

The structure of human diamine oxidase has been refined to 2.1 Å resolution in space group C2221.
Keywords: diamine oxidases, copper amine oxidases, topaquinone, histaminase, kidney amine oxidases
Abstract
Copper amine oxidases (CAOs) are ubiquitous in nature and catalyse the oxidative deamination of primary amines to the corresponding aldehydes. Humans have three viable CAO genes (AOC1–3). AOC1 encodes human diamine oxidase (hDAO), which is the frontline enzyme for histamine metabolism. hDAO is unique among CAOs in that it has a distinct substrate preference for diamines. The structure of hDAO in space group P212121 with two molecules in the asymmetric unit has recently been reported. Here, the structure of hDAO refined to 2.1 Å resolution in space group C2221 with one molecule in the asymmetric unit is reported.
1. Introduction
Histamine mediates a multitude of different processes and prompt inactivation of exogenous and endogenous histamine is crucial to avoid undesirable reactions. In humans, histamine can be metabolized in two ways (Schwelberger, 2004 ▶). The first occurs via methylation of the imidazole group to form the inactive Nτ-methylhistamine by the cytosolic protein N-methyltransferase. The second involves oxidative deamination of the primary amine to imidazole acetaldehyde by the secreted diamine oxidase (hDAO). hDAO is believed to be the frontline enzyme involved in clearing both endogenous and exogenous histamine and is speculated to be released from basolateral vesicles to clear histamine on response to an external stimulus (Schwelberger, 2004 ▶, 2007 ▶). An inherited or acquired decrease in hDAO activity is directly correlated with histamine intolerance (Maintz & Novak, 2007 ▶). Furthermore, the balance between hDAO and histamine is thought to be crucial for an uncomplicated pregnancy (Maintz et al., 2008 ▶).
Diamine oxidase (DAO) is a member of the copper-containing amine oxidase (CAO) family of enzymes that are distinguished by the presence of a type II copper ion and a protein-derived trihydroxyphenylalanine quinone (TPQ) cofactor. In the presence of water and molecular oxygen, CAOs catalyse the oxidative deamination of primary amines to the corresponding aldehyde with the concomitant production of hydrogen peroxide and ammonia.
Three genes, AOC1–3, code for viable CAOs in humans (Schwelberger, 2006 ▶). AOC3 codes for the membrane-bound vascular adhesion protein (VAP-1), from which a soluble form, sVAP-1, is produced, presumably by proteolytic cleavage of the anchored membrane domain from the soluble portion (Kurkijarvi et al., 1998 ▶). AOC2 codes for a retina-specific CAO (RAO). AOC1 codes for the diamine oxidase enzyme. Humans also have an AOC4 gene which, owing to a nonsense mutation at position 225, translates to a nonfunctional truncated protein (Schwelberger, 2006 ▶). While VAP-1 and RAO have a sequence identity of 65%, DAO has only an approximately 38% pairwise identity to either VAP-1 or RAO.
hDAO is unique among the structurally characterized CAOs in that it shows a distinct specificity for diamine substrates over the more ‘traditional’ monoamine CAO substrates such as methylamine and benzylamine (Elmore et al., 2002 ▶). In addition to histamine, the diamines putrescine (1,4-diaminobutane) and cadaverine (1,5-diaminopentane) are also excellent substrates of hDAO. DAO is highly expressed in the placenta (Elmore et al., 2002 ▶), kidney (Klocker et al., 2005 ▶; Schwelberger et al., 1998 ▶), gut (Bieganski et al., 1983 ▶; Raithel et al., 1999 ▶) and lung, and is found at somewhat lower levels in the brain (Elmore et al., 2002 ▶; Klocker et al., 2005 ▶).
To date, crystal structures of CAOs are known from all kingdoms of life with the exception of archaea. CAOs are homodimers consisting of subunits of around 700 residues (Guss et al., 2009 ▶). The first structure described for a CAO was that from Escherichia coli (ECAO; Parsons et al., 1995 ▶). ECAO was described as being shaped like a mushroom, with the D1 domain forming the stalk and linked to a cap made up from the remaining three domains (D2–4). Structures from other organisms, including the bacterium Arthrobacter globiformis, the yeasts Hansuela polymorpha and Pichia pastoris, the plant Pisum sativum and the mammals Homo sapiens and Bos taurus, have been reported. ECAO is the only structure to contain the stalk-like D1 domain, but all of the structures retain a high degree of structural similarity for domains 2–4.
