

The crystal structure of bovine pancreatic ribonuclease A in complex with uridine 5′-monophosphate was determined at 1.60 Å resolution. The uridine lies in the pyrimidine-specific binding site of the enzyme, with the phosphate group extended into the solvent without interacting with the protein.
Keywords: ribonuclease A, uridine 5′-monophosphate, silver bullets
Abstract
Bovine pancreatic ribonuclease A (RNase A) was crystallized from a mixture of small molecules containing basic fuchsin, tobramycin and uridine 5′-monophosphate (U5P). Solution of the crystal structure revealed that the enzyme was selectively bound to U5P, with the pyrimidine ring of U5P residing in the pyrimidine-binding site at Thr45. The structure was refined to an R factor of 0.197 and an R free of 0.253.
1. Introduction
Bovine pancreatic ribonuclease (EC 3.1.27.5) or RNase A, as it is commonly known, is one of the most thoroughly studied and best understood enzymes in terms of catalysis. The chemical and structural features of RNase A have been thoroughly reviewed by Richards & Wyckoff (1971 ▶), Blackburn & Moore (1982 ▶), Wlodawer (1985 ▶) and Raines (1998 ▶). RNase A degrades ribonucleic acid (RNA) by cleaving selective phosphodiester bonds along its length. The substrate must be single-stranded and the enzyme has a high specificity for a pyrimidine base on the 3′ side of the phosphodiester to be hydrolyzed. Either a pyrimidine or a purine can be present on the 5′ side.
RNase A is a single polypeptide chain of 124 amino acids with a molecular weight of 13 700 Da and a pI of 9.45. Its three-dimensional structure consists of a large twisted β-sheet fortified by four disulfide bridges. There is a short α-helix at the amino-terminus and two other short helices of only one turn. The active site resides in a pronounced cleft across the middle of the molecule and contains Lys41, the ∊-amino group of which pins the target phosphate group of the RNA substrate, Thr45, which binds selectively to pyrimidines, and two histidines, His12 and His119, that are responsible for catalyzing the hydrolysis of the phosphodiester bond. This cleft divides the molecule into two domains, defined by Radha Kishan et al. (1995 ▶) as consisting of amino acids 1–14, 49–80 and 103–124 in domain A, and amino acids 15–48 and 81–102 in domain B. In addition to the active-site residues, it has been shown that a system of arginine and lysine residues over the surface of the protein bind and immobilize a length of the RNA substrate of roughly a dozen nucleotides (McPherson et al., 1986 ▶).
In the course of an investigation (McPherson & Cudney, 2006 ▶; Larson et al., 2007 ▶, 2008 ▶) into the ability of mixtures, or ‘cocktails’, of conventional small molecules to promote the crystallization of proteins, viruses and nucleic acids, we obtained an at the time previously unreported nucleotide–RNase A complex in a known unit cell and space group. Here, we describe the mode of binding of the nucleotide to the enzyme.
2. Materials and methods
2.1. Crystallization
Bovine pancreatic ribonuclease A was purchased from Sigma Biochemical Co. (St Louis, Missouri, USA) in lyophilized form and dissolved in water to a concentration of about 30 mg ml−1. The crystal screening experiments that yielded the RNase A–nucleotide complex crystals investigated in this work have been described in detail elsewhere (Larson et al., 2008 ▶). The crystals were grown in sitting drops by vapor diffusion at room temperature (298 K) in 96-well Intelli-Plates (Hampton Research, Aliso Viejo, California, USA) with 90 µl reservoirs of 25%(w/v) PEG 3350 in water. The protein droplets were of 2 µl volume and consisted of equal parts of the stock protein solution and a ‘cocktail’ of small molecules at concentrations of about 1%(w/v). The concentrations of the small molecules in the crystallization drops were approximately 5–10 mM. The crystals reported here were grown in the presence of a small-molecule ‘cocktail’ composed of basic fuchsin, tobramycin and uridine 5′-monophosphate (U5P), the chemical structures of which are shown in Fig. 1 ▶. All solutions were buffered with 0.1 M HEPES and adjusted to pH 7.0. Crystals usually appeared and grew to full size within one to two weeks.
Figure 1.

Schematic diagram of the small molecules comprising the ‘cocktail’ that was used in this study as well as the buffer that was used.
2.2. Data collection
A crystal of approximately 0.3 × 0.1 × 0.1 mm in size was mounted in a cryoloop (Hampton Research, Aliso Viejo, California, USA) and flash-frozen in liquid-nitrogen vapor as it was mounted on a Rigaku RU200 generator equipped with a Cu anode, Osmic mirrors and image-plate detectors. X-ray diffraction data were collected at 100 K to 1.60 Å resolution. The data were integrated, scaled and merged using the program d*TREK (Pflugrath, 1999 ▶). The space group, unit-cell parameters and relevant data-collection statistics are presented in Table 1 ▶.
Table 1. Data-processing, refinement and model statistics.
