

Abstract
The Y chromosome of Drosophila melanogaster has <20 protein-coding genes. These genes originated from the duplication of autosomal genes and have male-related functions. In 1993, Russell and Kaiser found three Y-linked pseudogenes of the Mst77F gene, which is a testis-expressed autosomal gene that is essential for male fertility. We did a thorough search using experimental and computational methods and found 18 Y-linked copies of this gene (named Mst77Y-1–Mst77Y-18). Ten Mst77Y genes encode defective proteins and the other eight are potentially functional. These eight genes produce ∼20% of the functional Mst77F-like mRNA, and molecular evolutionary analysis shows that they evolved under purifying selection. Hence several Mst77Y genes have all the features of functional genes. Mst77Y genes are present only in D. melanogaster, and phylogenetic analysis confirmed that the duplication is a recent event. The identification of functional Mst77Y genes reinforces the previous finding that gene gains play a prominent role in the evolution of the Drosophila Y chromosome.
THE Y chromosome of Drosophila melanogaster is essential for male fertility and is entirely heterochromatic (Bridges 1916; Gatti and Pimpinelli 1983). Formal genetics identified six regions that are essential for male fertility (named kl-1, kl-2, kl-3, kl-5, ks-1, and ks-2), and each region seems to contain only 1 essential gene (Kennison 1981; Hazelrigg et al. 1982; Gatti and Pimpinelli 1983). Hence the Y chromosome of D. melanogaster contains six genes that are essential for male fertility (subsequent work identified additional, nonessential genes; see below). The first molecularly identified Y-linked fertility gene was kl-5, which encodes a motor protein that is a component of the sperm flagella (Gepner and Hays 1993). After the sequencing of the D. melanogaster genome (Adams et al. 2000) and the development of proper computational methods 11 additional Y-linked genes were molecularly identified; indirect evidence suggests that the total number of genes is <20 (Carvalho et al. 2000, 2001; Carvalho and Clark 2003; Vibranovski et al. 2008). The correspondence between the 12 molecularly identified genes and the six fertility factors has been partially elucidated. For example, the kl-2 and kl-3 fertility factors also encode axonemal dynein heavy chains, whereas PRY and PPr-Y are nonessential and hence do not correspond to any fertility factor (Carvalho et al. 2000, 2001).
All protein-coding genes of the D. melanogaster Y chromosome originated from duplications of autosomal genes (Carvalho et al. 2000, 2001, 2009; Koerich et al. 2008), which is an interesting evolutionary phenomenon. Sex chromosomes are believed to evolve from a normal pair of autosomes, after one homolog acquires a sex-determining gene and becomes a proto-Y. A combination of evolutionary forces would then lead to massive gene degeneration and loss in the proto-Y, the end result being a mature Y chromosome that has very few genes, most of them shared with the X (Rice 1996; Charlesworth and Charlesworth 2000). For example, the human Y chromosome encodes 27 proteins, and 18 of them are shared ancestrally with the X (which has ∼1100 genes) (Skaletsky et al. 2003; Ross et al. 2005). The lack of X–Y shared genes (other than the special case of rDNA) (Carvalho 2002) and other features of the Drosophila Y suggest that it is not homologous to the X and has not originated through the above pathway (reviewed in Carvalho et al. 2009). Hence, as in other species, identification of Drosophila Y-linked genes tells much about the origin and evolution of the Y chromosome itself (Lahn and Page 1999; Carvalho et al. 2000, 2009; Skaletsky et al. 2003).
The D. melanogaster genome contains ∼100 Mbp of heterochromatin (including 41 Mbp from the Y chromosome) and 120 Mbp of euchromatin. During genome sequencing the euchromatin assembled in large megabase pair-sized scaffolds that could be easily mapped and studied, but in the heterochromatin the assembly problems caused by its high content of repetitive DNA led to many small scaffolds that remain unmapped, forming the so-called chromosome U (for “unmapped”) (Carvalho et al. 2000, 2001; Hoskins et al. 2002; see Hoskins et al. 2007 and Smith et al. 2007 for recent progress). In the WGS3 assembly chromosome U amounts to 20.7 Mbp of sequence, scattered in 2597 scaffolds (average size: 8 kbp) (Hoskins et al. 2002). Besides its main part, the WGS3 assembly also contains 35,039 small “degenerate scaffolds” (size range: 300 bp–21 kb) amounting to 26 Mbp of sequence (Hoskins et al. 2002). Degenerate scaffolds contain large amounts of transposable elements and satellite sequences and were flagged as potential assembly errors (Myers et al. 2000; Hoskins et al. 2002). Nearly all known D. melanogaster Y-linked genes were discovered by investigating chromosome U sequences (Carvalho et al. 2000, 2001; Vibranovski et al. 2008), and parts of one gene were found among the degenerate scaffolds (Carvalho and Clark 2003).
Mst77F (Male-specific transcript 77F) is an autosomal gene located in the 3L chromosome (Kalderon and Rubin 1988; Russell and Kaiser 1993). It is essential for male fertility and encodes a protein that replaces the histones during chromatin condensation in spermatogenesis (Raja and Renkawitz-Pohl 2005). In 1993, Russell and Kaiser found three Mst77F- related sequences in the Y chromosome (Mst77-ψ1, Mst77-ψ2, and Mst77-ψ3). Two of them have defects in the coding region and clearly are pseudogenes, whereas the third sequence was incomplete, so its functional state could not be ascertained. The Y-linked copies of Mst77F mapped to region h18–hl9 on the cytogenetic map of the Y, which does not correspond to any of the fertility factors (Kennison 1981). This result might suggest that the Y-linked copies are nonfunctional, but as Russell and Kaiser (1993) pointed out, it could also be accounted for by full or partial complementation of fertility by the autosomal locus. Although they mentioned several times the possible existence of functional copies of Mst77F on the Y chromosome, this was largely ignored in the literature.
The present work started while we were searching for Y-linked genes in the degenerate scaffolds of D. melanogaster and found seven Y-linked Mst77F-like sequences. Several of them have complete coding sequences. The thorough investigation reported here confirmed that there are more than three copies of Mst77F genes in the Y chromosome and that several of them are functional.
