose hydroxyl groups form hydrogen bonds with S. tokodaii hexokinase G69, D71, H94, D95, G135, D140 at N-termini of the α-helices α3[72–84], α4[95–105], and α5[143–160] and Y189 within the helix α6[180–192] from the adjacent subunit. Water-mediated hydrogen bonds are formed by G11 (β1β2), N35 (α1), and G117 (β6β7) (Nishimazu et al. 2007). Corresponding elements of the human GNE/MNK would be S477, D499, N516, D517, S561, E566, associated with α-helices α3[500–509], α4[516–528], and α5[566–570] (Figure 1). S. tokodaii hexokinase amino acids G11 and G117 align with human GNE/MNK residues G416 and G545, which are located in the DXGGT ATP phosphate binding and GTG Zn2+ binding conserved motifs, respectively (Figure 1). The DXGGT ATP phosphate binding motif (residues 412–417) falls within the β-strand β1 of MNK, where residue D413 is proposed to interact with ATP-complexed Mg2+ and residue R420, located within β-strand β2, may interact with α and β phosphate oxygens (Effertz et al. 1999).
Besides these motifs, homology between S. tokodaii hexokinase and human MNK can be seen mainly in the region of MNK domain helix α4 and its interface with helix α10. In addition, the structure of very similar H. sapiens GlcNAc kinase in complex with GlcNAc (pdb code 2ch5) provides data on the N-acetyl portion of the ligand (Weihofen et al. 2006).
Comparison of human GNE/MNK with Arthrobacter sp. glucomannokinase (GMK, pdb code 1woq) reveals a high number of identical residues in the glucose binding helices α4 and α5 including their N-terminal loop regions (Figure 1B). Therefore, it suggests that E566 of human GNE/MNK, which corresponds to E168 of glucomannokinase, might be a glucose binding amino acid (Figure 3). In spite of low sequence homology (28% identical residues), the Arthrobacter sp. glucomannokinase and the human GNE/MNK enzyme have very similar tertiary structure (Figure 4A). The active site residues interacting with glucose hydroxyl groups and phosphates in Arthrobacter sp. glucomannokinase exhibit a high degree of conservation with residues proposed to carry out the same function in the human GNE/MNK enzyme (Figure 4B).
Fig. 4.

Tertiary structure of GNE/MNK. Superposition of the three-dimensional structures of the Arthrobacter sp. glucomannokinase (magenta ribbon) in complex with glucose and phosphates and the human GME/MNK enzyme (gray ribbon). Side chains of the active site residues (wire models) are shown. Oxygen – red, nitrogen – blue, carbon – gray/black, and phosphorus – yellow. (A) Overall fold. (B) Binding sites of the glucose and phosphates of the Arthrobacter sp. glucomannokinase (upper lines) and the corresponding residues of the human GME/MNK enzyme (lower lines).
As a result, the following residues of the human MNK domain may play a role in binding glucose hydroxyl groups in the kinase enzyme active site.
The N516 and D517 residues of GNE/MNK (see Figure 3), since they correspond to the H94 and D95 residues of S. tokodaii hexokinase (pdb code 2e2o), the N656 and D657 residues of H. sapiens hexokinase I (pdb code 1dgk), the N122 and D123 residues of Arthrobacter sp. glucomannokinase (pdb code 1woq) (Figure 4B), and the H194 and D195 residues of the E. coli Mlc protein (pdb code 1z6r). Aspartic acid is assigned a role of abstracting the proton from the 6-hydroxyl group of glucose as a catalytic base (Nishimazu et al. 2007).
E566, H569, E588 of GNE/MNK, which correspond to E168, H171, E180 of Arthrobacter sp. glucomannokinase (pdb code 1woq) (Figure 4B), and E244, H247, E266 of E. coli Mlc protein (1z6r).
Based on the above data, the following residues are part of the putative active site of the human ManNAc kinase enzyme activity of GNE/MNK: N516, D517, E566, H569, and E588. All residues contributing to catalytic activity are listed in Table III.
Human GNE/MNK mutations
All reported GNE/MNK mutations associated with HIBM and sialuria were collected using existing databases and extensive literature search (Nishino et al. 2002; Eisenberg et al. 2003; Tomimitsu et al. 2004; Huizing and Krasnewich 2009) and are listed in Table I. For the dominant disorder sialuria, only missense mutations are identified in the heterozygous state (Ferreira et al. 1999; Seppala et al. 1999; Enns et al. 2001; Leroy et al. 2001). For the recessive disorder HIBM, a missense mutation is present on at least one affected allele; severe mutations (frameshifts, nonsense mutations, indicated by gray background in Table I, likely resulting in nonsense-mediated GNE mRNA decay) on both alleles are likely not compatible with life (Schwartzkopf et al. 2002).