Values in parentheses are for the highest resolution shell.
| Crystal data | |
| Space group | P21 |
| Z | 4 |
| Unit-cell parameters (Å, °) | a = 30.73, b = 74.85, c = 50.52, β = 107.80 |
| Data-processing statistics | |
| Resolution (Å) | 29.54–1.60 (1.66–1.60) |
| No. of unique reflections | 26265 |
| Redundancy | 2.61 (2.15) |
| 〈I/σ(I)〉 | 16.8 (2.4) |
| Completeness | 91.2 (64.2) |
| Rmerge† | 0.027 (0.340) |
| Structure-refinement statistics | |
| Resolution (Å) | 29.54–1.60 (1.64–1.60) |
| No. of reflections (Fo = 0 removed) | 25900 (2256) |
| R/Rfree‡ (all data) | 0.197/0.253 (0.356/0.411) |
| Reflections in test set (%) | 9.80 (5.76) |
| No. of refined parameters | 8938 |
| No. of reflections | 23360 |
| No. of restraints | 14567 |
| Data-to-parameter ratio | 2.61 |
| Data/restraints-to-parameter ratio | 4.24 |
| Model statistics | |
| Non-H atoms | |
| Protein atoms (full/partial) | 1871/62 |
| Ligand atoms (full/partial) | 37/10 |
| Water atoms (full/partial) | 181/42 |
| Geometry: r.m.s. deviations from ideal values | |
| Bonds (Å) | 0.011 |
| Angles (°) | 1.55 |
| Planes (Å) | 0.010 |
| Chiral centers (Å3) | 0.09 |
| Average isotropic B factors (Å2) | |
| B estimate (Wilson) | 33.2 |
| Overall | 38.7 |
| Protein (full/partial) | 38.1/34.4 |
| Ligands (all) | 48.1/47.2 |
| Waters (full/partial) | 43.6/37.2 |
| Ramachandran plot | |
| Most favored region | 199 [86.5%] |
| Allowed region | 31 [13.5%] |
| Generously allowed and disallowed regions | 0 [0.0%] |
R
merge = 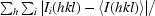
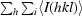 , where I
i(hkl) is the ith used observation for unique hkl and 〈I(hkl)〉 is the mean intensity for unique hkl.
, where I
i(hkl) is the ith used observation for unique hkl and 〈I(hkl)〉 is the mean intensity for unique hkl.
R = 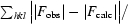
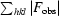 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes.
2.3. Structure solution and refinement
Calculation of the volume-to-mass ratio V M for the monoclinic unit cell (Matthews, 1968 ▶) yielded a value of 2.02 Å3 Da−1 if it were assumed that there were two protein molecules comprising the asymmetric unit. This is the most reasonable value for V M and is near the center of the range for most protein crystals (McPherson, 1999 ▶). Of the two isomorphous structures in the PDB, entry 1u1b (Beach et al., 2005 ▶; Berman et al., 2000 ▶) had the most similar unit cell and was the wild-type enzyme. After removing the ligands and water, the 1u1b model was refined as two rigid bodies, followed by simulated annealing using CNS (Brünger et al., 1998 ▶; R = 0.30, R free = 0.37). The small molecules were identified and built into the density of an F o − F c difference density map using the program O (Jones & Kjeldgaard, 1994 ▶) and refinement was continued with CNS. In order to utilize TLS capabilities, refinement was continued using REFMAC5 (Murshudov et al., 1997 ▶). Each enzyme–U5P complex was treated as three groups for TLS refinement based on domains A and B defined by Radha Kishan et al. (1995 ▶) and the bound U5P, giving a total of six TLS groups. Model rebuilding and addition of the water structure over the course of the final cycles of refinement was performed with the program Coot (Emsley & Cowtan, 2004 ▶). The initial F o − F c difference Fourier map was computed with CNS and all subsequent maps were generated from MTZ files from REFMAC5 using the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶) or Coot. Final refinement and model statistics are shown in Table 1 ▶.
2.4. Other program information
The program PyMOL (DeLano, 2002 ▶) was used to display maps and produce figures. The RCSB validation server (Berman et al., 2000 ▶) and PROCHECK (Laskowski et al., 1993 ▶) were used to assess the quality of the model and generate the Ramachandran plots and statistics. Geometrical target values were those contained in the CCP4 dictionary and consisted of the values from (i) Engh & Huber (1991 ▶) for the protein structure, (ii) Kennard & Taylor (1982 ▶) for the pyrimidine portion of U5P and (iii) Saenger (1983 ▶) for the sugar-phosphate structure.
3. Results
3.1. The model
After initial refinement of the 1u1b model against our data, an F o − F c difference Fourier map was calculated and analyzed. Two independent electron-density features, similar in shape and volume, were observed, one associated with each of the two protein molecules in the asymmetric unit. Of the three components of the ‘cocktail’, only U5P, a component of the natural substrate of the enzyme and a competitive inhibitor, could be readily fitted into both densities in the asymmetric unit.
