

Abstract
Plants possess many metabolic genes for the production of a wide variety of phytochemicals in a tissue-specific manner. However, the metabolic systems behind the diversity and tissue-dependent regulation still remain unknown due to incomplete characterization of phytochemicals produced in a single plant species. Thus, having a metabolome dataset in addition to the genome and transcriptome information resources would enrich our knowledge of plant secondary metabolism. Here we analyzed phytochemical accumulation during development of the model plant Arabidopsis (Arabidopsis thaliana) using liquid chromatography-mass spectrometry in samples covering many growth stages and organs. We also obtained tandem mass spectrometry spectral tags of many metabolites as a resource for elucidation of metabolite structure. These are part of the AtMetExpress metabolite accumulation atlas. Based on the dataset, we detected 1,589 metabolite signals from which the structures of 167 metabolites were elucidated. The integrated analyses with transcriptome data demonstrated that Arabidopsis produces various phytochemicals in a highly tissue-specific manner, which often accompanies the expression of key biosynthesis-related genes. We also found that a set of biosynthesis-related genes is coordinately expressed among the tissues. These data suggested that the simple mode of regulation, transcript to metabolite, is an origin of the dynamics and diversity of plant secondary metabolism.
The structural diversity of secondary metabolites is an important part of the nature of plants as a rich source of useful phytochemicals for humans. Comparison of the metabolite compositions of plant tissues investigated for specific secondary metabolites in Arabidopsis (Arabidopsis thaliana; Brown et al., 2003) and for primary metabolites in Lotus japonicus (Desbrosses et al., 2005) revealed that plants have evolved metabolic systems for producing a variety of metabolites in a tissue-dependent manner to improve plant fitness. Genome sequencing has uncovered the genetic background of the diversity of plant genomes, which encode large families of metabolism-related genes such as cytochrome P450s and glycosyltransferases (D'Auria and Gershenzon, 2005; Yonekura-Sakakibara and Saito, 2009). Recent progress in phytochemical genomics studies using the model plant Arabidopsis has enriched the list of functionally identified genes (Hirai et al., 2007; Saito et al., 2008; Yonekura-Sakakibara et al., 2008) with the aid of transcriptome resources (Craigon et al., 2004; Schmid et al., 2005; Kilian et al., 2007; Obayashi et al., 2007, 2009; Goda et al., 2008). However, the majority of metabolic gene functions as well as plant metabolic systems themselves remain unknown, because the phytochemicals produced in Arabidopsis have not been fully characterized. Indeed, metabolome analyses using liquid chromatography-mass spectrometry (LC-MS) led to the postulation that Arabidopsis produces a large number of unknown metabolites (Bottcher et al., 2008; Farag et al., 2008; Iijima et al., 2008; Matsuda et al., 2009). Thus, in addition to the genome and transcriptome resources, two types of metabolome resources are required to investigate metabolic systems in plants. The first is information for structural elucidation of metabolites (Matsuda et al., 2009). The second is metabolic profile data for integrated analyses with other omics datasets. In this study, to explore the structural diversity of secondary metabolites produced in one plant species, we analyzed the accumulation of known and unknown phytochemicals during development of the model plant Arabidopsis using LC-MS of samples covering many growth stages and diverse organs. We also acquired tandem mass spectral (MS/MS) data of many metabolites (MS/MS spectral tags [MS2Ts]) to elucidate the structures of Arabidopsis metabolites. The analyses of the dataset (AtMetExpress development) demonstrated that Arabidopsis has the capability of producing diverse metabolites with high tissue specificity.
The AtMetExpress development dataset also makes it possible to understand the mechanism behind the variations in metabolic profiles among plant tissues by investigating the relationship between gene expression and metabolite accumulation. For example, when metabolite accumulation patterns are distinct from the patterns of biosynthesis-related genes, several other factors such as catabolism, translocation, and feedback regulation are likely to play important roles in the metabolic systems. An underdetermined system requires the identification of novel components, based on which a more detailed analysis of the behavior of the metabolic system can be achieved. In contrast, a concerted regulation of biosynthesis-related gene expression and metabolites indicate a simple mode of regulation of the metabolic systems. In such systems, it is expected that coexpression/accumulation analysis of the gene and metabolite would reveal additional components of the system such as biosynthesis-related genes. To perform integrated analyses, metabolome data of the AtMetExpress development dataset were acquired by an experimental design compatible with that of the AtGenExpress development transcriptome dataset (Schmid et al., 2005). An analysis of the datasets revealed how transcriptional programs control the tissue-specific production of diverse phytochemicals. The results suggested that a simple mode of regulation employing transcript to metabolite is an origin of the dynamics and diversity of plant secondary metabolism.
RESULTS
Data Acquisition of AtMetExpress Development Dataset
For determining the metabolite levels, we obtained quadruplicate metabolic profiles of 36 distinct tissues by using LC coupled with electrospray ionization quadrupole time-of-flight MS/MS (LC-ESI-Q-TOF/MS) according to previously described methods (Matsuda et al., 2009). The obtained data matrix contains the relative peak intensity values of 1,589 metabolite signals from 144 samples (36 tissues by four replicates; Supplemental Data S1). The experimental design was compatible with that of the AtGenExpress developmental (Schmid et al., 2005) series for integrated analyses with transcriptome data (Supplemental Tables S1 and S2). The following statistical analyses were performed by using the metabolic profile dataset containing the 1,589 detected metabolite signals.
In addition to the metabolic profile data for determining the metabolite levels, the MS/MS spectral data of detectable metabolites were obtained by employing the specific method for structurally elucidating Arabidopsis metabolites. Crude extracts of nine representative tissues of Arabidopsis were analyzed using the survey mode of the LC-ESI-Q-TOF/MS to obtain the MS/MS spectral data of detectable metabolites automatically. Hereafter, the MS/MS spectral data obtained using this method are referred to as MS2Ts (Matsuda et al., 2009). In this study, 36 MS2T libraries with 476,120 accessions were created (Supplemental Table S3).
Because the metabolic profile data and MS2T libraries were acquired by using compatible analytical conditions, we could obtain MS/MS spectral data of a metabolite signal in the metabolic profile data by identifying the MS2T data acquired from the same metabolite. This means that metabolite signals in the data matrix could be tagged with the corresponding MS2T data. By using this method, approximately 95% of the metabolite signals in the data matrix were tagged with MS2Ts. By referring to the structural information available from the MS2Ts, among the 1,589 metabolite signals in the dataset, the structures of 167 metabolites were elucidated (37 metabolites were identified, two were annotated, and 128 were characterized; Supplemental Table S2; refer to “Materials and Methods” for details). For instance, a metabolite accumulating in roots (retention time, 4.15 min; mass-to-charge ratio [m/z] 389) was deduced to be guaiasylglycerol-β-feruloyl ether by interpreting the MS/MS spectral data (Supplemental Fig. S1). Hexosyl and malonyl derivatives of guaiasylglycerol-β-feruloyl ether were also newly identified as Arabidopsis metabolites (e.g. compound 12 in Fig. 1A). Most of the annotated metabolites were glucosinolates (GSLs; 1–4), phenylpropanoids (5–19), and nitrogen-containing products (1–4, 13, 15, 17–19, 24, 25; Fig. 1A). The accumulation patterns of GSLs were similar to those of published data (Petersen et al., 2002; Bringmann et al., 2005), suggesting the reproducibility of the metabolic profile data. It was also confirmed that 53 metabolites among the 75 seed metabolites previously reported by Böttcher et al. (2008) were reproductively found in this study. The metabolites detected in the Böttcher et al. (2008) study alone were unusual metabolites accumulating only in a transparent testa4 mutant in the Landsberg erecta background.