The common architecture observed in CAOs belies the vast differences shown in the substrate preferences and biological roles of these ubiquitous enzymes. The differences in substrate preference stem from the varying chemical properties and dimensions of the substrate channel. The exceptionally large and wide substrate cavity of the Pichia pastoris enzyme has been proposed to allow oxidation of peptidyl lysine residues (Duff et al., 2003 ▶) and is in contrast to the narrow channels of the E. coli (Parsons et al., 1995 ▶) and A. globiformis (Wilce et al., 1997 ▶) enzymes. We have previously shown that the hDAO substrate channel is much narrower than that of the other structurally characterized human CAO, hVAP-1. In hDAO the channel divides into two before reforming into one (McGrath et al., 2009 ▶). The residues in close proximity to the TPQ, however, are mostly conserved between VAP-1 and hDAO.
The TPQ has been observed in two distinct conformations. In one the TPQ coordinates the copper ion and is referred to as being ‘on-copper’. The ‘off-copper’ conformation results from the TPQ swinging out towards the substrate channel, making a carbonyl O atom (O5) of the TPQ available for substrate binding. In the A. globiformis, E. coli, Hansuela polymorpha and Pisum sativum enzymes the position of a tyrosine residue has been shown to ‘gate’ the access of substrates to the TPQ (Guss et al., 2009 ▶). Furthermore, a link between the presence of a molecule bound in the active-site channel and the TPQ being ‘off-copper’ has been described for the A. globiformis enzyme (AGAO; Langley, Brown et al., 2008 ▶). We have previously shown that hDAO does not follow either of these paradigms (McGrath et al., 2009 ▶). In hDAO the residue corresponding to the gate is in an intermediate position that does not block the channel regardless of whether or not an inhibitor is bound in the substrate channel.
We have previously described the crystal structure of native hDAO and the structures of hDAO in complex with the diamino inhibitors berenil [1,3-bis(4′-amidinophenyl)triazene] and pentamidine [1,5-bis(4-amidinophenoxy)pentane] in space group P212121 containing two molecules in the asymmetric unit (McGrath et al., 2009 ▶). These structures are all from crystals of form II. In this paper, we describe a new crystal form of native hDAO, which is referred to as crystal form I. The crystal form I structure of hDAO has been refined to 2.1 Å resolution in the orthorhombic space group C2221 with one molecule in the asymmetric unit.
2. Materials and methods
2.1. Crystallization, X-ray data collection and structure solution
Recombinant hDAO was expressed as a secreted enzyme in Drosophila S2 cells and purified using a previously published protocol (Elmore et al., 2002 ▶) with an additional dialysis step (10 kDa molecular-weight cutoff, Pierce) against 3 × 500 ml changes of 100 mM HEPES pH 7.2 and 150 mM KCl at 277 K. The purified enzyme was then concentrated to ∼10 mg ml−1 in preparation for protein crystallization experiments.
Hanging-drop vapour-diffusion crystallization experiments were prepared with a Mosquito robot (Molecular Dimensions) using drops consisting of 200 nl protein solution and 200 nl crystallant solution equilibrated over 75 µl reservoir solution at room temperature. Plate-like crystals were observed after eight weeks in 0.1 M MES pH 6.5 and 12%(w/v) PEG 20K in the NeXtal Classics screen (Qiagen). These initial conditions were optimized using manually dispensed hanging-drop crystallization experiments containing 2 µl each of protein and crystallant equilibrated over 500 µl reservoir solution. The best diffracting crystals were grown using 0.1 M MES pH 6.1 and 12%(w/v) PEG at room temperature over a period of two months. The protein crystals were cryoprotected for low-temperature data collection by transferring them briefly to a drop containing reservoir solution with the addition of 30%(v/v) glycerol prior to flash-cooling in a stream of N2.