MATERIALS AND METHODS
D. melanogaster genomic sequences and Blast searches:
Y-linked genes were searched in the unmapped and in the degenerate scaffolds of the D. melanogaster genome. The degenerate scaffolds have not been deposited in GenBank and were downloaded from the Drosophila Heterochromatin Genome Project (ftp://ftp.dhgp.org/pub/DHGP/Primary_Sequence/wgs3_degenerate_scaffolds.fasta). The unmapped portion of the main (i.e., “nondegenerate”) WGS3 assembly was downloaded from the Berkeley Drosophila Genome Project. We used release 3 (http://www.fruitfly.org/sequence/release3genomic.shtml; file WGS3_het_genomic_dmel_RELEASE3-0.FASTA), but the same results were obtained with the latest version (release 5).
All Blast searches were run locally in a Linux workstation, using the programs from the NCBI blast package (downloaded from ftp://ftp.ncbi.nih.gov/blast/).
D. melanogaster strain and nucleic acids extraction:
DNA and RNA were extracted from the same D. melanogaster strain used in the genome project (“y cn bw sp”) (Adams et al. 2000). Genomic DNA was extracted separately from males and females (in pools of five individuals from each sex) by standard phenol-chloroform extraction followed by ethanol precipitation. In the RNA preparation, testes from adult males were dissected in PBS buffer and immediately transferred to PBS buffer on ice. RNA was extracted with Trizol (Invitrogen, Carlsbad, CA; no. 15596-018) according to the manufacturer's instructions. Prior to cDNA preparation the RNA was treated with DNase to avoid contamination with genomic DNA. The cDNA was then prepared using the RetroScript kit (Ambion; no. AM1710) and random primers, according to the manufacturer's instructions.
Number and sequence of Y-linked copies of the Mst77F gene:
We initially found seven Y-linked copies of the Mst77F gene in the assembled D. melanogaster genome (hereafter called collectively “Mst77Y”). We took this number of copies and their sequences as approximations because in the beginning of our work we found signs of misassembly and missing genes (see results). We used three different methods to more precisely quantify the number of Mst77Y genes, which allowed us to overcome the inherent limitations of each method.
Method 1—de novo sequencing:
Inspection of the Mst77Y genes in the assembled D. melanogaster genome showed that all shared a 12-bp insertion in the 5′-UTR region, which is absent from Mst77F. This allowed us to design a pair of PCR primers that specifically amplify the Mst77Y genes (mst77Y_F1 ATTTTTGTCGATCAAGAATTTACCAA and mst77_R1 AGGGTGTGGGAGATGAAACCTC). These primers encompass the whole coding region of the genes. We did two PCR experiments using the standard protocol, with an annealing temperature of 55° and 30 PCR cycles. In the pilot experiment we used normal Taq DNA Polymerase (GeneChoice) but in the second (and larger) experiment we switched to an enzyme with low error rate (PfuUltra High-Fidelity; Stratagene, La Jolla, CA, no. 600380), which was desirable because we would sequence individual clones, and the PCR-induced mutations might lead to spurious results in this case (Hogrefe and Borns 2003). To further reduce the effect of PCR-induced mutations, we conservatively discarded haplotypes sampled only once. In the first experiment the PCR products were cloned with the PCR TOPO TA cloning kit (Invitrogen, no. K450001) and in the second one, we used the Zero Blunt TOPO PCR Cloning kit (Invitrogen, no. K2800-20). A total of 115 clones (45 in the first experiment and 70 in the second) were randomly picked for DNA sequencing. This number was chosen as follows. Assuming no bias in the PCR amplification and cloning, the number of clones needed to sample each Mst77Y gene at least once, at a given probability, can be calculated using a multinomial cumulative distribution function
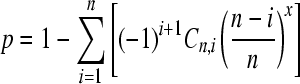 |
(1) |
(Degroot and Schervish 2002), where p is the probability that each gene will be sampled at least once, n is the number of genes, x is the number of clones, and C is the combination operator (supporting information, Figure S1 and File S1). We checked the above formula with a computer simulation written in R (R Development Core Team 2007; File S2), which also allowed the calculation of the probability that each gene was sampled at least twice (instead of at least once). As we mentioned above, this more stringent criterion is useful to deal with sequencing errors introduced by the PCR.
For example, if there are seven Mst77Y genes (which was our initial guess) and we aim at a 98% probability that all of them would be sampled at least once, we need 40 clones. We used this number in the first experiment, and when we found that the number of genes was higher than seven, we increased the sample size to 115 clones. Assuming 18 Mst77Y genes, these 115 clones would be enough for sampling each Mst77Y gene at least once with a probability of 97.5% and at least twice with 82% probability. These estimates are somewhat optimistic because they do not take into account the clones that would be discarded due to PCR-induced mutations (see results).
Method 1 produces two pieces of information: the number of Mst77Y genes (obtained by simple counting of haplotypes) and their accurate sequences. Basically the same method was used to identify the Mst77Y genes that are expressed (and to obtain a rough estimate of their expression level). In this case we used testis cDNA as the template for the PCR reaction and the same primer pair (mst77Y_F1 and mst77_R1) that specifically amplifies the Mst77Y genes. We sequenced 40 clones.
Method 2—restriction enzyme digestion:
After we got accurate nucleotide sequences of the Mst77Y genes by sequencing them de novo (as described above), we picked one of the Mst77Y genes and aligned it with Mst77F using Blast2. The Blast2 output file was then submitted to the BlastDigester program (Ilic et al. 2004; http://www.bar.utoronto.ca/ntools/cgi-bin/ntools_blast_digester.cgi) to identify sites that would be cut by restriction enzymes only in the Mst77F or in the Mst77Y genes. The potential restriction sites were then checked for conservation among all Mst77Y genes and for the availability of suitable flanking sequences for PCR amplification. We chose an XbaI site in exon 1 and a BstUI in exon 2, which are expected to be cut in all Mst77Y genes, but not in Mst77F. We then designed PCR primers that target conserved regions (i.e., that are identical between Mst77F and Mst77Y genes) surrounding the restriction enzyme sites. A 179-bp fragment of exon 1 was amplified from male genomic DNA with the primers mst77_F3 (5′-CAAAATGAGCAATCTGAAACAAAAGGATA-3′) and mst77_R3 (5′-CTCAGAAAATTAACAAAGCCAGACT-3′), using standard PCR protocol (annealing temperature: 48°). Five microliters of the PCR products were digested overnight at 37° with the XbaI enzyme (20 units; New England Biolabs, Beverly, MA). Three bands can be observed in agarose gels: a 179-bp (undigested) one from Mst77F and two smaller bands (69 and 110 bp) from the Mst77Y genes. The intensity of each band was measured with Kodak (Rochester, NY) 1D Image Analysis v. 3.6, using Invitrogen's Low Mass Ladder (no. 10068-013) as a standard. As detailed in the results, the ratio of the masses of digested to undigested bands estimates the number of Y-linked copies. The same general procedure was repeated for exon 2; in this case we used the primers mst77_F4 (5′-GCACTCCAAGAAAGGAGAACAAG-3′) and mst77_R4 (5′-GGCTTAAGACACCTTGGCTTTG- 3′) with a 52° annealing temperature and digestion with 10 units of BstUI at 60° (New England Biochemicals). The 209-bp PCR product of exon 2 is cleaved by BstUI (in the Mst77Y genes) in two fragments, of 69 and 140 bp.