Figure 1.

A, Structures of representative Arabidopsis phytochemicals with summary of biosynthetic pathways and enzymes referred to in text. B, Accumulation and expression patterns of spermidine-di-p-coumaroyl (17; left) and SHT gene (right). Gene expression patterns were obtained from the Bio-Array Resource eFP Browser (Winter et al., 2007).
Diversity of Metabolites Produced by Arabidopsis
The clustering of metabolites by MS/MS spectral similarity indicates that Arabidopsis produces groups of metabolites that are structurally uncharacterized. Based on the MS2Ts for each metabolite signal, the structural similarities among metabolites were examined by determining the similarity of MS/MS spectra using the dot product method (Stein and Scott, 1994). The structural similarity network showed several clusters of metabolite signals (Fig. 2; S > 0.35). The threshold level was arbitrarily selected to find the largest number of metabolite clusters. The relationships between the threshold values and the properties of the metabolite structural similarity networks are shown in Supplemental Table S4. Some of the clusters were characterized as known metabolites, including glycosides of flavonols (kaempferol, quercetin, and isorhamnetin), sinapoyl-containing metabolites, sinapoylcholine (SC) derivatives, GSLs, and lignans. Although many other clusters were poorly annotated in the databases, the results still indicate that Arabidopsis has the capability to produce known and unknown metabolites with wide structural diversity.
Figure 2.

Clustering of metabolite signals by structural similarity. The structural similarity values (S) were determined using dot product methods (0 ≤ S ≤ 1). Nodes in the graph represent metabolite signals detected in positive (red) and negative (blue) ion modes. Edges represent pairs of structurally similar metabolites above the threshold (S > 0.35). The network contains 79 clusters of 1,269 edges among 467 metabolites, of which 95 metabolites are structurally assigned. The filled circles and diamonds indicate structurally unassigned and assigned metabolites, respectively. The metabolite classes included in clusters are also shown.
High Tissue Specificity of Metabolite Accumulation
The dataset enabled us to compare the spatial distribution of phytochemicals with AtGenExpress gene expression data (Winter et al., 2007). For example, spermidine-di-p-coumaroyl (17) and its key biosynthetic gene (spermidine hydroxycinnamoyltransferase [SHT]; Grienenberger et al., 2009) are specifically expressed in flowers (Fig. 1B). However, the Pearson correlation coefficient (PCC) between them is weak (r = 0.41) because SHT (At2g19070) is expressed only transiently at the early stages of flower development, whereas 17 is also detected in mature flowers. Because of such inconsistencies, overall gene-metabolite (G-M) correlations were weaker than gene-gene (G-G) and metabolite-metabolite (M-M) correlations (Supplemental Fig. S2). Here, the global trend of metabolite accumulation across the tissues was investigated by statistical analyses and compared with gene expression data.
Analysis of the metabolome dataset revealed that organ systems, including roots, flowers, and seeds, had distinct and characteristic metabolic profiles as shown by principal component analysis (Fig. 3). Overall morphological similarity was reflected as the distances in principal component analysis. In contrast, intermediate metabolic profiles were obtained for rosette leaves and internodes because the data for these regions were plotted near the origin of the axes. A similar trend was observed in the graphical representation of the network of metabolites with very similar accumulation patterns (PCC r > 0.7; Supplemental Fig. S3). The high tissue specificity of metabolite accumulation was confirmed by a Venn diagram of detectable metabolites in five representative tissues, including roots, leaves, internodes, flowers, and seeds (Supplemental Fig. S4). Relatively large numbers of metabolites specifically accumulated in flowers (22%), seeds (13%), and roots (12%), whereas only 8% of metabolites were commonly detected in all five tissues, and rosette leaves showed the basal phenotype (4%). The annotation data indicated that hydroxycinnamoylspermidines (17, 18) and flavonol glycosides (8) in flowers and SC derivatives (11, 15) in seeds are tissue-characteristic metabolites (Yonekura-Sakakibara et al., 2007, 2008; Bottcher et al., 2008; Grienenberger et al., 2009).
Figure 3.

Principle component analyses of metabolic profiles across 36 tissues of Arabidopsis (n = 144). Despite the low variance (36%) shown in the figure, the remaining principal components, each describing a low variance, represent the noise (data not shown).
To compare the global trend of tissue specificity between gene expression and metabolite accumulation, Shannon entropy, H, were determined for the accumulation patterns of each metabolite and gene (Schug et al., 2005). Histograms of the entropy levels among metabolome and transcriptome data are shown in Figure 4. The results demonstrated that most genes are more evenly expressed among the 36 tissues (reflected by the higher entropy values) than the phytochemicals. Higher tissue specificity of the phytochemical accumulations (lower entropy values) was also observed in the subset of the structurally annotated metabolites (n = 167). However, small numbers of genes showed relatively low entropy values. The overrepresenting gene ontology (GO) in a subset of genes with low entropy (H < 4.0; 2,007 genes) were investigated (Supplemental Table S5), since these low-entropy genes may have some role in tissue-specific function of metabolic systems. In addition to genes possessing GO terms such as flower development and cell wall, the results indicated that genes belonging to GOs such as oxygen binding (including CYP P450), transferase, and secondary metabolism are significantly overrepresented in the subsets (P < 0.05). These results suggested that, in addition to the tissue-dependent accumulation of phytochemicals, genes likely responsible for secondary metabolism tend to be expressed in tissue-specific manner.
Figure 4.

Frequency distribution of tissue specificity of metabolite accumulation and gene expression. Degrees of specificity were evaluated by determining Shannon entropy levels (H) among 36 tissue samples of Arabidopsis. Higher and lower entropy levels, respectively, indicate lower and higher tissue specificity.
Comparison between Accumulation Patterns of Metabolites and Expression of Their Biosynthetic Genes
To clarify any possible similarity of expression pattern of each gene to the accumulation pattern of its associated metabolites, we conducted a clustering analysis using the batch-learning self-organizing map (BL-SOM) method. BL-SOM is an improved and a reproducible method of the original SOM (Kanaya et al., 2001), and thus applied to integrated analysis of transcriptome and metabolome, leading to successful prediction of genes' functions (Hirai et al., 2004, 2005). All 1,589 metabolite signals with 10,147 metabolism-related genes (selected by GO terms) were classified into a 30 × 26 lattice according to their relative expression level across 36 tissues (Fig. 5, A–D). The dataset also contains dummy profile data as tissue markers (Supplemental Table S6). For example, the seed marker has an artificially generated reference representing metabolic profile accumulating only in mature seeds (ATME84). Therefore, the metabolites and genes specifically expressed and accumulated in mature seeds should be classified near the seed marker position. Figure 5A also indicates lattices by circles in which the accumulated genes and metabolites are classified according to the tissues. These regions were arbitrarily defined by summarizing the feature maps of individual experiments (Supplemental Fig. S5).