All human GNE/MNK missense mutations were mapped on the predicted three-dimensional model and predicted active site residues of the GNE and MNK domains of GNE/MNK (Table I). Several mutations found in HIBM and sialuria patients are located in the proximity of proposed active sites. For example for GNE domain mutations, residues P27 and M29, which are mutated in HIBM (p.P27S, p.M29T), are involved in ligand binding and are part of the epimerase active site. These residues are located on the α1-helix [residues 18–34] of GNE, which also carries residues R19, D21, and K24 corresponding to R10, E12, and K15 of the E. coli enzyme where they function in stabilization of the structure and binding of the UDP portion of the substrate (Campbell et al. 2000). The human mutations p.R129Q, p.H132Q, p.G135V, p.V216A, and p.D225N are located at or near the predicted GNE active site residues H132, E134, G136, and H220, which were derived from homology to H115, E117, G119, and H213 of the E. coli enzyme (Campbell et al. 2000). Predicted structural positions of other reported GNE-epimerase domain mutations are listed in Table I.
Similar predictions can be made for mutations in the MNK enzymatic domain of GNE/MNK (Table I). For example, the human p.N519S mutation is located within the α4-helix [residues 516–528], which contains a glucose binding site at the N-terminal site of the α4-helix. It is possible that substrate binding is impaired in the human p.N519S mutant. A serine residue is present in the structurally equivalent position of the Mlc protein (Figure 1A and B), which does not bind glucose. Previous CD spectroscopy studies observed the largest structural changes for the p.N519S mutant, associated with loss of α-helical structures (Penner et al. 2006). Another human MNK mutation, p.A524V, is located at the interface of the α-helices α4 [residues 516–528] and α10 [residues 703–717]. The alanine residue is conserved among structurally similar proteins except HKI carrying a threonine in this position. It is possible that this residue is important in helix–helix interface contacts. Five MNK mutations are located at the site of the ROK consensus sequences (motifs 1 and 2). The ROK family of enzymes (Repressors, Open reading frames, and Kinases) contains two characteristic sequence motifs involved in zinc binding, a highly conserved motif 2 with the cysteine-rich consensus sequence CXCGXXGCXE and a less specific 28-residue sequence (motif 1) located nine residues upstream of motif 2 (Hansen et al. 2002). The MNK domain of the GNE/MNK enzyme being a member of the ROK family contains both motifs, which bind zinc and substrate (Figure 2B). It is 25% homologous to the Mlc protein, its closest homolog in the ROK family. The zinc binding site as observed in the crystallographic structure of the MNK domain is coordinated by the H569, C579, C581, and C586 residues (Tong et al. 2009). The human mutations p.G576E and p.I587T are located in the proximity of a characteristic zinc and substrate binding site (motif 2) with the consensus sequence CXCGXXGCXE of ROK proteins (Hansen et al. 2002). Mutations p.I557T and p.G559R are part of consensus sequence 1, whereas p.V572L is located in the 9-residue sequence connecting the two motifs.
Fig. 2.

Tertiary structure of the active site of the 2-epimerase (GNE) domain of the GNE/MNK enzyme. (A) Superposition of the three-dimensional structure of the E. coli 2-epimerase in complex with UDP-N-acetylglucosamine (pdb code 1vgv) and the model of the H. sapiens 2-epimerase domain of the GME/MNK enzyme. Side chains of the active site residues (wire models) and binding site of the UDP-N-acetylglucosamine of the E. coli 2-epimerase (upper lines) and the corresponding residues of the human 2-epimerase (GNE) domain of the GME/MNK enzyme (lower lines) are shown. (B) Sequence of the two ROK characteristic motifs (underlined) involved in zinc and substrate binding in human GNE/MNK /3eo3/ and the E. coli Mlc protein /1z6r/. Motif 2 contains the consensus sequence CXCGXXGCXE, whereas motif 1 is a 28-residue sequence N-terminal to motif 2.
In the glucose binding site, S158 of Schistosoma mansoni and Saccharomyces cerevisiae hexokinases (Kuser et al. 2000; pdb code 1ig8) and E. coli glucokinase (Lunin et al. 2004; pdb code 1sz2) bind glucose in the closed conformation. The mutant S158A impairs catalytic activity of S. mansoni hexokinase (Kleywegt and Jones 1996). The S158 residue is conserved among hexokinases and contains an autophosphorylation site (Heidrich et al. 1997; Kuser et al. 2000). In GNE/MNK, the corresponding residue is S473. Two HIBM mutations, p.I472T and p.S473Y, are associated with this site. However, no data are available on the possible phosphorylation of S473 in mammalian GNE/MNK.
The MNK domain mutation p.M712T is the most common mutation causing HIBM, due to a founder effect in the Persian-Jewish population (Eisenberg et al. 2001). The methionine at position 712 is located at the interface of two MNK domain α-helices α4[516–528] and α10[703–717] and is involved in interactions with strands β4 and β5 of a β3β2β1β4β5 β-sheet so that 85% of the M712 surface is removed from possible contacts with solvent (Figure 3). This β3β2β1β4β5 β-sheet contains the typical DXGGT motif of sugar kinases involved in ATP binding (Nishimazu et al. 2007). The MNK-domain helix α4[516–528] is equivalent to an α-helix [95–105] of hexokinase (pdb code 2e2n, 2e2p, 2e2o, 2e2q), which is involved in glucose and Mg2+ binding (Nishimazu et al. 2007). Previous NMR studies showed that GlcNAc binds to the ManNAc kinase site as a substrate in a similar way as ManNAc does. Active site atomic groups for both steps of the reaction are close to each other in space (Benie et al. 2004; Blume et al. 2004). Mutation p.M712T could affect the interface between the helices and therefore potential GlcNAc/ManNAc and/or Mg2+ binding. Since GlcNAc binding is needed for both the epimerase step and the kinase step of GNE/MNK activity, it could explain how MNK mutations result in reduced GNE activity. Predicted structural positions of other reported MNK-kinase domain mutations are listed in Table I.