The final model is composed of two molecules (designated A and B) of RNase A (residues 1–124), two U5P molecules and 211 fully or partially occupied water molecules. Multiple conformations were modeled for Met29, Ser59, Lys61, Arg85 and His119 in molecule A and for Asn27, Met30, Ser59 and Lys104 in molecule B. In addition, the phosphate group of the U5P bound to molecule B was modeled with disorder. Of the 42 partially occupied water molecules, 24 are paired as disordered positions and five are involved with the disordered groups His119, Asn27 and the phosphate. There was broken main-chain density at Ser21 for both molecules. The final working R factor was 0.197 and R free was 0.253 at 1.60 Å resolution for all data with F o > 0. Model statistics are given in Table 1 ▶.
Molecules A and B have nearly the same orientation in the unit cell but are translated by about half the c axis. The r.m.s. deviation between the Cα atoms after superpositon of the two molecules is 0.65 Å. When the two domains are superpositioned separately, the r.m.s. deviation is 0.34 Å for domain A and 0.64 Å for domain B. The largest deviation of 2.47 Å for the full superposition is found at Ser22, around which the density is poorly defined for both molecules. The average B factors for the two molecules are similar at 37.3 and 38.8 Å2 for molecules A and B, respectively. Likewise, the average B factors for the U5P molecules are similar at 49.3 and 46.2 Å2 in complexes A and B, respectively. However, for U5P bound to molecule B (U5P-B) the phosphate was treated as disordered, which reduced the average B factor for the group. Since the phosphate is entirely solvent-exposed in U5P bound to molecule A (U5P-A) this phospate is probably also disordered, but it was not obvious how to model the disorder.
3.2. U5P conformation
The pyrimidine base of the uridine 5′-monophosphate ligands have an anti conformation (χCN = −151.9° and −151.5° for molecules A and B, respectively) with respect to the ribose ring. This conformation is normal and necessary for docking of the uridine ring into the B1 subsite (Moodie & Thornton, 1993 ▶). The ribose rings of both U5P ligands have the expected 3′-endo conformation that is generally observed for ribonucleotides (Altona & Sundaralingam, 1972 ▶).
3.3. U5P binding
The U5P molecules, as seen in Figs. 2 ▶ and 3 ▶, occupy the pyrimidine-binding sites (subsite B1; Raines, 1998 ▶) at the active sites of the enzyme molecules. Thus, the main U5P interactions are through the U5P N3⋯Thr45 OG1 and U5P O2⋯Thr45 N hydrogen bonds. Although they can accommodate pyrimidine bases [Raines, 1998 ▶; PDB codes 8rsa (Nachman et al., 1990 ▶), 1rta (Birdsall & McPherson, 1992 ▶), 3dxg and 3dxh (Tsirkone et al., 2009 ▶)], the purine-binding sites were vacant. In addition, the ribose ring interacts with the enzyme through an O2′⋯Lys41 NZ hydrogen bond (2.61 and 2.98 Å in molecules A and B, respectively). This interaction is the same as that observed in the transition-state analog structures containing uridine vanadate [PDB codes 6rsa (Borah et al., 1985 ▶) and 1ruv (Ladner et al., 1997 ▶)]. In fact, most pyrimidine nucleotide–RNase A structures have a similar Lys41 NZ⋯O2′ interaction unless they are devoid of the O2′ atom (PDB code 1w4p, Jenkins et al., 2005 ▶), the ribose pucker is 2′-endo or 3′-exo [PDB codes 1o0n (Leonidas et al., 2003 ▶) and 1rta (Birdsall & McPherson, 1992 ▶)] or Lys41 is directed away from the ribose ring (PDB code 3dxg; Tsirkone et al., 2009 ▶). The only other hydrogen-bonding interaction involving U5P is seen in molecule B, in which Lys66 appears to hydrogen bond to either orientation of the phosphate group, as shown in Fig. 2 ▶(b). In molecule A, Lys66 is directed away from the phosphate group of the corresponding U5P.
Figure 2.

View of uridine 5′-monophosphate in the pyrimidine-binding site of RNase A. Electron density from F o − F c maps calculated with the respective U5P omitted from the structure-factor calculations are shown contoured at 2.5σ. (a) U5P bound to molecule A of RNase A. (b) U5P bound to molecule B of RNase A showing the disordered phosphate group. Hydrogen-bonding interactions are shown as dashed lines with distances given in Å.
Figure 3.

Overall view of RNase A molecule A, shown as an electrostatic surface, with U5P in the active-site cleft.
4. Discussion
4.1. Structural comparison: hinge angles and r.m.s. deviations
108 wild-type structures of bovine pancreatic RNase A have been deposited in the Protein Data Bank, many of which have multiple copies of the molecule in the asymmetric unit. A comparison of all these structures would be impractical. We will therefore limit our discussion to two subsets. The first subset contains the 29 apo wild-type structures and the second subset consists of wild-type structures in which a pyrimidine is bound in the B1 subsite, as found in the structure presented here. These two sets of RNase A structures represent six different unit cells and four different space groups and a resolution range of 1.15–2.50 Å for the apo structures and 1.25–2.32 Å for the ligated structures. It was hoped that such an analysis would reveal what changes occur in the enzyme structure upon substrate binding.