Figure 5.

Integrated analysis of transcriptome (AtGenExpress) and metabolome (AtMetExpress) data. A to D, BL-SOM clustering of 10,147 metabolism-related genes and 1,589 metabolite signals by expression and accumulation patterns across 36 tissues. In the BL-SOM analysis, the genes and metabolites with similar expression or accumulation profiles are clustered into neighboring cells. Positions of genes are indicated by red, and other colors represent positions of metabolites. A, Positions of tissue markers: filled circles roughly represent dominant tissues in each cell. Dummy metabolic profile data of each tissue marker and feature maps of BL-SOM analysis are shown in Supplemental Table S4 and Supplemental Figure S5, respectively. B to D, Mapping of aromatic amino acid (B), phenylpropanoid (C), and Met-derived GSL (D) biosynthesis-related genes and metabolites. E, Coexpression/accumulation network between hydroxycinnamoylspermidine (blue) and cytochrome P450 genes (orange). Positive correlations above threshold levels are indicated with connecting lines (r, PCC). A relatively low threshold level (r > 0.5) was employed to detect weak relationships. F, Coexpression/accumulation network between MS-n-alkylglucosinylates (GSLs; blue) and all genes (orange). Positive correlations (r > 0.7) are indicated with connecting lines. Gene symbols and metabolite abbreviations are shown in Supplemental Figure S7.
The results of BL-SOM analyses revealed that the regulation of gene expression and metabolite accumulation in primary metabolic pathway is rather complex. Although Tyr, Phe, and Trp (21–23) are synthesized from the shikimate pathway (Fig. 1A), the three aromatic amino acids are located at a different position on the map (blue cells; Fig. 5B). Mapping of these biosynthesis-related genes described in the metabolic pathway database, AraCyc (Mueller et al., 2003; Poole, 2007; red cells), showed poor similarity between both G-M and G-G relationships. Similar results were also observed for the cases of Met, Leu, and Ile biosynthesis (Supplemental Fig. S6).
In contrast, simpler modes of regulation are observed in the case of the phenylpropanoid pathway. As shown in Figure 1, Arabidopsis has the capability to produce various types of phenylpropanoids. The phenylpropanoid pathway looks like a tree structure in which many pathways branch from the common pathway for the biosynthesis of a C6-C3 (phenylpropanoid) unit. BL-SOM analysis demonstrated that 11 genes of the core phenypropanoid pathway were coordinately expressed (high G-G similarity) and that the most intense expression was observed in internode tissues, probably for the active lignin biosynthesis in vascular tissues (Fig. 5C). On the other hand, the accumulation patterns of phenylpropanoids such as SC derivatives (11) in seeds (yellow cells), hydroxycinnamoylspermidines (17, 18) in flowers (blue cells), and coumarins (5) in roots (green cells; Bottcher et al., 2008; Fellenberg et al., 2008; Kai et al., 2008) were tissue specific with relatively high M-M similarity. In addition, it has also been observed that the key enzyme genes responsible for these biosyntheses (SHT for hydroxycinnamoylspermidines [17, 18] and feruloyl-CoA 6′-hydroxylase for scopolins [5]; Kai et al., 2008; Grienenberger et al., 2009) were located near the product metabolites (red cells). In the case of SNG2, encoding sinapoylglucose:choline sinapoyltransferase (Shirley et al., 2001), gene expression was activated in siliques, indicating that SC (15) is actively biosynthesized during seed development. Similar results were observed for procyanidin (6, 7) and flavonol glycoside (8) biosynthesis (Supplemental Fig. S6C). These results indicated that the functional differentiation of the phenylpropanoid pathway among the tissues was attained by controlling the expression of a small number of key regulatory genes, leading to the proposal that the high M-G similarity results in the tissue-specific accumulation of phenylpropanoids.
We next considered Met-derived GSL biosynthesis (see Supplemental Text S1 for details). Despite the concerted regulation of biosynthesis-related genes (red; Hirai et al., 2004, 2005, 2007), GSLs accumulated predominantly in the dry seeds and the flowers as reported previously (in Fig. 5D; Brown et al., 2003). The difference in GSL profiles between the seeds and the maternal organs (flowers and leaves) raises the question of how the structure-dependent GSL accumulation pattern is controlled, because methylsulfinyl (MS)-GSLs (2, blue) are formed from methylthio (MT)-GSLs (1, orange; Hansen et al., 2007), and then converted into hydroxyalkyl (OH)-GSLs (3, green; Kliebenstein et al., 2001). The accumulation pattern of OH-GSLs in the seeds is explained by the expression of the responsible gene AOP3 during seed developing (Kliebenstein et al., 2001). On the other hand, as the known genes involved in MS- and MT-GSL synthesis are poorly expressed in seeds (see Supplemental Text S1 for details), a translocation of MS or MT GSLs from the maternal organs into the embryos has been postulated (Nour-Eldin and Halkier, 2009).
Correlation between Gene Expression and Metabolite Accumulation
BL-SOM analysis suggested that there are correlations between gene expression and metabolite accumulation in the case of phenylpropanoid pathway. However, the correlation is not expected to be strong, as demonstrated by the SHT gene and spermidine-di-p-coumaroyl (17) pair, where weak correlation was observed (PCC r = 0.41) due to the presumable time gap between gene expression and resultant metabolite accumulation (Fig. 1B). Although the weak correlations were statistically insignificant, from these results new hypotheses of metabolism-related gene functions could be generated by correlation analyses of a small subset of genes and metabolites.
The 5-hydroxyferuloyl moiety in hydroxycinnanoylspermidines (18) is synthesized by 5′-hydroxylation of the feruloyl moiety (Fellenberg et al., 2008). The G-M correlation network of CYP genes and hydroxycinnanoylspermidines revealed candidate CYPs responsible for 5′-hydroxylation steps by employing a lower threshold level (PCC r > 0.5) to detect weak correlations (Fig. 5E). Among them, CYP98A8 and CYP98A9 could be the candidates involved in this reaction and thus deserved further investigation, because a homologous gene, CYP98A3, is responsible for a similar reaction (Schoch et al., 2001). Recently, this hypothesis was independently confirmed by a reverse genetics study (Matsuno et al., 2009).