The final data set for refinement consisted of data collected at two sources from one crystal. X-ray diffraction data were first collected in-house using Cu Kα X-rays produced by a Rigaku RU200H rotating-anode generator with Osmic optics and were recorded on a MAR345 image plate (MAR Research) to 2.9 Å resolution. The data were indexed and scaled using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). A search model for molecular replacement was constructed with the program CHAINSAW (Stein, 2008 ▶) using a template model derived from the structure of VAP-1 (PDB code 2c10; Jakobsson et al., 2005 ▶) from which the solvent and metal ions had been removed. Side chains were pruned to common atoms based on a sequence alignment generated using ClustalW (Larkin et al., 2007 ▶). A clear molecular-replacement solution was found with the program Phaser (McCoy, 2007 ▶) in space group C2221 (Z score = 29.1, log-likelihood gain = 590). The resulting model was first refined as a rigid body with REFMAC5 (Murshudov et al., 1997 ▶), followed by multiple rounds of restrained refinement with simulated annealing in PHENIX (Afonine et al., 2005 ▶) interspersed with restrained refinement using REFMAC5. Between rounds of refinement the model was manually checked and corrected against the corresponding σA-weighted electron-density maps in Coot (Emsley & Cowtan, 2004 ▶). Solvent molecules were added as the refinement progressed either manually or automatically within Coot and were routinely checked for correct stereochemistry, for sufficient supporting density above a 2F o − F c σ threshold of 1.0σ and for a reasonable thermal factor. The highest peak in the difference electron density corresponded to the position of the copper ion. The next two highest peaks could be assigned as Ca2+ ions based on the stereochemistry of their coordination spheres and by homology with the closely related CAO structures of bovine serum amine oxidase (BSAO) and VAP-1. Carbohydrate residues were modelled at three of the four putative N-linked glycosylation sites identified by the presence of the sequon Asn-X-Thr, where X can be any residue.
The TPQ was only included in the model in the final stages of refinement to ensure that it was positioned within the difference density in the most unbiased manner. The quality of the model was regularly checked for steric clashes, incorrect stereochemistry and rotamer outliers using MolProbity (Davis et al., 2007 ▶).
Data to 2.11 Å resolution were subsequently recorded from the same crystal at the Australian Synchrotron. In-house data as well as the synchrotron-radiation diffraction data were integrated separately using MOSFLM (Leslie, 1992 ▶) and scaled together using SCALA (Collaborative Computational Project, Number 4, 1994 ▶). The refinement protocol described above was followed to produce a model with reasonably good refinement statistics. However, there were several regions of electron density that could not be interpreted in terms of a molecular model.
Subsequently, crystals in space group P212121 were grown and the structure was solved using the partially refined model from the form I crystals as reported previously (McGrath et al., 2009 ▶). Briefly, this model underwent a similar refinement protocol as the form I crystals and showed superior refinement statistics and electron density for portions of the structure not observed in form I. This refined structure was superposed on the form I model using secondary-structure matching and edited to reflect the form I model as closely as possible in order to avoid carryover model bias. This model underwent refinement as stated above using the form I data, resulting in the structure reported here.
3. Results and discussion
3.1. The structure of hDAO in C2221 (form I) and comparisons to form II hDAO
The data-processing and refinement statistics for the form I model are listed in Table 1 ▶. One monomer is present in the asymmetric unit, comprising residues 27–264 and 278–751. A crystallographic twofold relates the subunits of the biological dimer. There is no evidence of density before residue 27 and very little or no density between residues 265 and 277. Residues 20–26 and 265–277 are disordered in the form I structure and in both subunits of the form II structure. The model contains one type II Cu ion, two Ca2+ ions, one glycerol molecule, five N-acetylglucosamine units and 299 solvent molecules. The r.m.s. deviation between the subunit of form I hDAO and the A and B chains of form II hDAO are 0.41 Å (for 711 Cα atoms) and 0.38 Å (for 709 Cα atoms), respectively. The TPQ is in the ‘on-copper’ conformation in all structures of hDAO.
Table 1. Diffraction data and refinement statistics for hDAO form I.
Values in parentheses are for the highest resolution shell.