Mutations associated with sialuria are located in the allosteric site of the H. sapiens GME/MNK enzyme, within the region of an α10β10 loop (Figure 1A). This allosteric site is absent from prokaryotic GNE, and amino acid sequences within and surrounding this site have very low degree of homology with bacterial enzymes. It is therefore a matter of future work to make structural predictions on effects of human allosteric site mutations.
Interhelical angle calculations
Helix–helix interfaces form structural frames to help position protein atomic groups for their interactions with ligands. Interhelical angles influence the ability of molecules to bind ligands. Amino acids at key interior positions of helix–helix interfaces are important for the orientation of helices (Kurochkina 2007, 2008). Mutations away from the active site of the enzyme may affect catalytic activity via changes in the composition of secondary structure interfaces.
Alignment of amino acids at positions a and d of two interacting helices was made for the interface between MNK domain helices α4 [residues 113–126] and α10 [residues 249–260] (Figure 3B), which contain the human mutations p.M712T and p.A524V. Aspartic acid (assigned helix position e) at the N-terminus of helix α4 is the most conserved residue, which is preceded by either an N, H or T residue in other homologs (assigned helix position d), also contributing to catalytic activity (Figure 3B). However, interface residues at positions a and d as well as e and g are not conserved. As a consequence, interhelical angles vary from 69° to 90° (Figure 3B). Residue M712 (mutated in HIBM p.M712T) is located at position e of the interface and forms an interhelical angle of 69° with helix α4 in human GNE/MNK. In some other homologs (Figure 3B), this position is occupied by alanine. Interestingly, human hexokinase HKI does not bind GlcNAc, and HKI exhibits a threonine similarly located in position e (89° interhelical angle with helix α4), which may suggest that the GNE/MNK p.M712T mutation may loosen its ability to bind GlcNAc. Similarly, a mutated alanine in position e in helix α4, as is the case for the p.A524V HIBM mutation, may also result in reduced GlcNAc binding.
High variability of the interface residues in H. sapiens MNK, S. tokodaii, H. sapiens and S. cerevisiae hexokinases, Poly(P)/ATP glucomannokinase, H. sapiens GlcNAc kinase, E. coli Mlc protein, and E. coli glycerol kinase correlates with variation of the interhelical angle and indicates possible site of adjustment of ligand conformation to protein conformation.
Mutations associated with HIBM or sialuria (Table I) are largely located (1) inside active sites and binding motifs, (2) in the proximity of active sites, allosteric sites, and ligand binding motifs or (3) at the secondary structure interfaces, which influence positioning of atomic groups important for catalysis.
Discussion
In this study, we explored three-dimensional modeling of the human GNE/MNK enzyme and created a model of both GNE-epimerase and MNK-kinase enzymatic domains, as well as a prediction of the putative active sites of these enzymes. Most of the active site residues are conserved and could be assigned functions similar to bacterial homologs. Residues located at the secondary structure interfaces showed a higher degree of homology (42%) compared to overall homology (18–27%) and indicate similar folds.
We identified GNE-domain residues R19, A20, D21, K24, P27, M29, H45, G111, D112, R113, H132, E134, G136, D143, D144, R147, G182, D187, H220, S301, G302, and E307 as putative active site residues. Similarly, we identified residues D413, G416, R420, N516, D517, G545, E566, H569, C579, C581, C586, and E588, as putative MNK active site residues (Table III).
As expected, very low similarity in amino acid composition is observed at the site of bacterial allosteric regulation (which is absent from the mammalian enzyme), at the site of mammalian allosteric regulation (which is absent from the bacterial enzymes), and at the subunit interface.
The active site of the 2-epimerase (GNE) domain is located between domains I (N-terminal) and II (C-terminal), each exhibiting topology of Rossmann dinucleotide binding fold. Most of the secondary structure elements of domains I and II can be aligned (Table II) indicating similarity in their three-dimensional structure.
Differences in the active site residues between enzymes contribute to enzyme specificity toward substrates. For example, S. tokodaii hexokinase (pdb code 2e2o) phosphorylates glucose, mannose, glucosamine, and GlcNAc while H. sapiens hexokinase I (pdb code 1dgk) phosphorylates glucose, but does not accommodate GlcNAc. Arthrobacter sp. glucomannokinase (pdb code 1woq) phosphorylates glucose and mannose. However, not only active site residues but also residues distant from the active site can contribute to specificity. Four of the five residues involved in glucose binding (D195, E244, H247, and E266) are identical in Mlc, Arthrobacter sp. glucomannokinase, E. coli glucokinase, H. sapiens MNK, and ROK family member B. subtilis fructokinase. The fifth residue, H194 in Mlc, is an asparagine in Arthrobacter sp. glucomannokinase, H. sapiens MNK, and E. coli glucokinase, and a threonine in B. subtilis fructokinase. However, despite the high similarity of the binding site, Mlc does not bind glucose nor glucose 6-phosphate. Mutation of the histidine to asparagine does not result in glucose binding or glucokinase activity (Schiefner et al. 2005). Residues at helix–helix interfaces, which position active site residues, may be candidates for this role.