Basically the same method was used to quantify the mRNA expression of the Mst77Y genes in comparison to Mst77F. In this case we performed the PCR using testis cDNA as the template (instead of male genomic DNA) and used only the mst77_F3/mst77_R3 primers. The products were digested with XbaI and analyzed as described above.
Method 3—computational analysis of the sequencing traces:
The D. melanogaster genome was sequenced using unsexed embryos (presumably 50:50 male:female) and the whole-genome shotgun method, which randomly samples the genome (Myers et al. 2000). Hence we can estimate the number of Mst77Y genes by comparing the number of traces that came from them with those from the Mst77F genes. We first identified the traces that presumably belong to the Mst77F or Mst77Y genes by doing a BlastN search (BlastN parameters: expected value, 0.0001; word size, 20), using these genes as queries, against the trace database of D. melanogaster in the NCBI and downloaded them (in FASTA format) from the NCBI Trace Archive (ftp://ftp.ncbi.nih.gov/pub/TraceDB/misc/query_tracedb). We then classified each whole-genome shotgun trace as “autosomal” or “Y linked” by doing a BlastN alignment of each one against an extended sequence (500 bp upstream from start ATG and 500 bp downstream from the stop codon) of the autosomal Mst77F and of the Y-linked Mst77-ψ1 (accession no. Z19565). The extended sequences were used to improve the classification of the traces that aligned only to the edges of the genes. Traces were classified as autosomal or Y linked on the basis of which gene (Mst77F or Mst77-ψ1) they align better. The ratio of their numbers estimates the number of Mst77Y genes.
A variant of the same method was used to compare the mRNA expression of the Mst77Y genes with Mst77F. In this case we used 25 million cDNA traces from larvae, young males, and old males, downloaded from MachiBase (Ahsan et al. 2009).
Molecular evolution of Y-linked copies of Mst77F:
We extracted the coding sequences from the Mst77Y genes, from Mst77F (accession no. NM_079464) and from its orthologs in D. sechellia (XM_002040688), D. simulans (XM_002085675), D. erecta (XM_001973509), and D. yakuba (XM_002086611), and aligned them using ClustalW with manual correction to preserve the reading frame. One sequence was very short (Mst77Y-15) and was excluded from our analysis. Then, we obtained a cladogram with the neighbor-joining method using the software MEGA 4 (Tamura et al. 2007). The analysis of the synonymous (dS) and nonsynonymous (dN) distances along the branches of this cladogram was performed in the HyPhy program package (Kosakovsky Pond et al. 2005), using the MG94 model of codon substitution (Muse and Gaut 1994). HyPhy independently estimates dN and dS values, but for the sake of simplicity we report the results in the widely used dN/dS ratio (also called “ω”). Several testing strategies were used to verify the occurrence of purifying selection on Mst77Y genes (see results).
Timescale of the evolution of the Mst77Y gene family:
The timescale of the evolution of the Mst77Y gene family was inferred using the correlated Bayesian method (Rannala and Yang 2007) implemented in the mcmctree program of the PAML 4 package (Yang 2007). The relaxed clock was calibrated by adopting normal prior distributions for the ages of two nodes: the D. melanogaster/D. simulans split was set at 5.4 ± 1.1 million years (mean ± standard deviation), and the D. melanogaster/D. yakuba split was set at 12.8 ± 2.7 million years (Tamura et al. 2004).
RESULTS
Initial detection of Y-linked copies of the Mst77F gene:
During the search of Y-linked genes we found seven scaffolds that seem to encode a protein similar to Mst77F (∼87% identical at the protein level): the unmapped scaffold AABU01000419 and six degenerate scaffolds (211000022215023, 211000022226552, 211000022236579, 211000022241185, 211000022243199, 211000022246937, and 211000022246939). There are also smaller fragments, among both the unmapped and the degenerate scaffolds. These Mst77F-related sequences were also found in the D. melanogaster EST database of the NCBI (e.g., accessions BF505110, BF488351, BF499179, AI946428, and AI946506). Inspection of their nucleotide sequence disclosed a shared 12-bp insertion in the putative 5′-UTR that is absent from the autosomal Mst77F. A primer targeting this region (mst77Y_F1), in combination with a nondiscriminating primer (mst77_R1), gave the expected PCR product only in males, which shows that all copies of Mst77F carrying this 12-bp insertion are Y linked. We initially thought that they are the pseudogenes described by Russell and Kaiser (1993), but surprisingly none of these sequences had the premature stop codon and frameshift indels described there. When we searched the Trace Archive, we did find traces with these disruptive mutations (e.g., ti|180852087, with the 124G → T premature stop codon mutation); these traces were either left unassembled or got misassembled with traces from other Mst77F copies that lack the stop codons. In both cases, the conclusions are the same: there seem to be functional copies of the Mst77F gene in the Y chromosome, and we need to sequence them de novo to get accurate data.
