

Recombinant P. somniferum salutaridine reductase (SalR) was purified and crystallized with NADPH using the hanging-drop vapor-diffusion method. Crystals of the SalR–NADPH complex diffracted X-rays to a resolution of 1.9 Å.
Keywords: Papaver somniferum, salutaridine reductase, SDR family, plant secondary metabolism
Abstract
The opium poppy Papaver somniferum is the source of the narcotic analgesics morphine and codeine. Salutaridine reductase (SalR; EC 1.1.1.248) reduces the C-7 keto group of salutaridine to the C-7 (S)-hydroxyl group of salutaridinol in the biosynthetic pathway that leads to morphine in the opium poppy plant. P. somniferum SalR was overproduced in Escherichia coli and purified using cobalt-affinity and size-exclusion chromatography. Hexagonal crystals belonging to space group P6422 or P6222 were obtained using ammonium sulfate as precipitant and diffracted to a resolution of 1.9 Å.
1. Introduction
The opium poppy (Papaver somniferum L.) is one of the oldest known medicinal plants. The narcotic analgesic morphine and the antitussive codeine are the most important active alkaloids from this plant. The tetracyclic morphinan salutaridine is an intermediate in codeine and morphine biosynthesis. P. somniferum salutaridine reductase (SalR; EC 1.1.1.248) reduces the C-7 keto group of salutaridine to the C-7 (S)-configuration hydroxyl group of salutaridinol using the 4-pro-S hydride of NADPH. Only the (S)-configuration of salutaridinol is biologically active, as demonstrated by its transformation to codeine and morphine in vivo (Barton et al., 1965 ▶). SalR was purified from a plant cell culture and characterized by Gerardy & Zenk (1993 ▶). The SalR cDNA was identified during a cross-species comparison of gene-expression profiles between 16 different Papaver species (Ziegler et al., 2006 ▶). SalR was highly expressed in the morphine-producing P. somniferum.
SalR belongs to the short-chain dehydrogenase/reductase (SDR) family. The classical SDR NAD(P)(H)-dependent oxidoreductase is usually about 250 amino-acid residues in length (Jörnvall et al., 1995 ▶). The SDR constitutes a single-domain structure comprised of a parallel α/β-fold and a Rossmann-fold motif for NAD(P)(H) binding. The SDR family is present in animals, bacteria, viruses and plants. Genome studies have shown 71 nonredundant putative SDR genes in humans and 149 nonredundant putative SDR genes in Arabidopsis (Persson et al., 2009 ▶; Kavanagh et al., 2008 ▶). Among the cofactor and active-site motifs in the classical SDR family (Kallberg et al., 2002 ▶; Filling et al., 2002 ▶), SalR conserves the N-terminal cofactor-binding motif TGxxxGxG (Thr18–Gly25) and the isolated aspartic acid residue (Asp70). In addition, the NNAG motif (Asn97–Gly100), the catalytic tetrad (Asn152, Ser180, Tyr236, Lys240) and the C-terminal PG motif (Asn260, Pro264, Gly265, Thr269) are conserved in SalR. After residue Thr269, the C-terminal segment of SDR comprises the highly variable substrate-recognition site (Yamashita et al., 1999 ▶; Korman et al., 2004 ▶).
Of the protein structures that have been deposited in the Protein Data Bank, human carbonyl reductase 1 (CBR1) has the highest sequence identity to SalR (105 of 311 amino-acid residues; Tanaka et al., 2005 ▶; Bateman et al., 2007 ▶, 2008 ▶). SalR and CBR1 have a 42-residue insertion (Ser184–Glu225 in SalR) outside the α/β-fold structure of the prototypical SDR family. This insertion makes this type of carbonyl reductase a monomer by covering the hydrophobic surface of the symmetry-related helices (αE and αF) as reported in porcine carbonyl reductase (Ghosh et al., 2001 ▶). Furthermore, P. somniferum SalR has 29 residues of another insertion (Arg110–Met138) that does not exist in animal carbonyl reductases.
The medicinal significance of codeine and morphine makes their biosynthetic pathway an important biotechnological target. Optimization or alteration of the morphine-biosynthetic pathway requires detailed knowledge of the structures of the individual catalysts. Here, we report the crystallization and preliminary X-ray diffraction analysis of P. somniferum SalR heterologously expressed in Escherichia coli. The final expressed protein consisted of 317 amino acids with a molecular mass of 34.6 kDa.