Table 2 ▶ is a tabulation of the hinge angles between the A and B domains, as designated by Radha Kishan et al. (1995 ▶) and defined as the angle between the vectors from the center of mass of the Cα atoms of the designated hinge residues (residues 14, 15, 47, 48, 80, 81, 102, 103 and 104) to the centers of mass of the Cα atoms of the two domains. Changes to this angle upon substrate binding would quantify the overall change that occurs in the RNase A molecule upon substrate binding. The overlap of the ranges of these data suggest that there is essentially no difference between the apo structures and the pyrimidine-containing structures. Our structure alone has a difference of 2.9° between the two molecules in the asymmetric unit; the apo structures as a group have a range of 3.0°, while the pyrimidine-containing structures as a group have a range of 2.8°. Thus, using this definition for the hinge angle there is essentially no difference in hinge angles within two standard deviations of the mean.
Table 2. Hinge angles between the A and B domains of various wild-type RNase A structures.
Estimated standard deviations are given in parentheses.
| Hinge angles | ||
|---|---|---|
| Structure set | Range | Average |
| Using all Cα atoms | ||
| Our structure | 93.7–96.6 | 95.2 (2.1) |
| 1u1b | 93.0–94.1 | 93.6 (0.8) |
| 3dxg | 94.4–95.8 | 95.1 (1.0) |
| Average of 29 apo structures | 93.2–96.2 | 94.8 (0.7) |
| Average of 28 pyrimidine-containing structures | 93.0–95.8 | 94.2 (0.8) |
| Without five highly variable regions | ||
| Our structure | 95.5–98.1 | 96.8 (1.9) |
| 1u1b | 95.0–95.9 | 95.4 (0.6) |
| 3dxg | 95.7–97.4 | 96.6 (1.1) |
| Average of 29 apo structures | 94.3–98.0 | 96.2 (0.9) |
| Average of 28 pyrimidine-containing structures | 94.6–98.1 | 95.8 (0.9) |
The average pairwise r.m.s. deviations are presented in Table 3 ▶. Clearly, the r.m.s. deviation between the two molecules of our structure is similar to the averages within the apo group and the pyrimidine-containing group. Furthermore, in each set of structures the average r.m.s. deviation for the A domain is approximately half of that seen for the B domain. However, notwithstanding the narrow overall differences between structures, there are some residues that exhibit large deviations from one structure to the next, as demonstrated by the maximum deviations. Within the apo group, there is a maximum difference of 4.08 Å for Lys1 between the 1afu molecule II (Leonidas et al., 1997 ▶) and 1xps molecule B (Sadasivan et al., 1998 ▶) structures. In the pyrimidine-containing structures (excluding our structure), there is an even greater difference of 4.88 Å for Lys1 between the 1u1b molecule I and 1o0m molecule II (Leonidas et al., 2003 ▶) structures. Large deviations of this sort may distort the hinge-angle calculations and the conclusions derived therefrom.
Table 3. R.m.s. deviations between various subsets of wild-type RNase A structures.
Average r.m.s. deviations are followed by estimated standard deviations in parentheses.
| Single or average pairwise r.m.s. deviations (Å) | |||||
|---|---|---|---|---|---|
| Overall | A domain | B domain | Maximum deviation (Å) | Residue | |
| Our structure | 0.65 | 0.34 | 0.64 | 2.47 | 22 |
| 1u1b | 0.58 | 0.31 | 0.60 | 1.82 | 88 |
| 3dxg | 0.67 | 0.51 | 0.72 | 3.27 | 21 |
| Our structure versus3dxg | 0.77 (0.06) | 0.48 (0.20) | 0.77 (0.16) | 4.57 | 1 |
| Average of 29 apo structures | 0.45 (0.16) | 0.34 (0.12) | 0.42 (0.18) | 4.08 | 1 |
| Average of 28 ligated structures | 0.55 (0.16) | 0.37 (0.14) | 0.61 (0.19) | 5.37 | 1 |
| Range of 29 apo structures | 0.06–0.87 | 0.04–0.73 | 0.05–1.00 | ||
| Range of 28 ligated structures | 0.13–0.98 | 0.06–0.83 | 0.13–1.02 | ||
| Apo versus ligated structures | 0.55 (0.15) | 0.38 (0.11) | 0.57 (0.19) | 5.18 | 1 |
| Average of all 57 structures | 0.53 (0.16) | 0.37 (0.12) | 0.55 (0.20) | 5.37 | 1 |
| C2 structures (average cell: a = 100.5, b = 32.8, c = 72.7 Å, β = 90.4°) | |||||
| Average of all 24 structures | 0.50 (0.18) | 0.34 (0.16) | 0.56 (0.19) | 4.68 | 20 |
| Average of 8 apo structures | 0.44 (0.18) | 0.32 (0.14) | 0.48 (0.19) | 3.89 | 20 |
| Average of 16 ligated structures | 0.49 (0.18) | 0.32 (0.18) | 0.56 (0.20) | 4.50 | 20 |
| P21 (small) (average cell: a = 30.1, b = 38.0, c = 53.2 Å, β = 106.9°) | |||||
| Average of all 19 structures | 0.26 (0.12) | 0.21 (0.06) | 0.25 (0.16) | 1.91 | 1 |
| Average of 15 apo structures | 0.27 (0.13) | 0.21 (0.07) | 0.26 (0.18) | 1.90 | 21 |
| Average of 4 ligated structures | 0.19 (0.03) | 0.19 (0.04) | 0.19 (0.02) | 1.38 | 1 |
| P21 (large a) (average cell: a = 33.8, b = 100.8, c = 31.3 Å, β = 100.2°) | |||||
| Average of 4 apo structures | 0.47 (0.10) | 0.40 (0.08) | 0.30 (0.08) | 2.35 | 1 |
| P21 (large b) (average cell: a = 30.9, b = 75.4, c = 51.2 Å, β = 107.1°) | |||||
| Average of 4 ligated structures | 0.58 (0.16) | 0.30 (0.07) | 0.58 (0.17) | 2.47 | 22 |
| P3221 (average cell: a = b = 65.3, c = 65.4 Å) | |||||