Number and sequence of copies of the Mst77F gene in the Y chromosome:
Method 1—de novo sequencing:
Primers mst77Y_F1/mst77_R1 amplified the whole coding sequence of the Mst77Y genes. After cloning and sequencing, we got 115 sequences in two experiments (Table 1). Forty-six clones have haplotypes that were found only once (“singletons”). Several lines of evidence suggest that these singletons are artifacts introduced by PCR (Zylstra et al. 1998; Hogrefe and Borns 2003), either point mutations or chimerical sequences: (i) many fewer singletons occurred in the second experiment, where we switched to a low-error DNA polymerase; (ii) none of their unique sequence features could be found in the Trace Archive (not shown); and (iii) nearly all singletons differ from some valid sequence by one or two mismatches, and the frequency of these mismatches seems to be compatible with the error rate of the DNA polymerase employed. These 46 clones were discarded. Among the remaining 69 clones we found 18 different haplotypes (Figure 1, Figure S2 A, and Figure S3; accession nos. GQ868243–GQ868260). These 18 haplotypes must correspond to 18 different Mst77Y genes (instead of to allelic variants of a smaller number of genes) because we used a highly inbred line as the DNA source and additionally, the Y chromosome has very low polymorphism (Zurovcova and Eanes 1999). A limitation of this “haplotype-counting” method to estimate the number of Mst77Y genes is that two or more identical genes would be counted as one. This problem is addressed by the two other methods we used (next sections).
TABLE 1.
Identification of Mst77Y genes by de novo sequencing of PCR clones
| Male genomic DNA |
Features |
|||||
|---|---|---|---|---|---|---|
| Gene | Exp. 1 | Exp. 2 | Both | Testis cDNA | Sizea | Functionalityb |
| Mst77Y-1 | 1 | 6 | 7 | 2 | 214 | Intact ORF |
| Mst77Y-2 | — | 2 | 2 | — | 214 | Intact ORF |
| Mst77Y-3 Ψ | 1 | 5 | 6 | 3 | 118 | Fs (Δ366–376) |
| Mst77Y-4 | 5 | 2 | 7 | — | 213 | Intact ORF |
| Mst77Y-5 Ψ | — | 3 | 3 | — | 118 | Fs (Δ366–376) |
| Mst77Y-6 Ψ | 2 | 2 | 4 | 3 | 71 | Psc (214) |
| Mst77Y-7 | 1 | 9 | 10 | 5 | 214 | Intact ORF |
| Mst77Y-8 | — | 3 | 3 | — | 213 | Intact ORF |
| Mst77Y-9 | — | 2 | 2 | — | 214 | Intact ORF |
| Mst77Y-10 Ψ | — | 5 | 5 | 1 | 188 | Fs (Δ563) |
| Mst77Y-11 Ψ | — | 2 | 2 | — | 115 | Fs (Δ366–373) |
| Mst77Y-12 | 2 | 4 | 6 | 15 | 214 | Intact ORF |
| Mst77Y-13 | — | 2 | 2 | 8 | 214 | Intact ORF |
| Mst77Y-14 Ψ | — | 2 | 2 | — | 188 | Fs (Δ563) |
| Mst77Y-15 Ψ | 4 | 2 | 6 | — | 81 | Fs (complex) |
| Mst77Y-16 Ψc | 1 | — | 1 | — | 188 | Fs (Δ563) |
| Mst77Y-17 Ψ | 1 | — | 1 | 1 | 188 | Fs (Δ563) |
| Mst77Y-18 Ψ | — | — | — | — | 115 | Fs (Δ366–373) |
| Valid clones | 18 | 51 | 69 | — | ||
| Singletons | 27d | 19e | 46 | — | ||
| Total clones |
45 |
70 |
115 |
38 |
||
Sequencing of PCR clones identified 18 Mst77Y genes. Columns 2–5 show the number of clones per gene. Only haplotypes represented by two or more clones were validated as genes, and the remaining (“singletons”) were deemed as PCR errors. Note that Mst77Y-17 was included because it was observed in one cDNA clone; Mst77Y-16 and Mst77Y-18 were included because they were recovered in a preliminary experiment (data not shown).
Number of amino acids of the predicted protein that are homologous to Mst77F (the scrambled amino acids introduced after frameshifts were not counted).
Inferred functionality, based on the integrity of the coding regions. Disrupting mutations are marked as follows (coordinates of affected nucleotides shown in parentheses): Fs, frameshift deletions; Psc, premature stop codon caused by a point mutation. See Figure S3 for the alignment of the 18 genes.
Mst77Y-16 was previously annotated as CG40530 (Smith et al. 2007).
All singletons are explainable by point mutations induced by Taq, as they have few mismatches with one of the Mst77-Y genes. Specifically, 3 clones have 3 putative mutations; 9 clones have 2, 15 have 1, and the remaining 18 clones have 0 mutations.
Experiment 2 used a low error rate polymerase (Pfu Ultra High-Fidelity; Stratagene, La Jolla, CA). Six of the 19 singletons are explainable by single point mutations and the remaining 13 seem to be chimerical sequences [Pfu generates more recombination artifacts than Taq (Zylstra et al. 1998)].
Figure 1.—

Mst77Y genes identified by de novo sequencing. Transcribed genes are marked with arrows; genes in which no transcript was detected are represented by boxes; open arrows and boxes, genes with intact ORFs (“potentially functional”); shaded arrows and boxes, genes with disrupting mutations (“nonfunctional”). Neighbor-joining tree of aligned nucleotides of the coding sequence is shown; branch lengths and the outgroups are shown in Figure S2 A. Only bootstrap values >50% are shown.
The new data agree with and expand Russell and Kaiser's (1993) results. We recovered the three pseudogenes they described: Mst77-ψ1, Mst77-ψ2, and Mst77-ψ3 are identical to or closely match our Mst77Y-6, Mst77Y-15, and Mst77Y-10 genes, respectively. Mst77Y genes are on average 89% identical with Mst77F at the nucleotide level (Figure S3). Inspection of ESTs and genomic traces (from the Trace Archive) showed that all sequences that do not have the 12-bp insertion in the 5′-UTR (which was targeted by the mst77Y_F1 primer) belong to the Mst77F gene (data not shown). So it is unlikely that there are additional, divergent Mst77Y genes that were missed by our PCR approach. As summarized in Figure 1 and Table 1, 10 Mst77Y genes have disruptive mutations in their coding regions (including those described by Russell and Kaiser 1993) and were considered pseudogenes. However, the remaining 8 genes would be able to encode a protein similar to Mst77F and hence are potentially functional.