The occurrence of lignin- and neolignan-like metabolites (9–12) in roots and seeds suggests the participation of dirigent protein (DP) in their biosyntheses (Burlat et al., 2001; Davin and Lewis, 2005; Figs. 1A and 5C). The coexpression/accumulation network of lignin- and neolignan-like metabolites and 10 putative DP genes in Arabidopsis is shown in Figure 6A. The results indicated that eight DPs correlate redundantly with neolignans that accumulate in roots (guaiasylglycerol-β-feruloyl esters, 12) and lignans (lariciresionls, 10; Mir Derikvand et al., 2008; Nakatsubo et al., 2008). In contrast, no correlations were observed for the two homologs (At4g11180 and At1g55210), implying functional specialization of these two genes among DPs. Contrary to the tissue-nonspecific expression of At1g55210, the gene expression data indicated that At4g11180 (DP1) is expressed only in siliques, suggesting a role in neolignan accumulation in seeds (Fig. 6B). To test this hypothesis, the knockout T-DNA insertion mutant SAIL_60_D04, designated dp1, was used for reverse genetic analysis. T-DNA was inserted between +359 and +395 of the start codon of DP1 (Fig. 6C). LC-MS analysis revealed that the homozygous dp1 mutant lacks seed-specific neolignans such as 3-{4-[2-hydroxy-2-(4-hexosyloxy-3-methoxyphenyl)-1-hydroxymethylethoxy]-3,5-dimethoxyphenyl}acryloylcholine (Bottcher et al., 2008; 11; Fig. 6D), suggesting a critical role of DP1 in the biosynthesis of seed-specific neolignans. Although the function of the DP1 gene needs to be unequivocally characterized by further investigation, which is being undertaken in our laboratory, the approach using AtMetExpress is feasible for such hypothesis generation regarding an orphan gene's function.
Figure 6.

A T-DNA insertion mutant of At4g11180 (DP1). A, Coexpression and accumulation network between lignans (blue) and putative DP genes (orange). Positive correlations above threshold levels are indicated with connecting lines (r, PCC). A relatively low threshold level (r > 0.5) was employed to detect weak relationships. B, Gene expression patterns of DP1 obtained from Bar eFP browser. C, Schematic representation of DP1 with the T-DNA insertion mutant used. The thick black line indicates coding sequence. The DP1 gene has no intron, and the gene region containing 5′-untranslated region and 3′-untranslated region is shown in the figure. Numbers indicate the position of the T-DNA insertion. LB, Left border; RB, right border. D, Selected ion chromatograms of seed extracts from the wild type (Columbia-0, bottom section) and the homozygous dp1 mutant (top section). The peak for 3-{4-[2-hydroxy-2-(4-hexosyloxy-3-methoxyphenyl)-1-hydroxymethylethoxy]-3,5-dimethoxyphenyl}acryloylcholine [SC(4-O-β)G 4′-O-hexoside] is shown.
BL-SOM analysis also demonstrated that the accumulation of Met-derived GSLs (1, 2) was independent of the control of the biosynthesis-related genes (Fig. 5F). Coexpression/accumulation analyses between Met-derived GSLs and all Arabidopsis genes showed that five MS-GSLs (1) correlated with 27 genes even when a relatively higher threshold level was employed (PCC r > 0.7; Fig. 5D). Three of the genes were annotated as lipid transfer protein type 5 (At2g10940, At1g27950, and At1g55260), implying a role in aliphatic GSL transport. These results demonstrated that, despite the small number of data points for the calculation of G-M correlations (n = 36), further use of coexpression/accumulation analysis combined with other data should help in identifying novel genes related to secondary metabolism.
DISCUSSION
The AtMetExpress development LC-MS dataset obtained in this study consists of metabolic profiling data of 36 Arabidopsis tissues (Supplemental Table S1) and MS2T spectral libraries containing 476,120 accessions (Supplemental Table S3). The dataset is designed to fulfill two purposes required for understanding metabolic systems in plants. The first is to explore the structures of metabolites produced in Arabidopsis using mass spectral information stored in the MS2T libraries. Structural elucidation is a prerequisite for the functional characterization of metabolic systems and its related genes. In this study, structures of 167 metabolites were elucidated through intensive searching of metabolite databases (Fig. 1A; Supplemental Table S2). Although the majority of metabolite signals remain uncharacterized, the clustering of metabolites based on MS/MS spectral similarities successfully demonstrated that one plant species has the capability of producing metabolites with large structural diversity (Fig. 2; D'Auria and Gershenzon, 2005; Yonekura-Sakakibara and Saito, 2009). Since the MS2T libraries created in this study already include MS/MS data of many unknown metabolites, extending the MS/MS spectral databases of standard compounds would accelerate the structure elucidation of a wider range of metabolites such as alkaloids, polyketides, and terpenoids (Facchini et al., 2004; Samanani et al., 2004). For this purpose, we are constructing a MS/MS spectral database by collecting data reported in the literature (http://spectra.psc.riken.jp/). However, MS/MS data are insufficient for the strict identification of metabolites. Preparation of standard compounds and their detailed characterization by methods of natural product chemistry is essential for LC-MS metabolomics (Nakabayashi et al., 2009).
The second application of the AtMetExpress development dataset is an integrated analysis with other omics dataset, in particular, transcriptome, for an investigation of how different metabolites are produced among plant tissues and how transcriptional control develops distinct metabolite profiles. For this purpose, the dataset was acquired by an experimental design compatible with that of the AtGenExpress development transcriptome dataset (Schmid et al., 2005). Statistical analyses of the metabolic profile data indicated that the majority of metabolites accumulated unevenly among tissues (Fig. 4), and many types of metabolites were produced in a tissue-specific manner (Fig. 3). Since the metabolic profiling method employed in this study failed to detected many hydrophilic and hydrophobic metabolites such as sugars, organic acids, lipids, and volatiles (Fig. 1A), the high degree of tissue specificity does not necessarily reflect a general trend of plant metabolism. However, the results have highlighted that each tissue of Arabidopsis has distinct states of secondary metabolism. The accumulation of specific metabolites in reproductive and underground organs seems rational because these tissues require special constituents such as pigments to attract pollinators and toxic metabolites for protection from herbivores and pathogens (Tanaka and Ohmiya, 2008). In contrast, rosette leaves appear to be one of the most prototypic and undifferentiated organs in terms of secondary metabolism, because few metabolite signals only accumulated in leaves (Supplemental Fig. S4). This basal phenotype is likely to be due to differential gene expression since the level of expression of most genes in leaves is similar to their overall average (Schmid et al., 2005).
The drastic difference in plant secondary metabolism among tissues suggests a dynamic regulation of metabolic systems probably by the transcriptional control of those biosynthesis-related genes. Thus, we performed a combined analysis of metabolome and transcriptome data using the BL-SOM method to compare modes of regulation in three distinct metabolic pathways including aromatic amino acid biosynthesis, the phenylpropanoid pathway, and the Met-derived GSL biosynthetic pathway (Fig. 5).
It has been recognized that levels of free aromatic amino acids in plants are under the control of multiple factors, including protein biosynthesis, feedback regulation, transport, and distinct expression controls of each isomer of biosynthesis-related genes (Li and Last, 1996; Liu and Bush, 2006; Lee et al., 2007; Ueda et al., 2008; Yamada et al., 2008). Indeed, poor similarity between both G-M and G-G relationships was observed in BL-SOM results, indicating that expression of each gene is independently regulated and that other unidentified factors such as intertissue translocation play important roles in the regulation of amino acid levels. The complex and somewhat redundant regulation mechanism seems to contribute to the robustness of metabolic systems to ensure the stable supply of amino acids required for the steady growth of plants under any environment.