2. Materials and methods
2.1. Cloning and expression
Cloning and expression in E. coli of P. somniferum SalR have been described previously (Ziegler et al., 2006 ▶; Kempe et al., 2009 ▶; GenBank accession No. DQ316261). SalR was amplified by PCR from the recombinant plasmid SalR/pQE-30 using Pfu DNA polymerase (Stratagene, La Jolla, California, USA) with the following profile for 30 cycles after 2 min of preheating at 367 K: 30 s at 369 K, 1 min at 325 K and 1 min at 345 K. The following primers were used for the PCR amplification: 5′-CCCgctagcATGCCTGAAACATGTCCAAATACTGT-3′ as the forward primer (the NheI site is shown in lower case) and 5′-CCCgaattcTCAAAATGCAGATAGTTCTGAACAATC-3′ as the reverse primer (the EcoRI site in shown in lower case). The 0.9 kbp DNA fragment was subcloned into an NheI/EcoRI-digested pET28a expression vector (Novagen, Madison, Wisconsin, USA). The nucleotide sequence of the SalR gene was confirmed by Sanger sequencing. The recombinant protein contained a His-tag sequence (MGSSHHHHHHSSGLVPRGSHMAS) located at the N-terminus of the complete amino-acid sequence of SalR (Met1–Phe311). An N-terminal fragment (GSHMAS) remained after thrombin digestion.
The SalR/pET28a expression construct was transformed into E. coli BL21 (DE3) Codon Plus RIL cells. Transformed E. coli was grown at 310 K while shaking at 200 rev min−1 in 4 l LB medium containing 50 µg ml−1 kanamycin until the absorbance at 600 nm (A 600) reached 0.8. The culture was cooled to 277 K for 1 h. After protein induction with 1 mM isopropyl β-d-1-thiogalactopyranoside, the culture was grown for 16 h at 289 K with shaking. Compared with the previous report (Ziegler et al., 2006 ▶), the higher yield of 4.9 mg SalR from 1 l LB medium was accomplished by inducing expression at a lower temperature and with a longer incubation time (i.e. versus 310 K for 4 h).
2.2. Purification
Purification was performed at 277 K. E. coli cells were harvested by centrifugation at 8000g for 10 min and resuspended in lysis buffer containing 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM 2-mercaptoethanol and 10%(w/v) glycerol. After sonication and centrifugation at 20 000g for 20 min, the resulting supernatant was gently mixed for 30 min with 1 ml bed volume of cobalt-affinity resin (Talon, Clontech, Mountain View, California, USA), which was previously equilibrated with lysis buffer, per litre of LB medium. The resin was washed three times with 45 ml lysis buffer using a 50 ml centrifuge tube and centrifuged at 700g for 5 min. The resin was further washed with 2.5 ml lysis buffer containing 5 mM imidazole using a 2 ml disposable gravity column (Clontech). The elution buffer was 2 ml lysis buffer containing 100 mM imidazole. The enzyme was mixed with thrombin (1/6400 the amount of SalR by weight; Sigma–Aldrich, St Louis, Missouri, USA) and dialyzed twice against 1 l lysis buffer for 24 h. Uncleaved SalR and thrombin were removed on a column containing a mixture of Talon and Benzamidine-Sepharose (GE Healthcare). The enzyme was dialyzed overnight against 1 l 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 5 mM 2-mercaptoethanol and 30%(w/v) glycerol. The enzyme was loaded onto a gel-filtration column (HiLoad 16/60 Superdex 75 prep grade, GE Healthcare) and eluted in buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl and 5 mM 2-mercaptoethanol at a flow rate of 0.5 ml min−1 using an ÄKTA Purifier (GE Healthcare). Fractions containing SalR were concentrated to 10 mg ml−1 with a Centriprep YM-10 (Millipore, Billerica, Massachusetts, USA) and stored at 193 K. The purity of the SalR protein was judged by 12.5% SDS–PAGE stained with Coomassie Brilliant Blue (Fig. 1 ▶). The enzyme activity of highly purified SalR was analyzed as described previously (Ziegler et al., 2006 ▶). A total volume of 200 µl enzyme solution containing 150 mM potassium phosphate buffer pH 6.0, 100 nmol NADPH and 20 nmol salutaridine was incubated for 1–4 min at 303 K. The enzyme reaction was initiated by the addition of salutaridine. The enzymatic product was measured by HPLC using a LiChrospher 60 RP-select B HPLC column (250 × 4 mm, 5 µm; Merck, Darmstadt, Germany). The gradient was from 0% B to 40% B in 25 min with a hold for 5 min followed by an increase to 100% B in 2 min. The specific activity was 61.7 ± 3.9 nkat per milligram of protein.