Some additional comments about “method 1” may be useful for those planning a similar approach. PCR-induced errors remain a hard problem, particularly if there are many gene copies (as in Mst77Y): Pfu polymerase is known to induce fewer point mutations than Taq, but unfortunately it induces more chimeras (Zylstra et al. 1998), an effect we saw in our data (Table 1). Requiring two clones to validate each haplotype solves part of the problem, but unless they come from independent PCR amplifications and cloning experiments, the possibility remains that they trace to a single error induced in early PCR cycles. Perhaps the ideal approach would be to estimate the number of needed clones with Equation 1 (taking into account the mutant clones that will be discarded), run two independent PCR/cloning experiments with this number (possibly using Taq in one experiment and Pfu in the other), and validate only those haplotypes sampled in both experiments. Note also that we lost ∼40% of the clones due to polymerase-induced mutations (mostly by Taq), so the chance of missing some Mst77Y gene is substantially higher than our initial calculation.
Method 2—restriction enzyme digestion:
As expected, the PCR with the primers mst77_F3/mst77_R3 amplified a 179-bp fragment of exon 1 from the Mst77F and Mst77Y genes, and the XbaI enzyme digested exclusively the Mst77Y copies (Figure 2). Quantification of the digested (110 bp, from Mst77Y) and undigested (179 bp, from Mst77F) bands showed that there were on average 8.13 Mst77Y molecules for each Mst77F (Table 2). Since the initial PCR reaction used as template male genomic DNA (which is diploid for the autosomes, but haploid for the Y), the estimated number of Mst77Y genes is 16 (95% confidence interval: 14.7–17.9), which closely agrees with the previous estimate (18 genes). When we repeated the same procedure with exon 2 (using the mst77_F4/mst77_R4 primers and digestion with BstUI; data not shown) we consistently obtained an estimate of 8 Mst77Y genes, which certainly is incorrect (from the previous section we know that there are at least 18 genes). We repeated the experiment several times, checking for the completeness of the digestion and other possible sources of artifacts, and could not find any. Thus it seems that there was preferential PCR amplification of the Mst77F gene (Walsh et al. 1992), which resulted in underestimation of the number of Mst77Y genes.
Figure 2.—

Quantification of the number of Mst77Y genes and their mRNA expression through XbaI digestion. PCR amplification was carried with primers that amplify both Mst77F and Mst77Y genes, using as templates male genomic DNA (left) and testis cDNA (right). The products were digested with the XbaI restriction enzyme, which cuts the Mst77Y sequences, but not Mst77F. UD, undigested PCR product; D1–D3, three independent PCR/digestion replicas; positive control, digestion of PCR from a cloned Mst77Y gene. The relative intensity of the 110-bp band (compared to the 179-bp band) estimates the number of Mst77Y genes or their mRNA expression (see text and Table 2).
TABLE 2.
Quantification of the number of Mst77Y genes and their mRNA expression through XbaI digestion
| Template for PCR | Replica | Mass 179-bp band | Mass 110-bp band | Mst77Y/Mst77F ratioa | Mst77Y genes | % Mst77Y mRNAb |
|---|---|---|---|---|---|---|
| gDNA | 1 | 6.71 | 35.04 | 8.50 | 17.0 | — |
| gDNA | 2 | 6.75 | 32.67 | 7.88 | 15.8 | — |
| gDNA | 3 | 6.72 | 33.14 | 8.02 | 16.0 | — |
| Mean (95% C.I.)c | 16.3 (14.7–17.9) | |||||
| cDNA | 1 | 36.58 | 8.59 | 0.38 | — | 27.6 |
| cDNA | 2 | 33.20 | 8.46 | 0.41 | — | 29.3 |
| cDNA | 3 | 35.09 | 8.23 | 0.38 | — | 27.6 |
| Mean (95% C.I.)c |
28.2 (25.8–30.6) |
Number of Mst77Y molecules in relation to Mst77F, estimated as (column 4/column 3) × (179/110).
Contribution of Mst77Y genes to the Mst77F-like mRNA pool, estimated as column 5/(1 + column 5).
Confidence intervals are rough approximations, assuming normal distribution.
The sequence targeted by the PCR primers mst77_F4/mst77_R4 is identical between Mst77F and all Mst77Y, so this putative preferential amplification might have been caused by differences in the melting temperature or in secondary structure of the amplified fragments. Given its gross error, we dismissed the estimate obtained with exon 2. The result from exon 1 agrees with the previous estimate (18 copies) and also with the estimate described in the next section. However, it is clear that this method is not very reliable.
Method 3—computational analysis of the sequencing traces:
We found 156 “whole-genome shotgun” traces in the D. melanogaster Trace Archive that have a high similarity with the Mst77F and Mst77Y genes. After analysis of their sequences, we found that 121 traces came from the Mst77Y genes and 35 from Mst77F (Figure 3; ratio Mst77Y/Mst77F, 3.46; 95% confidence interval, 2.36–5.19). Since the D. melanogaster genome project used unsexed embryos as the DNA source, the autosomes are overrepresented in a 4:1 ratio in relation to the Y chromosome. So the trace data indicate that there are ∼14 Mst77Y genes (95% confidence interval: 9–21), which is similar to our previous estimates of 18 and 16 genes.
Figure 3.—

Quantification of the number of Mst77Y genes through computational analysis. The origin (Mst77F or Mst77Y) of each trace of the genome project that has similarity to these genes was identified by its best BlastN match. We quantified this by annotating for each trace the number of aligned bases (identity × alignment length) to Mst77F and to one of the Mst77Y genes (Mst77-ψ1) and subtracting the former from the latter (“Alignment difference”). Positive values indicate that the trace came from a Mst77Y gene, and negative values indicate Mst77F origin.
Transcription of Mst77Y genes:
We investigated the contribution of the Mst77Y genes to the pool of Mst77F-like mRNA by adapting the three methods used before to estimate the number of Mst77Y genes.
PCR using testis cDNA as the template and primers specific for the Mst77Y genes (mst77Y_F1 and mst77_R1) produced robust amplification (Figure 2), which shows, in accordance with Russell and Kaiser's (1993) results, that the Mst77Y genes are expressed. We cloned the PCR products and got 40 sequences. All match some of the previously identified 18 Mst77Y genes (Table 1), which again suggests that we identified all such genes. Thirty-eight clones are correctly spliced, and the remaining 2 retained the intron (both from Mst77Y-12). Two factors suggest that these 2 clones originated from immature transcripts rather than from contaminating genomic DNA: (i) the RNA was treated with DNase prior to reverse transcription and, (ii) as commented below, Mst77Y-12 is the most abundant Mst77Y transcript. We conservatively excluded these 2 clones from our analysis. The remaining 38 clones came from 8 Mst77Y genes (Table 1), 4 of which are pseudogenes (Mst77Y-3, Mst77Y-6, Mst77Y-10, and Mst77Y-17), and 4 are potentially functional (Mst77Y-1, Mst77Y-7, Mst77Y-13, and Mst77Y-12). These 4 potentially functional genes account for 30 of the 38 clones (79%). The majority of the spliced transcripts (15 of 38) came from the Mst77Y-12 gene, which is potentially functional and would encode a protein that has 86% identity with the Mst77F protein.