In the case of the phenylpropanoid pathway, the mode of regulation was simpler than that of primary metabolism since a clear similarity of the expression/accumulation pattern between G-G and M-M relationships was observed (Fig. 5C). Tissue-dependent production of phenylpropanoids was also regulated by expression of a small number of key enzyme genes (Figs. 1B and 5C). The simplicity of plant secondary metabolism is useful nature to generate a hypothesis regarding novel metabolism-related genes by coexpression/accumulation analysis as demonstrated for the cases of CYP98A8, CYP98A9, and DP1 genes in this study (Figs. 5E and 6). However, scarce G-M similarity was observed for the Met-derived GSL biosynthetic pathway despite clear G-G similarities being observed among the biosynthesis-related genes. This observation suggests the occurrence of other regulatory components such as intertissue translocation, especially for the seed-specific accumulation of MS-GSLs (see Supplemental Text S1 for details). The identification of these components using AtMetExpress development and other data (Fig. 6F) should uncover the dynamics of the metabolic system regulating GSL biosynthesis in detail.
As discussed above, roots, flowers, and seeds of Arabidopsis have distinct and characteristic metabolic profiles. This implies that some flexibility is needed for the regulation of secondary metabolism to perform dynamic control of metabolic activity among the tissues. In this regard, a simple mode of regulation by the concerted control of expression of a series of genes is preferable, because complex and redundant systems such as those used for the regulation of amino acid contents seems to be too stable to perform dynamic regulation. On the other hand, such a simple system should be unstable because a small number of errors would drastically change the metabolic composition. Indeed, it has been reported that variation in 3-hydroxy-n-propyl GSL (3) accumulation in rosette leaves among several Arabidopsis ecotypes is derived from a single polymorphism of a biosynthesis-related gene (Kliebenstein et al., 2001). This fragility, which in other words equates to the changeability of metabolic function, is also likely advantageous because it is presumably an origin for phytochemical diversity, which contributes to adaptation of plants to various environments.
CONCLUSION
AtMetExpress development is a dataset that facilitates the analysis of metabolic systems responsible for phytochemical diversity. The detection of novel Arabidopsis metabolites from MS2T data will enable us to find these biosynthesis-related genes by the integrated analyses of not only metabolome and transcriptome but proteome as well (Baerenfaller et al., 2008; Castellana et al., 2008). These findings could then be applied to understand the behavior of plant metabolic systems. The AtMetExpress development LC-MS dataset is a part of our AtMetExpress project, which is opened to the public through the PRIMe Web site (http://prime.psc.riken.jp/).
MATERIALS AND METHODS
Plant Materials
Arabidopsis (Arabidopsis thaliana; accession Columbia-0; Lehle Seeds) was used in this study. T-DNA-inserted knockout mutants of dp1 (SAIL_60_D04) were obtained from the Arabidopsis Biological Resource Center. The T-DNA insertion site was confirmed by sequencing the PCR fragment. The primers used for this study were DP1f (ACAATGACAAATCAAATCTACAAAC) and DP1r (GCCAACACACGAAGATCAATC). The primers for LBa1 and RBa1 were designed by following the Arabidopsis Biological Resource Center data (http://www.arabidopsis.org/abrc/pCSA110.pdf). Arabidopsis seedlings were grown under the conditions described in Supplemental Table S2. Collected sample tissues were weighed and stored at −80°C until analysis. The frozen tissues were homogenized in five volumes of 80% aqueous methanol containing 0.1% acetic acid, 0.5 mg/L of lidocaine, and d-camphor sulfonic acid (Tokyo Kasei) using a mixer mill (MM 300, Retsch) with a zirconia bead for 6 min at 20 Hz. Following centrifugation at 15,000g for 10 min and filtration (Ultrafree-MC filter, 0.2 μm, Millipore), the sample extracts were applied to an HLB μElution plate (Waters) equilibrated with 80% aqueous methanol containing 0.1% acetic acid. The eluates (3 μL) were subjected to metabolome analysis using LC-ESI-Q-TOF/MS.
Metabolome Analysis Using LC-ESI-MS
Metabolome analysis was performed with an LC-ESI-Q-TOF/MS system equipped with an ESI interface (HPLC: Waters Acquity UPLC system; MS: Waters Q-TOF Premier) operated under previously described conditions (Matsuda et al., 2009). In the negative ion mode, the MS conditions were as follows: capillary voltage: +3.0 keV; cone voltage: 22.5 V; source temperature: 120°C; desolvation temperature: 450°C; cone gas flow: 50 L/h; desolvation gas flow: 800 L/h; collision energy: 2 V; detection mode: scan (m/z 100–2,000; dwell time: 0.45 s; interscan delay: 0.05 s, centroid); dynamic range enhancement mode: on. The scans were repeated for 19.5 min in a single run. The raw data were recorded with the aid of MassLynx version 4.1 software (Waters). The raw chromatogram data were processed to produce a data matrix consisting of 1,589 metabolite signals (773 from positive and 816 from negative ion mode; Supplemental Data S1) using MetAlign (Lommen, 2009). The parameters used for data processing were as follows: maximum amplitude, 10,000; peak slope factor, 1; peak threshold factor, 6; average peak width at half weight, 8; scaling options, none; maximum shift per scan, 35; select min nr per peak set, 4. The data matrix generated by MetAlign was processed with in-house software written in Perl/Tk (Matsuda et al., 2009). By this procedure, the metabolite signals eluted before 0.85 min and after 12.0 min were discarded, original peak intensity values were divided with those of the internal standards (lidocaine: m/z = 235 [M + H]+, eluted at 4.19 min; camphor-10-sulfonic acid: m/z = 231 [M − H]−, eluted at 3.84 min, for the positive and negative ion modes, respectively) to normalize the peak intensity values, discarding low-intensity data (under signal-to-noise ratio < 5), and isotope peaks were removed by employing specific parameters (rthres > 0.8, ΔRt = 0.5 s, and Δm/z = 2 D). Metabolite signals were assigned unique accession codes, such as adn031026 (representing AtMetExpress Development negative ion mode data, peak number 31026).
MS2T data were acquired from nine tissues of Arabidopsis and processed to create 36 MS2T libraries using previously described methods (Matsuda et al., 2009). Each MS2T entry was assigned a unique accession code, such as ATH10n03690, in which ATH10n is the name of the library and 03690 is the entry number. A total of 36 MS2T libraries with 476,120 accession codes were created in this study (Supplemental Table S2). The MS2T libraries contain a high volume of redundant and low-quality data (Matsuda et al., 2009). Since the metabolic profile data and the MS2T libraries were acquired using compatible analytical conditions, a metabolite signal obtained in the profile can be tagged with MS2Ts obtained from a corresponding metabolite with identical unit mass eluting at a similar retention time. By this method, approximately 95% of the metabolite signals were tagged with at least one MS2T. The mean number of MS2Ts tagged to each metabolite peak was 13.5.