The proposed three-dimensional model of the active site of the kinase domain of the GNE/MNK enzyme assigns a structural role to all catalytic residues due to a high degree of similarity between hexokinases, ROK family kinases, and glucomannokinases.
Further modeling has to address possible orientations of the GNE domain relative to the MNK domain in the bifunctional GNE/MNK enzyme, possible arrangement of its subunits in the active hexameric state, composition of secondary structure interfaces with low homology, structure of the allosteric site, and exact location of ligands in the active sites and allosteric site.
Mutations associated with HIBM and sialuria were mapped onto the preliminary three-dimensional model of the GNE/MNK enzyme. Location of the mutations relative to active sites and secondary structure interfaces assists in predicting effects of these mutations on the enzymatic activity. Practically all mutations either directly interfere with the location of residues important for catalysis or affect secondary structure interfaces and indirectly contribute to the positioning of catalytic residues and binding sites of substrates.
Mutations associated with HIBM and sialuria have proximal (in the active sites and their vicinity) and distal (at the secondary structure interfaces) effects on the structure and function of the enzyme. Mutant p.M712T, being at the interface of α-helices α4 (catalytic) and α10, most likely affects GlcNAc, Mg2+, and/or ATP binding. Changes in secondary structure content were also observed for this mutant measured by CD spectroscopy (Penner et al. 2006).
Modeling of the GNE/MNK, the key enzyme in sialic acid biosynthesis, and related proteins contributes to further understanding of GNE/MNK function, its ligands, and the origin of substrate/inhibitor/regulator specificity of this family of enzymes. Furthermore, modeling of the mutations associated with HIBM or sialuria reveals why these mutations contribute to decreased/inhibited enzymatic activities. It is possible that further modeling studies reveal ligands (with a similar or different structure to ManNAc) that may be good candidates for rescuing the (misfolding) effects of mutated GNE/MNK, or suggest new possible metabolic pathways that may overcome these effects and may be applied for the treatment of HIBM or sialuria.
This work contributes to further understanding of GNE/MNK ligand binding and function, which may assist future studies for therapeutic options that target misfolded GNE/MNK in HIBM and/or sialuria.
Material and methods
Identification of GNE/MNK homologous structures
Atomic coordinates of proteins determined by X-ray crystallography were used from the Brookhaven Protein Databank (Bernstein et al. 1977). The following crystallographic structures of proteins homologous to the GNE/MNK enzyme including their complexes with ligands given by their PDB (Protein Data Bank) designation were used: 3eo3, 2e2n, 2w2p, 2e2o, 2e2q, 1dgk, 1hkc, 1qha, 1woq, 1ig8, 2aa4, 3b8a, 2ch5, 2ch6, 2bis, 1sz2, 1fxj, 1yqh, 1f6d, 1vgv, 1o6c, 1v4v, 3beo, 3dzc, 1z6r, 2hid, 2hro, 1glc, 2w41, 2zf5. In addition, proteins structurally related to GNE/MNK were included, such as sugar kinases, 2- and 4-epimerases, acyltransferases, pyrophosphorylases, glycogen synthase, glycogen phosphorylase, and histidines-containing phosphocarrier protein (HPr). Protein sequences were retrieved from GenBankTM including the human GNE/MNK enzyme (GenBank NM_005476).
Protein sequences of organisms (pro- and eukaryotes) with 2-epimerase and/or sugar kinase activities were selected using ontology gene annotations (Ashburner et al. 2000), and they were aligned using BLAST (Basic Local Alignment Search Tool, NCBI). For homology searches, the expected threshold E-value of 0.1 was used as a cut-off. This value is the statistical significance threshold for reporting matches against database sequences and is comparable with the p-value. These sequence alignments were then combined with structural alignments to identify structurally identical residues responsible for similar enzymatic functions (Figure 1).
Modeling of GNE/MNK interhelical angles
The secondary structure elements (α-helices, β-sheets, etc.) form a protein's three-dimensional structure. They associate in several distinct arrangements being parallel or antiparallel to each other. One characteristic parameter of this association is the angle between secondary structure elements, which is important for the description and prediction of protein structure (Chothia 1984). In this work, only interfaces between α-helices were considered. Interhelical angle describes the angle between two α-helices.
Interhelical angle α is calculated as an angle between vector X (x1, y1, z1) and vector Y (y1, y2, y3):
where X is the vector from the N-terminus to the C-terminus of the first α-helix. It connects the center of mass of the first four residues of the first helix with the center of mass of the last four residues of the same helix.
Y is the vector from the N-terminus to the C-terminus of the second α-helix.
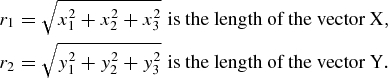 |
The positive (or negative) direction of the interhelical angle is chosen if the distant helix is rotated in a clockwise (or counterclockwise) direction relative to the proximal helix. For the viewer, the proximal helix is located in front of the distant helix. Values determined by this approach should agree with those described by Chothia et al. (1981).