The mst77_F3/mst77_R3 amplification of testis cDNA followed by XbaI digestion shows that 28% of the Mst77F-like mRNA came from the Mst77Y genes (95% confidence interval: 26–31%); the remaining 72% came from Mst77F (Figure 2 and Table 2).
For the computational analysis of expression we used the MachiBase 5′-end RNA Transcription Database (Ahsan et al. 2009), which consists of ∼26-bp tags from the transcription start site (TSS), from different developmental stages (embryos, unsexed larvae, young males, adult males, young females, adult females, and S2 cells). Their web page (http://machibase.gi.k.u-tokyo.ac.jp/) displays the count of 26-bp TSS tags matching each position of the genome. Querying it for Mst77F disclosed strong expression in larvae, young males, and adult males; very weak expression in young females (presumably caused by promoter leakage); and no expression in embryos and adult females (Figure S4). Except for the young females, this fully agrees with Russell and Kaiser's data, who additionally showed that Mst77F is expressed in male (but not in female) larvae and pupae. MachiBase also showed that this gene has a very broad distribution of TSSs (“slippery promoter”; Yasuhara et al. 2005) that starts 250 bp before the initial methionine codon and spans (surprisingly) to the middle of the second exon (Figure S4). This is not an artifact (caused, for example, by inefficient selection of the 5′ region of the mRNA), since many genes display sharp TSSs in MachiBase (Ahsan et al. 2009).
MachiBase attempts to eliminate false positives by excluding tags that mapped to multiple locations (Ahsan et al. 2009). However, as only one Mst77Y gene—CG40530, equivalent to Mst77Y-16—is present in the current release of the D. melanogaster genome (release 5), it is possible that the signal of several Mst77Y genes (i.e., number of matching tags) was partially mixed with Mst77F. To disentangle their signals, and measure the contribution of Mst77Y genes to the Mst77F-like mRNA pool, we compared each MachiBase 26-bp tag to the upstream regions of Mst77F and Mst77Y genes, as follows. First we retrieved the 250-bp upstream region of the Mst77Y genes (the sequences produced by our method 1 cover only the coding regions), with a BlastN search in the NCBI Trace Archive, using as query the Mst77-ψ1 sequence (Z19565). We identified 68 traces that have the Mst77Y-specific 12-bp insertion and that cover the 250-bp upstream region. Due to the 12-bp insertion, we are sure that they belong to one of the Mst77Y genes, although it is not possible to precisely identify which one. We trimmed all 68 traces, plus Mst77F and Mst77-ψ1, to the region between −250 bp and the start methionine codon. We then used these 70 sequences as a database and ran locally a BlastN search (word size: 20) of each MachiBase 26-bp tag (∼25 million sequences) against this database. As expected, the vast majority of the tags did not give any hit (they came from other genes); we found 644, 2844, and 908 Mst77F-like tags in the larvae, young male, and old male libraries, respectively. A small proportion of them (∼6%) matched equally well Mst77F and some (or all) Mst77Y upstream sequences; these tags were discarded. Among the classifiable tags, we found that 32, 18, and 11% of them came from the Mst77Y genes in the larvae, young male, and old male libraries, respectively (Figure S5 and Table S1). The difference among these three proportions is significant at the P < 10−6 level (heterogeneity chi-square test); it seems that the relative contribution of the Mst77Y genes steadily declines with age.
The values obtained with the MachiBase tags for adult males (young, 18%; old, 11%) are lower than our XbaI digestion estimate (28% of the total Mst77F-like mRNA came from Mst77Y genes). MachiBase libraries were prepared from Canton-S flies, aged for 5 days (“young males”) or 30 days (“old males”; B. Ahsan and S. Morishita, personal communication). We have not controlled the age of our males (we took them from the culture) but it is unlikely that age alone can explain the discrepancy (it would be necessary that our males were younger than 5 days and that these very young males have a larva-like expression pattern). More likely the discrepancy is due to differences between the two methods and strains used. Indeed, Russell and Kaiser (1993) and ourselves (data not shown) found interstrain variation in the number of Mst77Y genes. In summary, the XbaI digestion and the MachiBase data show that the Mst77Y genes account for between 11% (old males) and 32% (larvae) of the Mst77F-like mRNA. In natural conditions Drosophila seldom live 30 days (Dobzhansky and Wright 1943; Crumpacker and Williams 1973), so the Mst77Y expression at that age is less relevant, and it seems fairly safe to assume that Mst77Y genes account for ∼25% of the Mst77F-like mRNA.
Combining the result of the XbaI digestion/MachiBase database (∼25% of the Mst77F-like transcripts come from the Mst77Y genes) with the de novo sequencing (79% of the transcripts from the Mst77Y genes are potentially functional), we found that ∼20% of the functional Mst77F-like mRNA comes from the Y chromosome.
A final observation about the broad distribution of TSSs (Figure S4) is that many transcripts start after the first methionine codon, and we initially thought that it was due to aberrant transcription of some Mst77Y genes. However, when we repeated the MachiBase procedure using as a database the coding regions of the Mst77F and Mst77Y genes, we got almost exactly the same result (Table S1). Hence both Mst77F and Mst77Y genes produce transcripts that, if translated, would produce a much shorter protein.