Figure 1.

SDS–PAGE shows the two major steps in the purification of P. somniferum SalR. Lane 1, molecular-weight markers; lane 2, crude E. coli extract; lane 3, flowthrough of Talon affinity resin; lane 4, eluate of the affinity resin; lane 5, eluate of gel filtration after thrombin digestion; lane 6, molecular-weight markers. The arrow indicates untagged SalR.
2.3. Crystallization
The initial crystallization experiment was performed using Crystal Screen and Crystal Screen 2 from Hampton Research. Screening was performed using the hanging-drop method, in which 1 µl protein solution containing either apoprotein or SalR–NADPH complex was mixed with the same volume of reservoir solution and incubated at 277 K. Severe precipitation was observed at pH 5.6 and below. These initial experiments were followed by screening using a mixture of varying concentrations of PEG 1500 or PEG 3350 [from 10%(w/v) to 30%(w/v)], 100 mM buffer (MES pH 5.5, pH 6.5; Tris–HCl pH 7.5, pH 8.5) and an additional 200 mM ammonium sulfate or 200 mM LiCl. Additional screening consisted of a mixture of ammonium sulfate (from 1.0 to 2.2 M), 100 mM buffer (from sodium acetate pH 5.0 to Tris–HCl pH 9.0) and 5%(v/v) PEG 400. Ammonium formate, ammonium citrate, lithium sulfate and PEG 8000 were also examined as a primary precipitant. Crystallization was further optimized by varying the drop size, the temperature, the concentrations of protein and precipitant, the pH and additives.
Thin crystals were observed with reservoir solutions containing 25%(w/v) PEG 3350, 100 mM MES pH 6.5 and 200 mM ammonium sulfate, ammonium formate or ammonium acetate. Long rod-shaped crystals or hexagonal plate-shaped crystals (Fig. 2 ▶ a) were observed with a reservoir solution containing 2.0 M ammonium sulfate, 100 mM MES pH 5.5 and 5%(v/v) PEG 400. The addition of 100 mM LiCl improved the production efficiency of sharp-edged hexagonal rods (Fig. 2 ▶ b). Larger crystals were observed on the addition of 3%(v/v) glycerol or 1%(v/v) acetone. The best crystals of the SalR–NADPH complex were grown using the vapor-diffusion method in 4 µl hanging drops. 2 µl 6 mg ml−1 SalR with 4 mM NADPH was mixed with 2 µl of a reservoir solution containing 2.1 M ammonium sulfate, 100 mM MES pH 5.8, 5%(v/v) PEG 400, 100 mM LiCl, 3%(v/v) glycerol at 277 K. The SalR–NADPH solution was made by mixing six parts of 10 mg ml−1 SalR in buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl and 5 mM 2-mercaptoethanol with four parts of 10 mM NADPH in water. The volume of the reservoir solution was 800 µl. The crystals grew in two weeks to dimensions of 0.25 × 0.06 × 0.06 mm.
Figure 2.

Crystals of P. somniferum SalR grown under different conditions. (a) 2.0 M ammonium sulfate, 100 mM MES pH 5.5, 5%(v/v) PEG 400; (b) 2.1 M ammonium sulfate, 100 mM MES pH 5.8, 5%(v/v) PEG 400, 100 mM LiCl, 3%(v/v) glycerol.