Since some (mutational) changes in the composition of secondary structure interfaces are predicted to be located at key interior positions important for orientation of helices (Kurochkina 2007, 2008), we determined for each GNE/MNK missense mutation (Table I) whether it was located within helix–helix interfaces (and at which position, a–g) of two interacting α-helices.
Modeling of human GNE/MNK mutations associated with active sites and binding motifs
The position of each mutation was identified on the three-dimensional model of the GNE domain and on the three-dimensional structure of the MNK domain of GNE/MNK. Mutations were subdivided into several groups. The first group consists of mutations located inside active sites and binding motifs. The second group consists of mutations located in the proximity of active sites, allosteric sites, and binding motifs. The third group contains mutations located away from the active sites, mainly at the secondary structure interfaces. The position of mutations in each group in relation to interfaces between secondary structure elements was considered. For mutations located at helix–helix interfaces, change in composition of amino acids at core positions a and d and neighboring positions e and g was considered.
Modeling of human GNE/MNK mutations located at helix–helix interfaces
Nomenclature for coiled coils designates positions of two interacting α-helices by the letters a–g for the first helix and a′–g′ for the second helix (Hodges et al. 1972). The interface between helices is formed mainly by amino acids in positions d–d′ occupied by leucines and a–a′ where valines, asparagines, or other residues can occur (Rasmussen et al. 1991). Residues at positions a–a′ and d–d′ can be to a large extent shielded from contacts with solvent. These a–a′ and d–d′ residues have to fit together to pack inside the space between helices and they also have to be compatible with interactions of the entire interface. Another important interaction between the helices can occur at positions e–e′ and g–g′ (Krylov et al. 1994).
Since (mutational) changes in the composition of secondary structure interfaces are predicted to be located at key interior positions important for orientation of helices (Kurochkina 2007, 2008), we determined for each GNE/MNK missense mutation (Table I) whether it was located within helix–helix interfaces (and at which position, a–g) of two interacting α-helices. Positions a–g for interfaces of the GNE/MNK enzyme and its homologs were assigned by visual inspection of three-dimensional structures of proteins.
Funding
The Intramural Research programs of the National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA and Research Funds of The School of Theoretical Modeling, Chevy Chase, MD, USA.
Glossary
Abbreviations
- ATP
adenosine triphosphate
- BGT
bactriophage T4 glycosyltransferase
- BLAST
Basic Local Alignment Search Tool
- CMP
cytidine monophosphate
- GlcNAc
N-acetylglucosamine
- GMK
glucomannokinase
- GNE/MNK
uridine diphosphate-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase
- GP
glycogen phosphorylase
- HIBM
hereditary inclusion body myopathy
- HKI
Human hexokinase I
- ManNAc
N-acetylmannosamine
- pdb
protein data bank
- ROK motif
repressors, open reading frames, and kinases motif
- UDP
uridine diphosphate
References
- Amouri R, Driss A, Murayama K, Kefi M, Nishino I, Hentati F. Allelic heterogeneity of GNE gene mutation in two Tunisian families with autosomal recessive inclusion body myopathy. Neuromuscul Disord. 2005;15:361–363. doi: 10.1016/j.nmd.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. The Gene Ontology Consortium. Gene ontology: Tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger J, Sauder JM, Adams JM, Antonysami S, Bain K, Bergside MG, Buchanan SG, Buchanan MD, Batienko Y, Christopher JA, et al. Structural analysis of a set of proteins resulting from a bacterial genome project. Proteins. 2005;60:787–796. doi: 10.1002/prot.20541. [DOI] [PubMed] [Google Scholar]
- Behin A, Dubourg O, Laforet P, Pecheux C, Bernard R, Levy N, Eymard B. Distal myopathy due to mutations of GNE gene: Clinical spectrum and diagnosis. Rev Neurol (Paris) 2008;164:434–443. doi: 10.1016/j.neurol.2008.02.040. [DOI] [PubMed] [Google Scholar]
- Benie AJ, Blume A, Schmidt RR, Reutter W, Hinderlich S, Peters T. Characterization of ligand binding to the bifunctional key enzyme in the sialic acid biosynthesis by NMR. J Biol Chem. 2004;279:55722–55727. doi: 10.1074/jbc.M410239200. [DOI] [PubMed] [Google Scholar]