Molecular evolution of Y-linked copies of Mst77F:
Several Mst77Y genes seem to be functional, since they encode a protein similar to Mst77F and contribute one-fifth of the mRNA pool. We further investigated this possibility with a molecular evolutionary analysis. If these Mst77Y genes indeed are functional, we expect natural selection to be acting on them. The ratio of nonsynonymous to synonymous divergence (dN/dS, called ω) is a standard measure of selective pressure (Yang and Bielawski 2000). It is expected to be 1 in pseudogenes (due to the lack of selective constraints), whereas in most functional genes it is <1 (“purifying selection”). In rare cases where selection favored the fixation of beneficial alleles, ω can be >1 (“positive selection”). The ω-values (actually, dN and dS) for each branch of the cladogram (Figure 4 and Figure S6) were estimated with the HyPhy program package (Kosakovsky Pond et al. 2005) and used in four different tests. In test 1 (Figure 4, Table S2, and Table S3), we asked the question “Are the Mst77Y genes as a whole evolving under selective constraints?” by estimating a single ω-value for all Mst77Y branches and comparing the fit of this hypothesis against the null hypothesis ω = 1 for these branches. The null hypothesis was rejected (P = 0.027, LRT) and the best-fitting ω-value is <1 (ω = 0.61), which strongly suggests that at least some of the Mst77Y genes evolved under purifying selection. We further investigated this possibility as follows. The sequence data show that some Mst77Y genes are potentially functional, whereas others clearly are pseudogenes (Table 1). If the previous result (test 1) was indeed caused by natural selection, this selection should be detected mainly in the potentially functional genes. We verified this prediction in test 2, by estimating one ω for the potentially functional branches (ωpf) and one ω for the nonfunctional branches (ωnf) and comparing the fit of this hypothesis against the null hypothesis of a single ω for all Mst77Y branches. The null hypothesis was rejected (P = 0.015) and the estimated ω is much smaller in the potentially functional branches (ωpf = 0.41) than in the nonfunctional branches (ωnf = 0.85). These results again support the hypothesis that some Mst77Y genes are functional. We did two additional tests that are described below, which further support this conclusion.
Figure 4.—

Molecular evolution of Mst77Y genes. The four tests of neutral evolution of the Mst77F genes were based on dN/dS ratios (ω), which were constrained to 1 (“ω = 1,” dashed lines) or estimated from the data (“ω = free,” solid and shaded lines, one for each independently estimated ω). The estimated ω is shown in parentheses (Table S2 and Table S3 show the values of dN and dS). The result of the LRT and the biological interpretation are shown in column 4. “Ypf,” potentially functional Mst77Y genes (i.e., with intact ORF); “Ynf,” nonfunctional Mst77Y genes (with disrupting mutations). The figure is a simplification; the actual tests include the outgroups shown in Figure S6 and Table S3 (ω was free in all these branches). Tests were performed with the HyPhy package (Kosakovksy Pond et al. 2005), using Files S3 and S4.
There are two nonexclusive explanations for ω being <1 in the nonfunctional branches under test 2: (i) sampling variance, in which case the estimated ωnf will not be significantly different from 1, and (ii) depending on the time of inactivation of the pseudogenes, some of their branches might show a sign of selection, imparted by the time before the inactivation. We addressed these issues with test 3, which again estimates one ω for the potentially functional branches (ωpf) and one ω for the nonfunctional branches (ωnf), but now compares the fit against the null hypothesis ωnf = 1. The null hypothesis was not rejected (P = 0.54), which suggests that the nonfunctional copies are indeed evolving without selective constraints. Finally, in test 4 we tested the hypothesis of selective constraints in the potentially functional branches under the assumption of lack of selective constraints in the nonfunctional branches (ωnf = 1); we again found that ωpf is significantly <1 (P = 0.017).
The results from the four tests fully support the hypothesis that some Mst77Y genes are functional: the potentially functional genes evolved under purifying selection, whereas the nonfunctional genes evolved as pseudogenes.
Tempo and mode of Mst77Y origin:
All Mst77Y genes share a 12-bp insertion in the 5′-UTR (as well as an in-frame deletion of three nucleotides at position 381 in the coding sequence; Figure S3), which suggests that there was a single duplication of Mst77F to the Y chromosome, followed by repeated rounds of intrachromosomal duplications that produced the 18 Mst77Y genes. This single-origin scenario is confirmed by phylogenetic analysis (Figure 1 and Figure S2 A; note the 100% bootstrap support for the basal node of all Mst77Y genes). The same analysis (Figure S2 A) suggests that the original duplication occurred after the split between D. melanogaster and D. simulans (which happened 5.4 MYA), as there is some bootstrap support (74%) for the cluster that includes all Mst77Y genes and the D. melanogaster Mst77F, to the exclusion of all other species. Using the correlated Bayesian method (Rannala and Yang 2007), the original duplication was dated at 4.3 MYA (Figure S2 B). In accordance with these data, a BlastN search in the genome of sequenced species of the melanogaster subgroup (D. yakuba, D. erecta, D. sechellia, and D. simulans) failed to identify any Mst77F-like gene, other than the autosomal one.
It is interesting to know the size of the region that was duplicated to the Y chromosome. We investigated this issue by performing a BlastN search against the whole-genome shotgun traces of D. melanogaster, using as query a 30-kb sequence of chromosome 3L sequence (NT_037436.3) surrounding the Mst77F gene. The duplicated region shows up as a huge increase in trace number (technically, in sequence depth) and also by the presence of several mismatches between the traces and the autosomal sequence (Figure S7). Using this procedure we inferred that the duplicated region (or at least what survived from it) spans ∼3.5 kb (coordinates 20,809,000–20,812,389 of the 3L chromosome), encompassing the whole Mst77F gene, the two first two exons of Pka-R1, and the 5′-UTR of CG3618. The finding that the duplicated region contains more than one transcription unit shows that it did not originate through retrotransposition (not even from immature mRNA) and must have occurred through a DNA-based mechanism.
DISCUSSION
There are functional Mst77Y genes in the D. melanogaster Y chromosome. This conclusion is supported by a coherent set of data. De novo sequencing and two other methods identified 18 Mst77Y genes and showed that 8 of them are able to encode a protein that is similar to Mst77F (88% identity). Four of these genes are transcribed and collectively account for one-fifth of the functional Mst77F-like mRNA. Similarly to the previously identified Drosophila Y-linked genes (Carvalho et al. 2000, 2001; Carvalho and Clark 2003; Vibranovski et al. 2008), the Mst77Y genes have a male-specific function (Russell and Kaiser 1993; Raja and Renkawitz-Pohl 2005). Finally, molecular evolutionary analysis showed that the Mst77Y genes that have intact coding regions evolved under purifying selection, whereas no selective constraint could be detected in those with disrupting mutations. Mst77Y genes are the 13th protein-coding function identified in the D. melanogaster Y. The same as the 12 other genes, they are a duplication of an autosomal gene, which reinforces the previous finding that gene gains play a prominent role in the evolution of the Drosophila Y chromosome (Koerich et al. 2008).