| Space group | C2221 |
| Unit-cell parameters (Å) | a = 94.8, b = 97.0, c = 178.1 |
| X-ray source | AUS 3BM1/rotating anode |
| Wavelength (Å) | 0.95663/1.5418 |
| Detector | ADSC Quantum 210r/MAR 345 |
| Resolution range (Å) | 178.1–2.1 (2.21–2.10) |
| Observed reflections | 332912 |
| Unique reflections | 45200 |
| Completeness (%) | 95.2 (96.2) |
| Multiplicity | 7.4 (3.0) |
| 〈I/σ(I)〉 | 15.7 (2.5) |
| Rmerge† | 0.11 (0.3) |
| Rp.i.m.‡ | 0.03 (0.19) |
| Reflections in working set | 42911 |
| Reflections in test set | 2288 |
| Protomers per ASU | 1 |
| Total atoms (non-H) | 5983 |
| Protein atoms | 5605 |
| Metal atoms | 3 |
| Water atoms | 299 |
| Atoms in alternate conformers | 34 |
| Other atoms | 76 |
| Rcryst | 0.236 (0.321) |
| Rfree | 0.290 (0.367) |
| R.m.s.d. bond lengths (Å) | 0.009 |
| R.m.s.d. bond angles (°) | 1.2 |
| 〈B〉 (Å2) | 26.0 |
| Cruickshank’s DPI§ (Å) | 0.3 |
| PDB code | 3k5t |
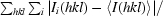

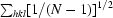
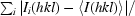
hDAO has the archetypal CAO fold (Fig. 1 ▶). Each subunit of hDAO is comprised of domains D2 (residues 27–135), D3 (residues 144–258) and D4 (residues 310–751). These domains are linked by loop regions consisting of residues 136–143 between D2 and D3 and residues 259–309 linking D3 to D4. D2 and D3 are similar in composition, containing relatively equal amounts of β-sheet and α-helices, and have been proposed to be the result of a gene-duplication event (Parsons et al., 1995 ▶). D2 apparently plays a purely structural role as it has not been assigned a catalytic function. In addition to playing a structural role similar to that of D2, D3 provides elements of the substrate channel, including an aspartic acid residue (Asp186) which we have proposed to be involved in binding the second amino group of the preferred diamine substrates (McGrath et al., 2009 ▶). The largest domain, D4, contains the active site and two extended β-hairpin ‘arms’ which interlock the two subunits, burying just under half of the solvent-accessible surface at the dimer interface. The total buried solvent-accessible interface was calculated to be 8692 Å2 per subunit using PISA (Krissinel & Henrick, 2007 ▶). The dimer interface forms an internal ‘lake’ which is proposed to allow the passage of smaller products and substrates (Duff et al., 2003 ▶; Wilce et al., 1997 ▶). A central β-sheet sandwich dominates D4 and consists of twisted eight-stranded and six-stranded β-sheets, both of which contribute elements of the active site.
Figure 1.

Stereoviews of the structure of hDAO in two orientations. One subunit is coloured by domain. Domain 2 (D2) is shown in red, domain 3 (D3) in green and domain 4 (D4) in blue. The linkers joining D2 to D3 and D3 to D4 are coloured yellow. The symmetry-related subunit of the homodimer is shown in wheat. Bound metals are shown as spheres in both subunits, with coppers shown in orange and calcium ions in green. The TPQ, the histidine residues coordinating the copper and carbohydrates are shown as sticks.
The active-site copper ion is coordinated in a distorted tetrahedral geometry by the N∊2 atoms of the imidazole side chains of His510 and His512 and the Nδ1 atom of His675 and apically by the O4 atom of the ‘on-copper’ TPQ cofactor. The TPQ cofactor is modelled as being 50% tyrosine, which is consistent with earlier experiments indicating that approximately one mole of TPQ per dimer was formed in recombinant hDAO in the insect-cell expression system (Elmore et al., 2002 ▶). The TPQ makes contact with a glycerol molecule in the active site. The equivalent glycerol in the form II crystals is in a slightly different orientation and does not directly contact the TPQ. A hydroxyl group of the glycerol is within hydrogen-bonding distance of O2 of the TPQ (Fig. 2 ▶). The TPQ also makes a contact with Thr685, which is observed in a single conformation (Fig. 2 ▶). In the form II crystals the side chain of Thr685 is observed in two conformations.
Figure 2.

Stereoview of the active site of hDAO. Contacts made by the TPQ to the active-site Cu2+, Thr685 and to a glycerol molecule within the active-site channel are shown as dashed lines.
Two secondary cation-binding sites have been modelled as Ca2+ ions. The first is present in all CAOs except for HPAO and is likely to play an important structural role (Guss et al., 2009 ▶). Oxygen ligands provided by the carboxylate groups of three aspartic acid residues, Asp519, Asp521, Asp664, the carbonyl O atoms of Leu520 and Leu665 and one water molecule provide octahedral geometry for the bound calcium (Fig. 3 ▶ a). These residues are completely conserved in the other mammalian amine oxidase structures, BSAO and VAP-1. The second calcium is coordinated by one water atom from the side chains of Asn656 and Glu658 and twice (bidentate) by the carboxyl group of Glu562 and the carbonyl O atom of Phe653 (Fig. 3 ▶ b). The residues coordinating the second calcium are also completely conserved in BSAO and VAP-1. However, in the form I structure one water ligand present in the form II structure is completely absent from the electron density. This water is also missing in the two available native VAP-1 structures. Its absence is most likely attributed to the lower resolution of these structures compared with that of the native form II hDAO structure.