| Average of all 4 structures | 0.36 (0.14) | 0.23 (0.06) | 0.34 (0.12) | 2.42 | 21 |
| Average of 3 ligated structures | 0.24 (0.03) | 0.18 (0.05) | 0.23 (0.01) | 1.32 | 21 |
| P212121 (average cell: a = 44.4, b = 73.4, c = 43.6 Å) | |||||
| R.m.s. deviation between 2 structures | 0.50 | 0.45 | 0.49 | 2.15 | 1 |
| Structure series (all one molecule per asymmetric unit) | |||||
| Average in temperature structure series† | 0.22 (0.06) | 0.21 (0.06) | 0.19 (0.06) | 1.08 | 1 |
| Average in pH structure series‡ | 0.08 (0.03) | 0.06 (0.02) | 0.09 (0.04) | 0.52 | 102 |
| Average in ionic strength structure series§ | 0.19 (0.04) | 0.12 (0.03) | 0.19 (0.04) | 1.82 | 21 |
Using structure 1kf5 (Berisio et al., 2002 ▶) as the reference molecule because it is the highest resolution apo wild-type RNase A structure in the PDB and because it was crystallized at neutral pH, we superimposed the other 56 structures in our sample set (including our structure). Fig. 4 ▶(a) shows a plot of the average and maximum deviations by residue and illustrates the fact that there are several regions that are highly variable in structure. Fig. 4 ▶(b) displays a plot of the frequency with which each residue in the 56 structures deviates by 1 Å or more from the reference structure, 1 Å being about twice the average r.m.s. deviation of the structures as a whole. Maximal variation occurs at the N-terminal Lys1 and in the loop around Ala20-Ser21-Ser22 (which is easily cleaved by proteases to form RNase S), with frequencies exceeding 50% of all structures. However, three other loop regions, residues 34–39, 66–69 and 87–94, are also quite variable, with the last two having frequencies of around 50% of structures deviating by more than 1 Å. As seen in Fig. 4 ▶(a), these deviations are not isolated to either the apo group or the pyrimidine-containing group of structures. Except for the additional region 66–69, the flexible regions identified here are consistent with those identified by Radha Kishan et al. (1995 ▶) using difference distance matrices.
Figure 4.

(a) Plot of the average and maximal deviations of the Cα atoms of each residue of RNase A upon superposition of 56 structures onto the reference structure 1kf5. The maximal deviations for the group of apo structures and the group of ligated structures are also shown, which demonstrates that the high variability of certain regions are independent of the ligation state of the enzyme. (b) Plot of the frequency of deviations greater than 1 Å. For four regions, more than 50% of the structures have deviations from the reference exceeding 1 Å (twice the average r.m.s. deviation of the structures as a group).
We posed the question whether elimination of these highly variable regions would affect the hinge angles and the interpretation thereof. Thus, the second part of Table 2 ▶ lists recalculated hinge angles with the suspect regions removed from the calculations of the centers of mass. However, the conclusion that there is essentially no difference between apo and ligated structures remains the same: the range of hinge angles in the ligated set is essentially the same as the range of the ligand-free set.
There are six different unit cells represented in the 57 structures analyzed here (see Table 3 ▶ for unit-cell parameters). There are no significant differences between hinge angles for structures obtained from various crystal forms (data not shown). Likewise, the average pairwise r.m.s. deviations found in Table 3 ▶ shed little light on whether significant conformational changes occur upon substrate binding. Some observations based on these statistics, without regard to the estimated standard deviations that are listed, are the following. (i) Although there is an exception for cell P21 (large a), the B domain generally exhibits greater variation than the A domain. (ii) The apo structures have about the same variability as the ligated structures. (iii) Apart from the P212121 structures, structures with only one molecule per asymmetric unit have less variability than those with two molecules per asymmetric unit. (iv) The average r.m.s. deviations between pairs, where the pairs are composed of one apo structure and one ligated structure, are comparable to those in which both structures of the pairs are either apo structures or ligated structures, but not mixed. (v) There is considerably less variation between structures in a series where there is basically only one variable such as temperature or pH. From these observations, it would appear that RNase A undergoes very little conformational change upon substrate binding. In a broader perspective, structural comparisons are clouded by the many variables underlying each experiment producing the structures being compared. Thus, the most valid comparisons will be between structures produced by a single laboratory using exactly the same conditions, reagents and protein samples, with the only variable being the variable under study such as temperature or pH in the studies cited above or a particular set of ligands.