- Bernstein FC, Koetzle TF, Williams GJB, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The protein data bank: A computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- Blume A, Benie AJ, Stolz F, Schmidt RR, Reutter W, Hinderlich S, Peters T. Characterization of ligand binding to the bifunctional key enzyme in the sialic acid biosynthesis by NMR. J Biol Chem. 2004;279:55715–55721. doi: 10.1074/jbc.M410238200. [DOI] [PubMed] [Google Scholar]
- Bo RD, Baron P, Prelle A, Serafini M, Moggio M, Fonzo AD, Castagni M, Bresolin N, Comi GP. Novel missense mutation and large deletion of GNE gene in autosomal-recessive inclusion-body myopathy. Muscle Nerve. 2003;28:113–117. doi: 10.1002/mus.10391. [DOI] [PubMed] [Google Scholar]
- Broccolini A, Pescatori M, D’Amico A, Sabino A, Silvestri G, Ricci E, Servidei S, Tonali PA, Mirabella M. An Italian family with autosomal recessive inclusion-body myopathy and mutations in the GNE gene. Neurology. 2002;59:1808–1809. doi: 10.1212/01.wnl.0000031808.04545.e0. [DOI] [PubMed] [Google Scholar]
- Broccolini A, Ricci E, Cassandrini D, Gliubizzi C, Bruno C, Tonoli E, Silvestri G, Pescatori M, Rodolico C, Sinicropi S, et al. Novel GNE mutations in Italian families with autosomal recessive hereditary inclusion-body myopathy. Hum Mutat. 2004;23:632. doi: 10.1002/humu.9252. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Mosimann SC, Tanner ME, Strynadka NC. The structure of UDP-N-acetylglucosamine 2-epimerase reveals homology to phosphoglycosyl transferases. Biochemistry. 2000;39:14993–15001. doi: 10.1021/bi001627x. [DOI] [PubMed] [Google Scholar]
- Chothia C. Principles that determine the structure of proteins. Annu Rev Biochem. 1984;53:537–572. doi: 10.1146/annurev.bi.53.070184.002541. [DOI] [PubMed] [Google Scholar]
- Chothia C, Levitt M, Richardson D. Helix to helix packing in proteins. J Mol Biol. 1981;145:215–250. doi: 10.1016/0022-2836(81)90341-7. [DOI] [PubMed] [Google Scholar]
- Chu CC, Kuo HC, Yeh TH, Ro LS, Chen SR, Huang CC. Heterozygous mutations affecting the epimerase domain of the GNE gene causing distal myopathy with rimmed vacuoles in a Taiwanese family. Clin Neurol Neurosurg. 2007;109:250–256. doi: 10.1016/j.clineuro.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Darvish D, Vahedifar P, Huo Y. Four novel mutations associated with autosomal recessive inclusion body myopathy (MIM: 600737) Mol Genet Metab. 2002;77:252–256. doi: 10.1016/s1096-7192(02)00141-5. [DOI] [PubMed] [Google Scholar]
- Effertz K, Hinderlich S, Reutter W. Selective loss of either the epimerase or kinase activity of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase due to site-directed mutagenesis based on sequence alignments. J Biol Chem. 1999;274:28771–28778. doi: 10.1074/jbc.274.40.28771. [DOI] [PubMed] [Google Scholar]
- Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–89. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- Eisenberg I, Grabov-Nardini G, Hochner H, Korner M, Sadeh M, Bertorini T, Bushby K, Castellan C, Felice K, Mendell J, et al. Mutations spectrum of GNE in hereditary inclusion body myopathy sparing the quadriceps. Hum Mutat. 2003;21:99–100. doi: 10.1002/humu.9100. [DOI] [PubMed] [Google Scholar]
- Enns GM, Seppala R, Musci TJ, Weisiger K, Ferrell LD, Wenger DA, Gahl WA, Packman S. Clinical course and biochemistry of sialuria. J Inherit Metab Dis. 2001;24:328–336. doi: 10.1023/a:1010588115479. [DOI] [PubMed] [Google Scholar]
- Ferreira H, Seppala R, Pinto R, Huizing M, Martins E, Braga AC, Gomes L, Krasnewich DM, Sa Miranda MC, Gahl WA. Sialuria in a Portuguese girl: Clinical, biochemical, and molecular characteristics. Mol Genet Metab. 1999;67:131–137. doi: 10.1006/mgme.1999.2852. [DOI] [PubMed] [Google Scholar]
- Fisher J, Towfighi J, Darvish D, Simmons Z. A case of hereditary inclusion body myopathy: 1 patient, 2 novel mutations. J Clin Neuromuscul Dis. 2006;7:179–184. doi: 10.1097/01.cnd.0000211406.94445.f0. [DOI] [PubMed] [Google Scholar]
- Ghaderi D, Strauss HM, Reinke S, Cirak S, Reutter W, Lucka L, Hinderlich S. Evidence for dynamic interplay of different oligomeric states of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase by biophysical methods. J Mol Biol. 2007;369:746–758. doi: 10.1016/j.jmb.2007.03.037. [DOI] [PubMed] [Google Scholar]
- Hansen T, Reichstein B, Schmid R, Schönheit P. The first archaeal ATP-dependent glucokinase, from the hyperthermophilic crenarchaeon Aeropyrum pernix, represents a monomeric, extremely thermophilic ROK glucokinase with broad hexose specificity. J Bacteriol. 2002;184:5955–5965. doi: 10.1128/JB.184.21.5955-5965.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich K, Otto A, Behlke J, Rush J, Wenzel KW, Kriegel T. Autophosphorylation-inactivation site of hexokinase 2 in Saccharomyces cereviciae. Biochemistry. 1997;36:1960–1964. doi: 10.1021/bi9623643. [DOI] [PubMed] [Google Scholar]