Structure Elucidation of Metabolites
Structures of metabolites were elucidated by the following procedure. The retention time (3.06 min) and m/z value (420 D) of metabolite peaks (adn031026) were searched against the data obtained for in-house standard compounds in our previous study (Matsuda et al., 2009). This peak was matched with an entry for 4-methylthiobutylglucosinolate due to identical unit mass numbers and similar retention times (threshold <0.05 min). In addition, 58 MS2Ts with identical unit mass numbers and similar retention times (threshold <0.15 min) were found from MS2T libraries, providing putative structural information for adn031026. For each MS2T, the high-resolution m/z value of the precursor ion was compared with the theoretical values of metabolites listed in the KNApSAcK database of phytochemicals (Shinbo et al., 2006; Takahashi et al., 2008). The query was considered to match a metabolite when the measured data were very similar to the theoretical value (threshold 10 mD). This process was repeated for all MS2Ts with results indicating that 43 MS2T queries matched the molecular formula entry “C12H24N1O9S3:4-Methylthiobutyl glucosinolate;Glucoerucin.” Similarly, MS/MS spectra data for each MS2T were searched against ReSpect (a collection of literature and in-house MSn spectra data for plant metabolomics research) using the dot product method (see below). ReSpect data were available from our PRIMe Web site (http://prime.psc.riken.jp/; Akiyama et al., 2008). Among 58 MS2Ts tagged to adn031026, 50 queries matched the MS/MS spectra “4-methylthiobutyl glucosinolate_Ramp5-45 V” (threshold for searching, S > 0.8). Since an identical metabolite was suggested by three different methods, the metabolite signals were identified as 4-MT-n-butylglucosinolate. In this study, the four levels for metabolite annotation nomenclature proposed by the Metabolome Standard Initiative were employed as follows. (1) Identified: A minimum of two independent and orthogonal data relative to an authentic compound analyzed under identical experimental conditions are proposed as necessary to validate nonnovel metabolite identifications; (2) annotated: without chemical reference standards, based upon physicochemical properties and/or spectral similarity with public/commercial spectral libraries; (3) characterized: based upon characteristic physicochemical properties of a chemical class of compounds or by spectral similarity to known compounds of a chemical class; and (4) unknown: although unidentified or unclassified, these metabolites can still be differentiated and quantified based upon spectral data (Sumner et al., 2007).
In addition, manual annotation using metabolite information from the literature was also performed. For example, an occurrence of scopolin in Arabidopsis root has been reported (Kai et al., 2006). Although a scopolin standard is not commercially available, searching for its predicted MSn spectra data against MS2T using the spectra search function of the AtMetExpress database (query: 193.05:100;133.0289:20;) indicated that the peak adp009805 (retention time 3.54 min, m/z 355) was annotated as scopolin. To find structurally related metabolites, the similarity of assigned MS2T data was assessed among metabolite signals using the dot product method (see below for details; Stein and Scott, 1994). Consequently, adp013943 was characterized as malonylated scopolin.
All structural data assigned to each metabolite signal can be displayed in the AtMetExpress database in searchable form (http://prime.psc.riken.jp/). From a list of search results, a metabolite signal of interest can be selected to provide detailed information, such as lists of tagged MS2Ts, results of database searching, final annotation, annotation levels, heat map representation of metabolite levels in each tissue (Fig. 1), and raw chromatogram data of representative peaks in all samples (Supplemental Fig. S8).
Evaluation of Structural Similarity
For MS/MS spectra X and Y, a spectral similarity between them, sXY, was determined by the following equation (dot product method; Stein and Scott, 1994):
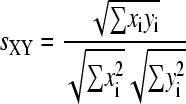 |
where xi represents the intensity of a fragment ion detected at m/z = i in MS/MS spectra X. The m/z values of xi and yi were considered to be the same when the difference between them was less than 10 mD.
To calculate the structural similarity between two metabolite signals, five representative MS2Ts with the most intense base peaks were selected for each metabolite signal. Metabolite signals tagged with less than four MS2Ts were discarded from the structure similarity calculations. The average dot product (S) values between two metabolite signals were defined as the mean s of the total number of 25 MS2T pairs. The metabolite similarity networks were visualized by means of Cytoscape 2.6 software (Shannon et al., 2003).
Data Mining
Microarray data were downloaded from the Gene Expression Omnibus Web site (http://www.ncbi.nlm.nih.gov/geo/) and normalized using MAS5 methods. Log2-transformed and Z-scored intensity values of metabolome and transcriptome data were presented for principal component analysis, performed by MeV4.2 (Saeed et al., 2003, 2006) and for calculation of PCCs. PCC values between metabolite data were calculated using a total of 144 samples. The mean intensity value of each tissue was used for the calculation of PCC between gene expression and a set of metabolite accumulation data (n = 36). Shannon entropy values (H) were calculated using a previously described procedure (Schug et al., 2005). For BL-SOM analysis, 10,647 genes annotated with GO terms by TAIR9 (Poole, 2007), including transporter activity, transferase activity, transcription factor activity, other metabolic processes, other enzyme activity, oxygen binding, secondary metabolic process, and hydrolase activity, were selected as metabolism-related genes. The intensity values were normalized by dividing the mean intensity value of each tissue with an overall mean value. BL-SOM analysis was performed with the aid of simple BL-SOM software (http://kanaya.naist.jp/SOM/; Kanaya et al., 2001; Abe et al., 2003). The number of cells along the x axis was set to 30. GO categories statistically overrepresented in a set of genes were investigated with BiNGO 2.3 using TAIR9 GO data (GOSlim_Plants) and hypergeometric testing with Benjamini and Hochberg's technique of false discovery rate correction (Maere et al., 2005).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Fragmentation patterns of representative MS2T assigned to metabolite signals and those expression profile among tissues.
Supplemental Figure S2. Frequency distribution of PCC values.
Supplemental Figure S3. The graph representation of M-M similarities in terms of accumulation patterns across 36 tissues of Arabidopsis.
Supplemental Figure S4. The nature of tissue-specific phytochemicals.
Supplemental Figure S5. Feature maps of BL-SOM analysis.
Supplemental Figure S6. BL-SOM clustering of metabolites and those biosynthesis-related genes.
Supplemental Figure S7. Detailed description of gene symbols and metabolites in Figure 5.
Supplemental Figure S8. A record example of AtMetExpress developmental database.
Supplemental Table S1. List of tissues analyzed in AtMetExpress developmental dataset.
Supplemental Table S2. List of annotated metabolites.
Supplemental Table S3. Lists of MS2T libraries created in this study.
Supplemental Table S4. Properties of metabolite structural similarity networks.
Supplemental Table S5. List of GO categories overrepresented in the set of low-entropy genes.
Supplemental Table S6. Dummy profile data of eight tissue markers for BL-SOM analysis.
Supplemental Text S1. An example: GSL metabolism.
Supplemental Data S1. Data matrix of AtMetExpress development dataset.
Supplementary Material
Acknowledgments
We wish to thank Prof. A. Ishihara (Kyoto University, Japan), Dr. R. Nakabayashi, and Prof. H. Takayama (Chiba University, Japan) for providing us with authentic Arabidopsis metabolites. We thank Drs. Y. Sawada, R. Niida, A. Takahashi, K, Takano, and M. Suzuki (RIKEN Plant Science Center, Japan) for their useful comments on this manuscript and technical support.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Kazuki Saito (ksaito@psc.riken.jp).
The online version of this article contains Web-only data.
Open Access articles can be viewed online without a subscription.