4.2. Comparison between the four RNase A–U5P structures
One other structure of an RNase A–U5P complex has recently been reported (PDB code 3dxg; Tsirkone et al., 2009 ▶). It was obtained by soaking U5P into C2 crystals of RNase A, which have two molecules per asymmetric unit. The U5P bound in the B1 site in both molecules and also bound to the B2 site but only in molecule A. As has been proposed to explain the absence of ligands in the B1 site in several other structures (Leonidas et al., 2003 ▶; Hatzopoulos et al., 2005 ▶), adverse lattice constraints around the active site of molecule B were proposed to be the basis for the absence of U5P in the B2 site. However, inspection of the lattice suggests that the B2 site of molecule B is more accessible than the B1 site of molecule B and is even more accessible than the B2 site of molecule A in which U5P has actually bound. We would suggest that it is the lattice contacts that in fact provide the additional interactions to bind U5P to the B2 site of molecule A. These include a direct hydrogen bond between O2′ and Thr70 OG1 and a water-mediated interaction between O3′ and Gly88 O and Asn62 OD1. Such interactions are not available at the B2 site of molecule B because the lattice neighbors are not sufficiently close to provide them. This observation further supports the proposition that the B2 site is preferential for purines and, more specifically, for adenine nucleotides.
Generally, the variation in protein structure between our structure and 3dxg parallels that presented in the previous discussion on hinge angles and r.m.s. deviations and is presented in Tables 2 ▶ and 3 ▶. With regard to hinge angles, the range for 3dxg is slightly narrower than for our structure but the average is the same. The r.m.s. deviations between molecules of 3dxg are slightly higher than in our structure and the maximum deviation of 3.3 Å occurs at Ser21; in our structure, the maximum deviation occurs at Ser22. The average r.m.s. deviations between the various RNase A–U5P structures are larger than the averages over all 57 structures analyzed on a pairwise basis. The backbone differences are illustrated in Fig. 5 ▶(a) and correspond to the highly variable regions identified in Fig. 4 ▶.
Figure 5.

(a) Superposition based on Cα atoms of the four independently determined structures of RNase A in complex with uridine 5′-monophosphate. Highly variable regions are identified. (b) U5P molecules from 3dxg and the B molecule of the present study. Relative positioning is based on the superpositioning that produced (a). (c) U5P from all four complexes. In all three images, U5P-A and U5P-B from 3dxg are shown in green and blue, respectively, and U5P-A and U5P-B from the present study are shown in magenta and red, respectively.
Fig. 5 ▶(b) illustrates the differences between the U5P molecules in the four complexes. The important torsion angles are listed in Table 4 ▶. The base rings of all U5P molecules have the anti conformation. All ribose rings have the C3′-endo conformation, except for U5P-B of 3dxg, which has the C2′-endo conformation. The β and γ angles dictate the position of the phosphate group. In 3dxg these angles vary, giving rise to two distinct orientations, both of which are represented by the disordered phosphate group of U5P-B of our structure; this suggests that the pucker of the ribose ring of U5P-B of 3dxg does not dictate the orientation of its phosphate. U5P-A of our structure resembles U5P-A of 3dxg.
Table 4. U5P torsion angles (°).
| Our molecule B | |||||
|---|---|---|---|---|---|
| Torsion angle | Our molecule A | Conformer A | Conformer B | 3dxg molecule A | 3dxg molecule B |
| α1 | 108.2 (O3P) | 56.8 (O2P) | 99.7 (O2P) | 49.5 (O1P) | 65.7 (O1P) |
| α2 | −131.8 (O2P) | 180.0 (O1P) | −140.6 (O1P) | 170.3 (O3P) | −173.3 (O3P) |
| α3 | −12.0 (O1P) | −60.7 (O3P) | −19.8 (O3P) | −71.5 (O2P) | −53.1 (O2P) |
| β | −178.0 | −180.0 | −141.5 | −174.8 | −145.1 |
| γ | 108.1 | 74.6 | 33.8 | 71.2 | 60.3 |
| δ | −152.4 | −156.7 | −156.7 | −157.8 | −105.9 |
| ∊ | 29.7 | 38.8 | 38.8 | 31.0 | −31.1 |
| χ | −151.9 | −151.5 | −151.5 | −148.7 | −140.5 |
Specific enzyme–U5P interactions at the B1 site in the two structures are listed in Table 5 ▶. There are only four conserved interactions in the four molecules: the pyrimidine-specific interactions between Thr45 and U5P and the water-mediated interactions between O2′ and Phe120 N and His12 NE2. In our structure Lys41 interacts directly with O2′, but in 3dxg U5P-A the interaction is water-mediated and in 3dxg U5P-B Lys41 interacts with O3′ owing to the different pucker of its ribose ring. In 3dxg U5P-A there are four interactions with the U5P molecule in the B2 site which are unique among the four complexes. Only U5P-B of our structure has a phosphate–Lys66 interaction; the other three U5P molecules have a water-mediated interaction between the phosphate group and Asp121 O.