No crystals were observed in the absence of NADPH under conditions where 8.8 mg ml−1 SalR in a buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl was equilibrated against reservoir solutions containing ammonium sulfate (from 1.5 to 2.25 M), 100 mM MES pH 6.0, 5%(v/v) PEG 400 and 150 mM NaCl. A mixture of small crystals and heavy protein precipitate was observed in the absence of 2-mercaptoethanol under conditions where 8.2 mg ml−1 SalR with 1 mM NADPH in a buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl was equilibrated against a reservoir solution containing 2.0 M ammonium sulfate, 100 mM MES pH 6.0, 5%(v/v) PEG 400 and 150 mM NaCl.
2.4. Diffraction data collection and processing
For diffraction studies, the crystals were vitrified in liquid nitrogen and data were collected in a 100 K nitrogen stream (Oxford Cryosystems). To prepare the crystals for freezing, they were serially transferred into increasing concentrations of glycerol in a synthetic mother liquor composed of 25 µl saturated ammonium sulfate added to 100 µl of solutions containing 2.0 M ammonium sulfate, 100 mM MES pH 6.1, 5%(v/v) PEG 400 and 4 mM NADPH to which 5, 10, 15 or 20 µl glycerol [corresponding to 3–14%(v/v)] was added. The additional ammonium sulfate was necessary to stabilize the crystals against the solubilization caused by the glycerol cryoprotectant. Crystals were incubated in each solution for ∼7 min and then plunged into liquid nitrogen until transfer to the cryostream for diffraction studies. Diffraction maxima were collected using a SBC3 3k × 3k CCD built by ANL-ECT with a 210 × 210 mm active area and a 1.8 s readout on the Structural Biology Center 19-BM beamline (Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois, USA). X-ray data were processed with HKL-3000 and scaled with SCALEPACK (Otwinowski & Minor, 1997 ▶). Data-collection statistics are reported in Table 1 ▶.
Table 1. X-ray diffraction data statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P6422 or P6222 |
| Unit-cell parameters (Å, °) | a = b = 139.9, c = 100.2, α = β = 90, γ = 120 |
| Resolution (Å) | 50–1.91 (1.94–1.91) |
| Total reflections | 836117 (7928) |
| Unique reflections | 40176 (1802) |
| Average redundancy | 19.6 (4.4) |
| Completeness (%) | 94.3 (68.0) |
| Rmerge† (%) | 5.9 (27.9) |
| 〈I/σ(I)〉 | 77.5 (4.7) |
| Solvent content (%) | 69.9 |
| Matthews coefficient (Å3 Da−1) | 4.09 |
| Molecules per ASU | 1 |
R
merge = 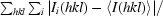
 , where Ii(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is its average.
, where Ii(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is its average.
3. Results and discussion
In the absence of salt, purified SalR lost activity (Gerardy & Zenk, 1993 ▶). In addition, when this enzyme was purified and stored at 193 K without 2-mercaptoethanol, it eluted from a gel-filtration column as two peaks at elution volumes of 43 and 60 ml. The protein in the 43 ml fraction gave the same single band of molecular mass 35 kDa on SDS–PAGE as that in the 60 ml fraction, although it was inactive (0.0 nkat per milligram of protein). The inactive SalR (43 ml fraction) regained activity on incubation overnight at 277 K with 2-mercaptoethanol at concentrations of 0.5 mM (0.0 nkat per milligram of protein), 5 mM (15.7 ±2.5 nkat per milligram of protein, 25% of the activity of the 60 ml fraction) and 50 mM (30.0 ± 2.4 nkat per milligram of protein, 49% of the activity of the 60 ml fraction). SalR is susceptible to inactivation and soluble aggregation in an oxidative environment, suggesting the presence of catalytically or structurally reactive cysteine residues. A reactive cysteine residue has been identified in a human carbonyl reductase (Tinguely & Wermuth, 1999 ▶).