Figure 3.

Stereoviews of the two calcium-binding sides of hDAO. Contacts made by coordinating-residue side chains and carbonyl O atoms are shown as dashed lines.
Two disulfides have been modelled in the present structure. The first, between Cys391 and Cys417, is present in all CAO structures apart from ECAO. The second disulfide linking Cys177 and Cys181 stabilizes a short loop between residues 174 and 181. In the current structure this region shows good density for the disulfide linkage but poor density for the surrounding loop residues. In form II hDAO this region was well ordered; however, there is a slight displacement between the two subunits, which are not related by crystallographic symmetry in the form II structure. Owing to the movement in this region between the two subunits in the form II structure and the poor density in the form I structure, it is presumed that this region is inherently flexible. An intermolecular disulfide observed in the form II structure between symmetry-related Cys736 residues in the two subunits appears to be partially reduced in the form I structure. The predominant conformation of Cys736 in the form I structure is in the reduced state and has been modelled as such. There is insufficient density to model the partial conformation which is involved in the intermolecular disulfide. The reduced state of this bond would seem to be the result of synchrotron-radiation damage.
Recombinant hDAO has been reported to contain approximately 20–26% carbohydrate by weight (Elmore et al., 2002 ▶). As in the form II structure, well resolved oligosaccharide chains are present at three of the four putative N-linked glycosylation sites on asparagine residues 110, 538 and 745, which are conserved in all reported mammalian diamine oxidases. In the form II structure the fourth site, Asn168, showed no electron density to support the presence of glycosylation even though it is well placed to allow unhindered access for glycosylation. In the form I structure spurious positive density is present that suggests the possibility of glycosylation occurring at this site; however, when the N-acetylglucosamine residue was modelled it lacked sufficient 2F o − F c density to be confidently included in the final model. This putative site is not conserved in mammalian diamine oxidases.
We have previously identified two residues as being important in substrate binding in hDAO: Ser380 and Asp186 (McGrath et al., 2009 ▶). Ser380 is seen to be in the same dual conformation in the form I structure as in the form II native structure and the form II hDAO–berenil complex structure. When bound to the diamino inhibitor pentamidine (PNT), Ser380 binds the terminal amidinium group of PNT and therefore adopts a single conformation. This residue has been proposed to be involved with binding longer substrates such as spermidine and spermine (McGrath et al., 2009 ▶). Asp186 has been implicated in orientating shorter diamine substrates within the substrate channel by binding to the second amino group. This residue has also been shown to be in an ideal position to bind to an imidazole N atom of histamine (McGrath et al., 2009 ▶). This residue is in much the same conformation in the form I and form II structures.
In previous CAO structures a tyrosine residue was seen to gate entry to the TPQ buried at the bottom of the active-site cavity. The residue corresponding to the ‘gate’ residue identified in other CAO structures is Tyr459 in hDAO. In both the form I and form II structures this residue is observed to be well ordered halfway between the ‘open’ and ‘closed’ conformations observed in various AGAO structures (Langley, Brown et al., 2008 ▶; Langley, Trambaiolo et al., 2008 ▶; O’Connell et al., 2004 ▶). We have noted previously that the corresponding residue is in the same conformation in all mammalian CAO structures (VAP-1 and BSAO). In this position the Tyr residue does not block access to the TPQ and cannot therefore function as a gate.
4. Conclusion
We have reported a second crystal form of human diamine oxidase at 2.1 Å resolution. The form I structure contains one molecule in the asymmetric unit. Overall, the structure is very similar to the form II structure of hDAO reported previously, with minor differences in loop regions and at a C-terminal disulfide bond, which appears to be reduced in the form I structure.
Supplementary Material
PDB reference: human diamine oxidase, 3k5t
Acknowledgments
This research was supported by a grant from the Australian Research Council (LP0669658 to CC and JMG) and from the NIH (GM 27659 to DMD). APM was supported by an Australian Postgraduate Award for Industry. Some of the data for this research were recorded on the 3BM1 beamline at the Australian Synchrotron, Victoria, Australia.
References
- Afonine, P. V., Grosse-Kunstleve, R. W. & Adams, P. D. (2005). CCP4 Newsl.42, contribution 8.