References
- Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuki T, Ikemura T (2003) Informatics for unveiling hidden genome signatures. Genome Res 13 693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama K, Chikayama E, Yuasa H, Shimada Y, Tohge T, Shinozaki K, Hirai MY, Sakurai T, Kikuchi J, Saito K (2008) PRIMe: a Web site that assembles tools for metabolomics and transcriptomics. In Silico Biol 8 339–345 [PubMed] [Google Scholar]
- Baerenfaller K, Grossmann J, Grobei MA, Hull R, Hirsch-Hoffmann M, Yalovsky S, Zimmermann P, Grossniklaus U, Gruissem W, Baginsky S (2008) Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320 938–941 [DOI] [PubMed] [Google Scholar]
- Böttcher C, Roepenack-Lahaye EV, Schmidt J, Schmotz C, Neumann S, Scheel D, Clemens S (2008) Metabolome analysis of biosynthetic mutants reveals diversity of metabolic changes and allows identification of a large number of new compounds in Arabidopsis thaliana. Plant Physiol 147 2107–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann G, Kajahn I, Neususs C, Pelzing M, Laug S, Unger M, Holzgrabe U (2005) Analysis of the glucosinolate pattern of Arabidopsis thaliana seeds by capillary zone electrophoresis coupled to electrospray ionization-mass spectrometry. Electrophoresis 26 1513–1522 [DOI] [PubMed] [Google Scholar]
- Brown PD, Tokuhisa JG, Reichelt M, Gershenzon J (2003) Variation of glucosinolate accumulation among different organs and developmental stages of Arabidopsis thaliana. Phytochemistry 62 471–481 [DOI] [PubMed] [Google Scholar]
- Burlat V, Kwon M, Davin LB, Lewis NG (2001) Dirigent proteins and dirigent sites in lignifying tissues. Phytochemistry 57 883–897 [DOI] [PubMed] [Google Scholar]
- Castellana NE, Payne SH, Shen Z, Stanke M, Bafna V, Briggs SP (2008) Discovery and revision of Arabidopsis genes by proteogenomics. Proc Natl Acad Sci USA 105 21034–21038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craigon DJ, James N, Okyere J, Higgins J, Jotham J, May S (2004) NASCArrays: a repository for microarray data generated by NASC's transcriptomics service. Nucleic Acids Res 32 D575–D577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Auria JC, Gershenzon J (2005) The secondary metabolism of Arabidopsis thaliana: growing like a weed. Curr Opin Plant Biol 8 308–316 [DOI] [PubMed] [Google Scholar]
- Davin LB, Lewis NG (2005) Lignin primary structures and dirigent sites. Curr Opin Biotechnol 16 407–415 [DOI] [PubMed] [Google Scholar]
- Desbrosses GG, Kopka J, Udvardi MK (2005) Lotus japonicus metabolic profiling: development of gas chromatography-mass spectrometry resources for the study of plant-microbe interactions. Plant Physiol 137 1302–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchini PJ, Bird DA, St-Pierre B (2004) Can Arabidopsis make complex alkaloids? Trends Plant Sci 9 116–122 [DOI] [PubMed] [Google Scholar]
- Farag MA, Huhman DV, Dixon RA, Sumner LW (2008) Metabolomics reveals novel pathways and differential mechanistic and elicitor-specific responses in phenylpropanoid and isoflavonoid biosynthesis in Medicago truncatula cell cultures. Plant Physiol 146 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellenberg C, Milkowski C, Hause B, Lange PR, Bottcher C, Schmidt J, Vogt T (2008) Tapetum-specific location of a cation-dependent O-methyltransferase in Arabidopsis thaliana. Plant J 56 132–145 [DOI] [PubMed] [Google Scholar]
- Goda H, Sasaki E, Akiyama K, Maruyama-Nakashita A, Nakabayashi K, Li W, Ogawa M, Yamauchi Y, Preston J, Aoki K, et al (2008) The AtGenExpress hormone and chemical treatment data set: experimental design, data evaluation, model data analysis and data access. Plant J 55 526–542 [DOI] [PubMed] [Google Scholar]
- Grienenberger E, Besseau S, Geoffroy P, Debayle D, Heintz D, Lapierre C, Pollet B, Heitz T, Legrand M (2009) A BAHD acyltransferase is expressed in the tapetum of Arabidopsis anthers and is involved in the synthesis of hydroxycinnamoyl spermidines. Plant J 58 246–259 [DOI] [PubMed] [Google Scholar]
- Hansen BG, Kliebenstein DJ, Halkier BA (2007) Identification of a flavin-monooxygenase as the S-oxygenating enzyme in aliphatic glucosinolate biosynthesis in Arabidopsis. Plant J 50 902–910 [DOI] [PubMed] [Google Scholar]
- Hirai MY, Klein K, Fujikawa Y, Yano M, Goodenowe DB, Yamazaki Y, Kanaya S, Nakamura Y, Kitayama M, Suzuki H, et al (2005) Elucidation of gene-to-gene and metabolite-to-gene networks in Arabidopsis by integration of metabolomics and transcriptomics. J Biol Chem 280 25590–25595 [DOI] [PubMed] [Google Scholar]
- Hirai MY, Sugiyama K, Sawada Y, Tohge T, Obayashi T, Suzuki A, Araki R, Sakurai N, Suzuki H, Aoki K, et al (2007) Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc Natl Acad Sci USA 104 6478–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai MY, Yano M, Goodenowe DB, Kanaya S, Kimura T, Awazuhara M, Arita A, Fujiwara T, Saito K (2004) Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc Natl Acad Sci USA 101 10205–10210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima Y, Nakamura Y, Ogata Y, Tanaka K, Sakurai N, Suda K, Suzuki T, Suzuki H, Okazaki K, Kitayama M, et al (2008) Metabolite annotations based on the integration of mass spectral information. Plant J 54 949–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai K, Mizutani M, Kawamura N, Yamamoto R, Tamai M, Yamaguchi H, Sakata K, Shimizu B (2008) Scopoletin is biosynthesized via ortho-hydroxylation of feruloyl CoA by a 2-oxoglutarate-dependent dioxygenase in Arabidopsis thaliana. Plant J 55 989–999 [DOI] [PubMed] [Google Scholar]
- Kai K, Shimizu B, Mizutani M, Watanabe K, Sakata K (2006) Accumulation of coumarins in Arabidopsis thaliana. Phytochemistry 67 379–386 [DOI] [PubMed] [Google Scholar]
- Kanaya S, Kinouchi M, Abe T, Kudo Y, Yamada Y, Nishi T, Mori H, Ikemura T (2001) Analysis of codon usage diversity of bacterial genes with a self-organizing map (SOM): characterization of horizontally transferred genes with emphasis on the E. coli O157 genome. Gene 276 89–99 [DOI] [PubMed] [Google Scholar]
- Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, D'Angelo C, Bornberg-Bauer E, Kudla J, Harter K (2007) The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J 50 347–363 [DOI] [PubMed] [Google Scholar]
- Kliebenstein DJ, Lambrix VM, Reichelt M, Gershenzon J, Mitchell-Olds T (2001) Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell 13 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Foster J, Chen J, Voll LM, Weber AP, Tegeder M (2007) AAP1 transports uncharged amino acids into roots of Arabidopsis. Plant J 50 305–319 [DOI] [PubMed] [Google Scholar]
- Li J, Last RL (1996) The Arabidopsis thaliana trp5 mutant has a feedback-resistant anthranilate synthase and elevated soluble tryptophan. Plant Physiol 110 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Bush DR (2006) Expression and transcriptional regulation of amino acid transporters in plants. Amino Acids 30 113–120 [DOI] [PubMed] [Google Scholar]