Consistent with the crystal habit, analysis of the X-ray diffraction data indicated that the crystals of SalR belonged to the hexagonal space group P6422 or P6222, with unit-cell parameters a = b = 139.9, c = 100.2 Å. From initial SAD phasing experiments using crystals of SeMet-substituted SalR, there was a single copy of SalR in the hexagonal asymmetric unit and the space group is likely to be P6422. The calculated Matthews coefficients (V M) for one and two copies of SalR in the asymmetric unit were 4.09 and 2.04 Å3 Da−1, respectively. Interestingly, the V M values are at both extremes of the observed values (for a review, see Kantardjieff & Rupp, 2003 ▶). With one copy of SalR per asymmetric unit, the solvent content of the crystals was ∼70% and the relatively high resolution observed in the diffraction studies is not typical of this rather loose packing arrangement (Kantardjieff & Rupp, 2003 ▶). The fact that NADPH is required for crystallization strongly suggests that NADPH is bound to SalR in the crystal and that there may be structural rearrangements and/or stabilization upon coenzyme binding. At a resolution of ∼1.9 Å, the details of substrate specificity and the catalytic mechanism of this enzyme along the morphine-biosynthetic pathway are expected to largely be elucidated.
Acknowledgments
We thank Dr Howard R. Berg for help in photographing the crystals. This work was supported by the Mallinckrodt Foundation, St Louis and by a Uehara Memorial Foundation postdoctoral fellowship (YH).
References
- Barton, D. H. R., Kirby, G. W., Steglich, W., Thomas, G. M., Battersby, A. R., Dobson, T. A. & Ramuz, H. (1965). J. Chem. Soc., pp. 2423–2438. [DOI] [PubMed]
- Bateman, R., Rauh, D. & Shokat, K. M. (2007). Org. Biomol. Chem.5, 3363–3367. [DOI] [PMC free article] [PubMed]
- Bateman, R. L., Rauh, D., Tavshanjian, B. & Shokat, K. M. (2008). J. Biol. Chem.283, 35756–35762. [DOI] [PMC free article] [PubMed]
- Filling, C., Berndt, K. D., Benach, J., Knapp, S., Prozorovski, T., Nordling, E., Ladenstein, R., Jörnvall, H. & Oppermann, U. (2002). J. Biol. Chem.277, 25677–25684. [DOI] [PubMed]
- Gerardy, R. & Zenk, M. H. (1993). Phytochemistry, 34, 125–132.
- Ghosh, D., Sawicki, M., Pletnev, V., Erman, M., Ohno, S., Nakajin, S. & Duax, W. L. (2001). J. Biol. Chem.276, 18457–18463. [DOI] [PubMed]
- Jörnvall, H., Persson, B., Krook, M., Atrian, S., Gonzàlez-Duarte, R., Jeffery, J. & Ghosh, D. (1995). Biochemistry, 34, 6003–6013. [DOI] [PubMed]
- Kallberg, Y., Oppermann, U., Jörnvall, H. & Persson, B. (2002). Eur. J. Biochem.269, 4409–4417. [DOI] [PubMed]
- Kantardjieff, K. & Rupp, B. (2003). Protein Sci.12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Kavanagh, K. L., Jörnvall, H., Persson, B. & Oppermann, U. (2008). Cell. Mol. Life Sci.65, 3895–3906. [DOI] [PMC free article] [PubMed]
- Kempe, K., Higashi, Y., Frick, S., Sabarna, K. & Kutchan, T. M. (2009). Phytochemistry, 70, 579–589. [DOI] [PubMed]
- Korman, T. P., Hill, J. A., Vu, T. N. & Tsai, S. C. (2004). Biochemistry, 43, 14529–14538. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Persson, B. et al. (2009). Chem. Biol. Interact.178, 94–98. [DOI] [PMC free article] [PubMed]
- Tanaka, M., Bateman, R., Rauh, D., Vaisberg, E., Ramachandani, S., Zhang, C., Hansen, K. C., Burlingame, A. L., Trautman, J. K., Shokat, K. M. & Adams, C. L. (2005). PLoS Biol.3, e128. [DOI] [PMC free article] [PubMed]
- Tinguely, J. N. & Wermuth, B. (1999). Eur. J. Biochem.260, 9–14. [DOI] [PubMed]
- Yamashita, A., Kato, H., Wakatsuki, S., Tomizaki, T., Nakatsu, T., Nakajima, K., Hashimoto, T., Yamada, Y. & Oda, J. (1999). Biochemistry, 38, 7630–7637. [DOI] [PubMed]
- Ziegler, J., Voigtländer, S., Schmidt, J., Kramell, R., Miersch, O., Ammer, C., Ges, A. & Kutchan, T. M. (2006). Plant J.48,177–192. [DOI] [PubMed]