Table 5. The ligand–enzyme interactions in the four RNase A–U5P complexes.
Conserved interactions over all four complexes are shown in bold. U5P2 is the U5P at the B2 site of molecule A of 3dxg.
| U5P atom at B1 subsite | Interaction | Our molecule A | Our molecule B | 3dxg molecule A | 3dxg molecule B |
|---|---|---|---|---|---|
| N3 | ⋯Thr45 OG1 | Y | Y | Y | Y |
| O2 | ⋯Thr45 N | Y | Y | Y | Y |
| O2′ | ⋯wat⋯Phe120 N | Y | Y | Y | Y |
| ⋯wat⋯His12 NE2 | Y | Y | Y | Y | |
| ⋯Lys41 NZ | Y | Y | N | N | |
| ⋯Phe120 O | N | N | N | Y | |
| ⋯wat⋯Gln11 NE2 | N | Y | N | N | |
| ⋯wat⋯U5P2 O2P | N | N | Y | N | |
| ⋯wat⋯Asn22 OD1 | N | N | Y | N | |
| ⋯wat⋯Lys41 NZ | N | N | Y | N | |
| O3′ | ⋯Lys41 NZ | N | N | N | Y |
| ⋯wat⋯Phe120 O | Y | N | N | N | |
| ⋯wat⋯Gln11 NE2 | N | Y | N | N | |
| ⋯U5P2 O3P | N | N | Y | N | |
| ⋯wat⋯U5P2 O1P | N | N | Y | N | |
| ⋯wat⋯U5P2 O2P | N | N | Y | N | |
| O1P | ⋯Lys66 NZ | N | Y | N | N |
| O3P | ⋯wat⋯Asp121 O | Y | N | Y | Y |
4.3. Invariant waters
Sadasivan et al. (1998 ▶) identified 14 invariant water molecules based on the common water molecules found in eight wild-type structures. For molecule A, we found 12 water molecules within 1.8 Å of these 14 invariant water molecules in 7rsa (Wlodawer et al., 1988 ▶), the reference molecule Sadasivan and coworkers used for their comparison; for molecule B, we found 11 water molecules. One conserved water position that is missing in each molecule is occupied by Asn113 OD1 of a symmetry-related molecule.
4.4. Chloride ions
Since we introduced chloride ions into the crystallization solutions with the basic fuchsin component of the small-molecule cocktail, we looked at the chloride-ion locations in nine chloride-containing structures of RNase A found in the PDB. There were six independent locations among these structures, two of which were in the active-site cleft and were occupied by the uridine ring and a disordered water molecule. At the other four sites we found nothing in the difference electron-density maps.
4.5. Note on TLS-group refinement
The following is an observation that may be of interest to those that use TLS refinement. We initially set up TLS groups for each molecule in the asymmetric unit, based on visual inspection of the structure, as follows: group 1, residues 1–48 and 80–102; group 2, residues 49–79 and 103–124. The refinement converged to R and R free values of 0.204 and 0.261, respectively. Revising the TLS groupings to conform to the domains identified by Radha Kishan et al. (1995 ▶) and dividing the so-called hinge residues between those domains, we set the two groups for each molecule as follows: group 1, residues 15–48 and 81–102; group 2, residues 1–14, 49–80 and 103–124. This reassignment basically switches the S peptide, residues 1–14, from one TLS group to the other. The R and R free values resulting from refinement under the new TLS groupings were 0.197 and 0.253, respectively. This seems like a dramatic effect for an essentially refined model and suggests that TLS refinement can be highly sensitive to the TLS groupings.
5. Conclusions
In the course of our investigations over the past three years, it has become increasingly evident that the inclusion of small molecules in crystallization samples of macromolecules can have profound effects. This is true for conventional small chemical compounds as well as for small biological molecules. U5P falls into the latter category. However, in this case the small molecule does not provide intermolecular lattice interactions that might stabilize the crystal lattice. On the other hand, under the crystallization conditions used here and where other small-molecule ‘cocktails’ failed to yield any crystals of RNase A, that containing U5P provided crystals of a rarely seen crystal form of RNase A.
Supplementary Material
PDB reference: ribonuclease A–uridine 5′-monophosphate complex, 3jw1, r3jw1sf
Acknowledgments
This work was supported by NIH Grant GM074899 for the establishment of the Center for High Throughput Structural Biology and NIH Grant GM080412 to AM.