- Lommen A (2009) MetAlign: interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Anal Chem 81 3079–3086 [DOI] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21 3448–3449 [DOI] [PubMed] [Google Scholar]
- Matsuda F, Yonekura-Sakakibara K, Niida R, Kuromori T, Shinozaki K, Saito K (2009) MS/MS spectral tag-based annotation of non-targeted profile of plant secondary metabolites. Plant J 57 555–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno M, Compagnon V, Schoch GA, Schmitt M, Debayle D, Bassard JE, Pollet B, Hehn A, Heintz D, Ullmann P, et al (2009) Evolution of a novel phenolic pathway for pollen development. Science 325 1688–1692 [DOI] [PubMed] [Google Scholar]
- Mir Derikvand M, Sierra JB, Ruel K, Pollet B, Do CT, Thevenin J, Buffard D, Jouanin L, Lapierre C (2008) Redirection of the phenylpropanoid pathway to feruloyl malate in Arabidopsis mutants deficient for cinnamoyl-CoA reductase 1. Planta 227 943–956 [DOI] [PubMed] [Google Scholar]
- Mueller LA, Zhang P, Rhee SY (2003) AraCyc: a biochemical pathway database for Arabidopsis. Plant Physiol 132 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi R, Kusano M, Kobayashi M, Tohge T, Yonekura-Sakakibara K, Kogure N, Yamazaki M, Kitajima M, Saito K, Takayama H (2009) Metabolomics-oriented isolation and structure elucidation of 37 compounds including two anthocyanins from Arabidopsis thaliana. Phytochemistry 70 1017–1029 [DOI] [PubMed] [Google Scholar]
- Nakatsubo T, Mizutani M, Suzuki S, Hattori T, Umezawa T (2008) Characterization of Arabidopsis thaliana pinoresinol reductase, a new type of enzyme involved in lignan biosynthesis. J Biol Chem 283 15550–15557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nour-Eldin H, Halkier BA (2009) Piecing together the transport pathway of aliphatic glucosinolates. Phytochem Rev 8 53–67 [Google Scholar]
- Obayashi T, Hayashi S, Saeki M, Ohta H, Kinoshita K (2009) ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res 37 D987–D991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Kinoshita K, Nakai K, Shibaoka M, Hayashi S, Saeki M, Shibata D, Saito K, Ohta H (2007) ATTED-II: a database of co-expressed genes and cis elements for identifying co-regulated gene groups in Arabidopsis. Nucleic Acids Res 35 D863–D869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen BL, Chen S, Hansen CH, Olsen CE, Halkier BA (2002) Composition and content of glucosinolates in developing Arabidopsis thaliana. Planta 214 562–571 [DOI] [PubMed] [Google Scholar]
- Poole RL (2007) The TAIR database. Methods Mol Biol 406 179–212 [DOI] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J (2006) TM4 microarray software suite. Methods Enzymol 411 134–193 [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34 374–378 [DOI] [PubMed] [Google Scholar]
- Saito K, Hirai MY, Yonekura-Sakakibara K (2008) Decoding genes by coexpression networks and metabolomics—“majority report by precogs”. Trends Plant Sci 13 36–43 [DOI] [PubMed] [Google Scholar]
- Samanani N, Liscombe DK, Facchini PJ (2004) Molecular cloning and characterization of norcoclaurine synthase, an enzyme catalyzing the first committed step in benzylisoquinoline alkaloid biosynthesis. Plant J 40 302–313 [DOI] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Scholkopf B, Weigel D, Lohmann JU (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37 501–506 [DOI] [PubMed] [Google Scholar]
- Schoch G, Goepfert S, Morant M, Hehn A, Meyer D, Ullmann P, Werck-Reichhart D (2001) CYP98A3 from Arabidopsis thaliana is a 3′-hydroxylase of phenolic esters, a missing link in the phenylpropanoid pathway. J Biol Chem 276 36566–36574 [DOI] [PubMed] [Google Scholar]
- Schug J, Schuller WP, Kappen C, Salbaum JM, Bucan M, Stoeckert CJ Jr (2005) Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol 6 R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinbo Y, Nakamura Y, Altaf-Ul-Amin M, Asahi H, Kurokawa K, Arita M, Saito K, Ohta D, Shibata D, Kanaya S (2006) KNApSAcK: a comprehensive species-metabolite relationship database. In K Saito, RA Dixon, L Willmitzer, eds, Biotechnology in Agriculture and Forestry 57 Plant Metabolomics, Vol 57. Springer, Berlin, pp 165–181
- Shirley AM, McMichael CM, Chapple C (2001) The sng2 mutant of Arabidopsis is defective in the gene encoding the serine carboxypeptidase-like protein sinapoylglucose:choline sinapoyltransferase. Plant J 28 83–94 [DOI] [PubMed] [Google Scholar]
- Stein SE, Scott DR (1994) Optimization and testing of mass-spectral library search algorithms for compound identification. J Am Soc Mass Spectrom 5 859–866 [DOI] [PubMed] [Google Scholar]
- Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TWM, Fiehn O, Goodacre R, Griffin JL, et al (2007) Proposed minimum reporting standards for chemical analysis. Metabolomics 3 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Kai K, Shinbo Y, Tanaka K, Ohta D, Oshima T, Altaf-Ul-Amin M, Kurokawa K, Ogasawara N, Kanaya S (2008) Metabolomics approach for determining growth-specific metabolites based on Fourier transform ion cyclotron resonance mass spectrometry. Anal Bioanal Chem 391 2769–2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Ohmiya A (2008) Seeing is believing: engineering anthocyanin and carotenoid biosynthetic pathways. Curr Opin Biotechnol 19 190–197 [DOI] [PubMed] [Google Scholar]
- Ueda A, Shi W, Shimada T, Miyake H, Takabe T (2008) Altered expression of barley proline transporter causes different growth responses in Arabidopsis. Planta 227 277–286 [DOI] [PubMed] [Google Scholar]
- Winter D, Vinegar B, Nahal H, Ammar R, Wilson GV, Provart NJ (2007) An “electronic fluorescent pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS One 2 e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Matsuda F, Kasai K, Fukuoka S, Kitamura K, Tozawa Y, Miyagawa H, Wakasa K (2008) Mutation of a rice gene encoding a phenylalanine biosynthetic enzyme results in accumulation of phenylalanine and tryptophan. Plant Cell 20 1316–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Saito K (2009) Functional genomics for plant natural product biosynthesis. Nat Prod Rep 26 1466–1487 [DOI] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Tohge T, Matsuda F, Nakabayashi R, Takayama H, Niida R, Watanabe-Takahashi A, Inoue E, Saito K (2008) Comprehensive flavonol profiling and transcriptome coexpression analysis leading to decoding gene-metabolite correlations in Arabidopsis. Plant Cell 20 2160–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura-Sakakibara K, Tohge T, Niida R, Saito K (2007) Identification of a flavonol 7-O-rhamnosyltransferase gene determining flavonoid pattern in Arabidopsis by transcriptome coexpression and reverse genetics. J Biol Chem 282 14932–14941 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.