- Hinderlich S, Stasche R, Zeitler R, Reutter W. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. 1997;272:24313–24318. doi: 10.1074/jbc.272.39.24313. [DOI] [PubMed] [Google Scholar]
- Hodges RS, Sodak J, Smillie LB, Jurasek L. Tropomyosin: Amino acid sequence and coiled-coil structure. Cold Spring Harb Symp Quant Biol. 1972;37:299–310. [Google Scholar]
- Huizing M. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Drug Discov Today: Dis Mech. 2005;2:519–527. doi: 10.1016/j.ymgme.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Huizing M, Krasnewich D. Hereditary inclusion body myopathy: A decade of progress. Biochim Biophys Acta. 2009;1792:881–887. doi: 10.1016/j.bbadis.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, Rakocevic G, Sparks SE, Manoli I, Shatunov A, Goldfarb L, Krasnewich D, Gahl WA, Dalakas MC. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab. 2004;81:196–202. doi: 10.1016/j.ymgme.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Kayashima T, Matsuo H, Satoh A, Ohta T, Yoshiura K, Matsumoto N, Nakane Y, Niikawa N, Kishino T. Nonaka myopathy is caused by mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannoseamine kinase gene (GNE) J Hum Genet. 2002;47:77–79. doi: 10.1007/s100380200004. [DOI] [PubMed] [Google Scholar]
- Kim BJ, Ki CS, Kim JW, Sung DH, Choi YC, Kim SH. Mutation analysis of the GNE gene in Korean patients with distal myopathy with rimmed vacuoles. J Hum Genet. 2006;51:137–140. doi: 10.1007/s10038-005-0338-5. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA. Phi/psi-chology: Ramachandran plot revisited. Structure. 1996;4:1395–1400. doi: 10.1016/s0969-2126(96)00147-5. [DOI] [PubMed] [Google Scholar]
- Krause S, Schlotter-Weigel B, Walter MC, Najmabadi H, Wiendl H, Muller-Hocker J, Muller-Felber W, Pongratz D, Lochmuller H. A novel homozygous missense mutation in the GNE gene of a patient with quadriceps-sparing hereditary inclusion body myopathy associated with muscle inflammation. Neuromuscul Disord. 2003;13:830–834. doi: 10.1016/s0960-8966(03)00140-8. [DOI] [PubMed] [Google Scholar]
- Krylov D, Michailenko I, Vinson C. A thermodynamic scale for leucine zipper stability and dimerization specificity: e and g interhelical interactions. EMBO J. 1994;13:2849–2861. doi: 10.1002/j.1460-2075.1994.tb06579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurochkina N. Amino acid composition of parallel helix–helix interfaces. J Theor Biol. 2007;247:110–121. doi: 10.1016/j.jtbi.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Kurochkina N. Specific sequence combinations at parallel and antiparallel helix–helix interfaces. J Theor Biol. 2008;255:188–198. doi: 10.1016/j.jtbi.2008.08.020. [DOI] [PubMed] [Google Scholar]
- Kuser PR, Krauchenco S, Antunes OAC, Polikarpov I. The high resolution crystal structure of yeast hexokinase PII with the correct primary sequence provides new insights into its mechanism of action. J Biol Chem. 2000;275:20814–20821. doi: 10.1074/jbc.M910412199. [DOI] [PubMed] [Google Scholar]
- Leroy JG, Seppala R, Huizing M, Dacremont G, De Simpel H, Van Coster RN, Orvisky E, Krasnewich DM, Gahl WA. Dominant inheritance of sialuria, an inborn error of feedback inhibition. Am J Hum Genet. 2001;68:1419–1427. doi: 10.1086/320598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liewluck T, Pho-Iam T, Limwongse C, Thongnoppakhun W, Boonyapisit K, Raksadawan N, Murayama K, Hayashi YK, Nishino I, Sangruchi T. Mutation analysis of the GNE gene in distal myopathy with rimmed vacuoles (DMRV) patients in Thailand. Muscle Nerve. 2006;34:775–778. doi: 10.1002/mus.20583. [DOI] [PubMed] [Google Scholar]
- Lunin VV, Li Y, Schrag JD, Iannuzzi P, Cygler M, Matte A. Crystal structures of Escherichia coli ATP-dependent glucokinase and its complex with glucose. J Bacteriol. 2004;186:6915–6927. doi: 10.1128/JB.186.20.6915-6927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motozaki Y, Komai K, Hirohata M, Asaka T, Ono K, Yamada M. Hereditary inclusion body myopathy with a novel mutation in the GNE gene associated with proximal leg weakness and necrotizing myopathy. Eur J Neurol. 2007;14:e14–15. doi: 10.1111/j.1468-1331.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- Mukai T, Kawai S, Mori S, Mikami B, Murata K. Crystal structure of bacterial inorganic polyphosphate/ATP-glucomannokinase: Insights into kinase evolution. J Biol Chem. 2004;279:50591–50600. doi: 10.1074/jbc.M408126200. [DOI] [PubMed] [Google Scholar]