- Bieganski, T., Kusche, J., Lorenz, W., Hesterberg, R., Stahlknecht, C. D. & Feussner, K. D. (1983). Biochim. Biophys. Acta, 756, 196–203. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cruickshank, D. W. J. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B. III, Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res.35, W375–W383. [DOI] [PMC free article] [PubMed]
- Duff, A. P., Cohen, A. E., Ellis, P. J., Kuchar, J. A., Langley, D. B., Shepard, E. M., Dooley, D. M., Freeman, H. C. & Guss, J. M. (2003). Biochemistry, 42, 15148–15157. [DOI] [PubMed]
- Elmore, B. O., Bollinger, J. A. & Dooley, D. M. (2002). J. Biol. Inorg. Chem.7, 565–579. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Guss, J. M., Zanotti, G. & Salminen, T. (2009). Copper Amine Oxidases: Structures, Catalytic Mechanisms and Role in Pathophysiology, edited by G. Floris & B. Mondovi, pp. 119–141. Boca Raton: Taylor & Francis.
- Jakobsson, E., Nilsson, J., Ogg, D. & Kleywegt, G. J. (2005). Acta Cryst. D61, 1550–1562. [DOI] [PubMed]
- Klocker, J., Matzler, S. A., Huetz, G. N., Drasche, A., Kolbitsch, C. & Schwelberger, H. G. (2005). Inflamm. Res.54, S54–S57. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- Kurkijarvi, R., Adams, D. H., Leino, R., Mottonen, T., Jalkanen, S. & Salmi, M. (1998). J. Immunol.161, 1549–1557. [PubMed]
- Langley, D. B., Brown, D. E., Cheruzel, L. E., Contakes, S. M., Duff, A. P., Hilmer, K. M., Dooley, D. M., Gray, H. B., Guss, J. M. & Freeman, H. C. (2008). J. Am. Chem. Soc.130, 8069–8078. [DOI] [PMC free article] [PubMed]
- Langley, D. B., Trambaiolo, D. M., Duff, A. P., Dooley, D. M., Freeman, H. C. & Guss, J. M. (2008). Acta Cryst. F64, 577–583. [DOI] [PMC free article] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Maintz, L. & Novak, N. (2007). Am. J. Clin. Nutr.85, 1185–1196. [DOI] [PubMed]
- Maintz, L., Schwarzer, V., Bieber, T., van der Ven, K. & Novak, N. (2008). Hum. Reprod. Update, 14, 485–495. [DOI] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- McGrath, A. P., Hilmer, K. M., Collyer, C. A., Shepard, E. M., Elmore, B. O., Brown, D. E., Dooley, D. M. & Guss, J. M. (2009). Biochemistry, 48, 9810–9822. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- O’Connell, K. M., Langley, D. B., Shepard, E. M., Duff, A. P., Jeon, H. B., Sun, G., Freeman, H. C., Guss, J. M., Sayre, L. M. & Dooley, D. M. (2004). Biochemistry, 43, 10965–10978. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Parsons, M. R., Convery, M. A., Wilmot, C. M., Yadav, K. D., Blakeley, V., Corner, A. S., Phillips, S. E., McPherson, M. J. & Knowles, P. F. (1995). Structure, 3, 1171–1184. [DOI] [PubMed]
- Raithel, M., Kufner, M., Ulrich, P. & Hahn, E. G. (1999). Inflamm. Res.48, S75–S76. [DOI] [PubMed]
- Schwelberger, H. G. (2004). Histamine: Biology and Medical Aspects, edited by A. Falus, pp. 43–52. Budapest: SpringMed Publishing.
- Schwelberger, H. G. (2006). Inflamm. Res.55, S57–S58. [DOI] [PubMed]
- Schwelberger, H. G. (2007). J. Neural Transm.114, 757–762. [DOI] [PubMed]
- Schwelberger, H. G., Hittmair, A. & Kohlwein, S. D. (1998). Inflamm. Res.47, S60–S61. [DOI] [PubMed]
- Stein, N. (2008). J. Appl. Cryst.41, 641–643.
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135.
- Wilce, M. C., Dooley, D. M., Freeman, H. C., Guss, J. M., Matsunami, H., McIntire, W. S., Ruggiero, C. E., Tanizawa, K. & Yamaguchi, H. (1997). Biochemistry, 36, 16116–16133. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human diamine oxidase, 3k5t
PDB reference: human diamine oxidase, 3k5t