References
- Altona, C. & Sundaralingam, M. (1972). J. Am. Chem. Soc.94, 8205–8212. [DOI] [PubMed]
- Beach, H., Cole, R., Gill, M. L. & Loria, J. P. (2005). J. Am. Chem. Soc.127, 9167–9176. [DOI] [PubMed]
- Berisio, R., Sica, F., Lamzin, V. S., Wilson, K. S., Zagari, A. & Mazzarella, L. (2002). Acta Cryst. D58, 441–450. [DOI] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res.28, 235–242. [DOI] [PMC free article] [PubMed]
- Birdsall, D. L. & McPherson, A. (1992). J. Biol. Chem.267, 22230–22236. [PubMed]
- Blackburn, P. & Moore, S. (1982). The Enzymes, 3rd ed., edited by P. D. Boyer, Vol. 15, pp. 317–340. New York: Academic Press.
- Borah, B., Chen, C. W., Egan, W., Miller, M., Wlodawer, A. & Cohen, J. S. (1985). Biochemistry, 24, 2058–2067. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). The PyMOL Molecular Viewer. DeLano Scientific, San Carlos, California, USA. http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Fedorov, A. A., Joseph-McCarthy, D., Fedorov, E., Sirakova, D., Graf, I. & Almo, S. C. (1996). Biochemistry, 35, 15962–15979. [DOI] [PubMed]
- Hatzopoulos, G. N., Leonidas, D. D., Kardakaris, R., Kobe, J. & Oikonomakos, N. G. (2005). FEBS J.272, 3988-4001. [DOI] [PubMed]
- Jenkins, C. L., Thiyagarajan, N., Sweeney, R. Y., Guy, M. P., Kelemen, B. R., Acharya, K. R. & Raines, R. T. (2005). FEBS J.272, 744–755. [DOI] [PubMed]
- Jones, T. A. & Kjeldgaard, M. (1994). O – The Manual, v.5.10. Uppsala University Press.
- Kennard, O. & Taylor, R. (1982). J. Am. Chem. Soc.104, 3209–3212.
- Ladner, J. E., Wladkowski, B. D., Svensson, L. A., Sjölin, L. & Gilliland, G. L. (1997). Acta Cryst. D53, 290–301. [DOI] [PubMed]
- Larson, S. B., Day, J. S., Cudney, R. & McPherson, A. (2007). Acta Cryst. D63, 310–318. [DOI] [PubMed]
- Larson, S. B., Day, J. S., Nguyen, C., Cudney, R. & McPherson, A. (2008). Cryst. Growth Des.8, 3038–3052.
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Leonidas, D. D., Chavali, G. B., Oikonomakos, N. G., Chrysina, E. D., Kosmopoulou, M. N., Vlassi, M., Frankling, C. & Acharya, K. R. (2003). Protein Sci.12, 2559–2574. [DOI] [PMC free article] [PubMed]
- Leonidas, D. D., Shapiro, R., Irons, L. I., Russo, N. & Acharya, K. R. (1997). Biochemistry, 36, 5578–5588. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- McPherson, A. (1999). Crystallization of Biological Macromolecules. New York: Cold Spring Harbor Laboratory Press.
- McPherson, A., Brayer, G., Cascio, D. & Williams, R. (1986). Science, 232, 765–768. [DOI] [PubMed]
- McPherson, A. & Cudney, B. (2006). J. Struct. Biol.156, 387–406. [DOI] [PubMed]
- Moodie, S. L. & Thornton, J. M. (1993). Nucleic Acids Res.21, 1369–1380. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nachman, J., Miller, M., Gilliland, G. L., Carty, R., Pincus, M. & Wlodawer, A. (1990). Biochemistry, 29, 928–937. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Radha Kishan, K. V., Chandra, N. R., Sudarsanakumar, C., Suguna, K. & Vijayan, M. (1995). Acta Cryst. D51, 703–710. [DOI] [PubMed]
- Raines, R. T. (1998). Chem. Rev.98, 1045–1065. [DOI] [PubMed]
- Richards, F. M. & Wyckoff, H. W. (1971). The Enzymes, edited by P. D. Boyer, Vol. 4, pp. 647–806. New York: Academic Press.
- Sadasivan, C., Nagendra, H. G. & Vijayan, M. (1998). Acta Cryst. D54, 1343–1352. [DOI] [PubMed]
- Saenger, W. (1983). Principles of Nucleic Acid Structure, pp. 70–86. New York: Springer.
- Tilton, R. F. Jr, Dewan, J. C. & Petsko, G. A. (1992). Biochemistry, 31, 2469–2481. [DOI] [PubMed]
- Tsirkone, V. G., Dossi, K., Drakou, C., Zographos, S. E., Kontou, M. & Leonidas, D. D. (2009). Acta Cryst. F65, 671–677. [DOI] [PMC free article] [PubMed]
- Wlodawer, A. (1985). Biological Macromolecules and Assemblies, edited by F. A. Jurnak & A. McPherson, Vol. 2, pp. 393–439. New York: John Wiley & Sons.
- Wlodawer, A., Svensson, L. A., Sjölin, L. & Gilliland, G. L. (1988). Biochemistry, 27, 2705–2717. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ribonuclease A–uridine 5′-monophosphate complex, 3jw1, r3jw1sf
PDB reference: ribonuclease A–uridine 5′-monophosphate complex, 3jw1, r3jw1sf