- Nishimazu H, Fushinobu S, Shoun H, Wakagi T. Crystal structures of an ATP-dependent hexokinase with broad substrate specificity from the hyperthermophilic archaeon. J Biol Chem. 2007;272:9923–9931. doi: 10.1074/jbc.M610678200. [DOI] [PubMed] [Google Scholar]
- Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, Nagata T, Chida K, Takahashi T, Takusa Y, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59:1689–1693. doi: 10.1212/01.wnl.0000041631.28557.c6. [DOI] [PubMed] [Google Scholar]
- Penner J, Mantey LR, Elgavish S, Ghaderi D, Cirak S, Berger M, Krause S, Lucka L, Voit T, Mitrani-Rosenbaum S, et al. Influence of UDP-GlcNAc 2-epimerase/ManNAc kinase mutant proteins on hereditary inclusion body myopathy. Biochemistry. 2006;45:2968–2977. doi: 10.1021/bi0522504. [DOI] [PubMed] [Google Scholar]
- Rao ST, Rossmann MG. Comparison of super-secondary structure in proteins. J Mol Biol. 1973;76:241–256. doi: 10.1016/0022-2836(73)90388-4. [DOI] [PubMed] [Google Scholar]
- Rasmussen R, Benvegnu D, O’Shea EK, Kim PS, Alber T. X-ray scattering indicates that the leucine zipper is a coiled coil. Proc Natl Acad Sci. 1991;88:561–564. doi: 10.1073/pnas.88.2.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro LS, Lee-Chen GJ, Wu YR, Lee M, Hsu PY, Chen CM. Phenotypic variability in a Chinese family with rimmed vacuolar distal myopathy. J Neurol Neurosurg Psychiatry. 2005;76:752–755. doi: 10.1136/jnnp.2004.048876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefner A, Gerber K, Seitz S, Welte W, Diederichs K, Boos W. The crystal structure of Mlc, a global regulator of sugar metabolism in Escherichia coli. J Biol Chem. 2005;280:29073–29079. doi: 10.1074/jbc.M504215200. [DOI] [PubMed] [Google Scholar]
- Schwartzkopf M, Knobeloch KP, Rohde E, Hinderlich S, Wiechens N, Lucka L, Horak I, Reutter W, Horstkorte R. Sialylation is essential for early development in mice. Proc Natl Acad Sci. 2002;99:5267–5270. doi: 10.1073/pnas.072066199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppala R, Lehto VP, Gahl WA. Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am J Hum Genet. 1999;64:1563–1669. doi: 10.1086/302411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppala R, Tietze F, Krasnewich D, Weiss P, Ashwell G, Barsh G, Thomas GH, Packman S, Gahl WA. Sialic acid metabolism in sialuria fibroblasts. J Biol Chem. 1991;266:7456–7461. [PubMed] [Google Scholar]
- Sparks SE, Ciccone C, Lalor M, Orvisky E, Klootwijk R, Savelkoul PJ, Dalakas MC, Krasnewich DM, Gahl WA, Huizing M. Use of a cell-free system to determine UDP-N-acetylglucosamine 2-epimerase and N-acetylmannoseamine kinase activities in human hereditary inclusion body myopathy. Glycobiology. 2005;15:1102–1110. doi: 10.1093/glycob/cwi100. [DOI] [PubMed] [Google Scholar]
- Tajima Y, Uyama E, Go S, Sato C, Tao N, Kotani M, Hino H, Suzuki A, Sanai Y, Kitajima K, et al. Distal myopathy with rimmed vacuoles: Impaired O-glycan formation in muscular glycoproteins. Am J Pathol. 2005;166:1121–1130. doi: 10.1016/S0002-9440(10)62332-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimitsu H, Ishikawa K, Shimizu J, Ohkoshi N, Kanazawa I, Mizusawa H. Distal myopathy with rimmed vacuoles: Novel mutations in the GNE gene. Neurology. 2002;59:451–454. doi: 10.1212/wnl.59.3.451. [DOI] [PubMed] [Google Scholar]
- Tomimitsu H, Shimizu J, Ishikawa K, Ohkoshi N, Kanazawa I, Mizusawa H. Distal myopathy with rimmed vacuoles: Novel mutations in the GNE gene. Neurology. 2004;62:1607–1610. doi: 10.1212/wnl.59.3.451. [DOI] [PubMed] [Google Scholar]
- Tong Y, Tempel W, Nedyalkova L, Mackenzie F, Park HW. Crystal structure of the N-acetylmannosamine kinase domain of GNE. PloS One. 2009;4:e7165. doi: 10.1371/journal.pone.0007165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos OM, Raju R, Dalakas MC. GNE mutations in an American family with quadriceps-sparing IBM and lack of mutations in s-IBM. Neurology. 2002;59:1776–1779. doi: 10.1212/01.wnl.0000039780.13681.ad. [DOI] [PubMed] [Google Scholar]
- Weihofen WA, Berger M, Chen H, Saenger W, Hinderlich S. Structure of human N-acetylglucosamine kinase in two complexes with N-acetylglucosamine and with glucose: Insights into substrate specificity and regulation. J Mol Biol. 2006;364:388–399. doi: 10.1016/j.jmb.2006.08.085. [DOI] [PubMed] [Google Scholar]
- Yabe I, Higashi T, Kikuchi S, Sasaki H, Fukazawa T, Yoshida K, Tashiro K. GNE mutations causing distal myopathy with rimmed vacuoles with inflammation. Neurology. 2003;61:384–386. doi: 10.1212/01.wnl.0000061520.63546.8f. [DOI] [PubMed] [Google Scholar]
- Yarema KJ, Goon S, Bertozzi CR. Metabolic selection of glycosylation defects in human cells. Nat Biotechnol. 2001;19:553–558. doi: 10.1038/89305. [DOI] [PubMed] [Google Scholar]