All evidence indicates that the multiple Mst77Y genes originated from a single duplication from an autosome to the Y chromosome, followed by duplications inside the Y (rather than from multiple autosome-to-Y duplications): (i) all Mst77Y genes map to a single location of the Y chromosome [bands h18–h19, in the pericentromeric region (Russell and Kaiser 1993; Abad et al. 2004)]; (ii) they share several unique indels; and (iii) in phylogenetic analysis they cluster together, with 100% bootstrap support. The initial duplication was a DNA-mediated event that spanned at least 3.5 kb of the 3L chromosome, encompassing the whole Mst77F gene (including the putative promoter region) and also parts of two neighbor genes (Figure S7). The phylogenetic position of the Mst77Y cluster, as well as the lack of additional copies of Mst77F in other species, strongly suggests that the initial duplication occurred after the split between D. melanogaster and its close relatives of the D. simulans clade (Figure S2 A). These data imply that Mst77Y is <5.4 MY old; the molecular clock dated its origin to 4.3 ± 1.4 MYA (Figure S2 B).
How many Mst77Y genes exist in the D. melanogaster genome?
We found seven Mst77Y genes in the assembled genome of D. melanogaster, and there is evidence in the Trace Archives of missing genes and misassemblies. These assembly problems are not surprising: in multicopy genes and other repetitive regions, different copies may be collapsed into a single sequence due to their high sequence identity (Bailey et al. 2002) (some Mst77Y genes have 99.8% identity at the nucleotide level). Furthermore, the shotgun coverage for the Y chromosome is low [∼3×, whereas at the autosomes it is 12× (Carvalho et al. 2003)] and several missing copies probably were represented by isolated traces (which are ignored by assembly programs) or just fell into sequence gaps. For example, there is no sign of the Mst77Y-18 gene among the traces, but it appeared in our experiments. Despite these limitations, these heterochromatic scaffolds (including the “degenerate scaffolds”) contain much valuable information, and they can also be used as a starting point for more detailed analysis (e.g., we used them to design the PCR primers for de novo sequencing).
By de novo sequencing we found 18 Mst77Y genes, and similar estimates were obtained by restriction enzyme digestion (16 genes) and by computational analysis of the genomic traces (14 genes). Each method has some limitations and possible bias, but the good agreement among them makes it very unlikely that our estimate is substantially wrong. Of course the real number of Mst77Y genes in the strain we used (y cn bw sp) can be, say, 16 or 20; two haplotypes could have been spuriously introduced by PCR mutations or missed by the de novo sequencing, and these values would be within the error margin of the two other methods. Still, from the biological point of view it would make little difference. We should also keep in mind that the number of copies seems to differ among strains (Russell and Kaiser 1993; our unpublished data). At any rate, a more precise answer to this question may be given by Alfredo Villasante and co-workers, who are sequencing heterochromatic BACs from the Y chromosome (Abad et al. 2004; Mendez-Lago et al. 2009): the BAC clones BACR17J08 and BACR11H07 contain the Mst77Y region.
Functionality of the Mst77Y genes and their molecular evolution:
Eight Mst77Y genes have intact open reading frames, have appropriate splice junctions, and potentially encode proteins that are similar to previously known gene products. Hence, they would be considered functional under the usual gene annotation procedures of genome projects. Their functional status is strengthened by other evidence. First, at least four of them are expressed at the mRNA level and collectively account for one-fifth of the functional Mst77F-like mRNA. The Mst77Y mRNAs most likely are translated, since they are similar to the Mst77F mRNA; unfortunately none of these proteins (Mst77F or Mst77Y) were detected in the D. melanogaster sperm proteome (Dorus et al. 2006) or in the whole male reproductive system (Takemori and Yamamoto 2009), so we cannot prove (or disprove) the expression of the Mst77Y genes at the protein level.
Molecular evolutionary analysis provides a second and independent evidence of functionality. We found that the potentially functional copies evolved under purifying selection for protein-coding function, since their dN/dS ratio was significantly smaller than one, whereas in the nonfunctional copies, which are expected to evolve without selective constraints, the dN/dS ratio was not significantly different from one. These findings deserve additional comments. Recent work showed that dN/dS ratios may be spuriously influenced by mutational bias related to GC composition (Berglund et al. 2009; Galtier et al. 2009). This might be problematic for our analysis of Mst77Y genes, since they moved from a GC-rich (euchromatin) to an AT-rich genomic environment (heterochromatin; indeed the third position GC content is 57% for Mst77F and 55% for Mst77Y-12). However, we found in the Mst77Y genes a precise matching between coding potential and evolutionary pattern, such that potentially functional copies have dN/dS < 1 and nonfunctional copies have dN/dS ∼ 1. This pattern rules out the mutational bias hypothesis and confirms that the potentially functional Mst77Y genes evolved under natural selection for protein-coding function.
An additional test of functionality of the Mst77Y genes would be to measure their ability to complement null mutations of Mst77F. Two mutants of Mst77F are available (Mst77Fc06969 and Mst77F1 ), but unfortunately none of them seem to be null. We measured mRNA production in males homozygous for Mst77Fc06969 (a piggyBac insertion near the putative promoter) using our method 2, and the preliminary data suggest that the mutant Mst77Fc06969 gene still accounts for 60% of the Mst77F-like mRNA. So Mst77Fc06969 is not a null mutant (probably is hypomorphic) and the known fertility of homozygous Mst77Fc06969 males does not imply complementation by Mst77Y. Regarding Mst77F1 [also known as nc3 (Fuller 1986)], the data from Raja and Renkawitz-Pohl (2005) strongly suggest that it is antimorphic, since Mst77Fc06969/Del is fertile and Mst77Fc06969/Mst77F1 is sterile. So the known sterility of homozygous Mst77F1 males does not imply absence of complementation by Mst77Y. In summary, a proper complementation test will have to await the availability of a Mst77F null mutant.
Explanations and consequences of multiple copies:
The Mst77Y genes look like the other D. melanogaster Y-linked genes in many respects (e.g., male-specific function, autosomal origin), but they are multicopy, whereas the 12 known Y-linked protein-coding genes of D. melanogaster are single copy. The Y chromosome of D. melanogaster has other multicopy genes, but they encode RNA [namely, the 18S/28S rDNA cluster and the Suppressor of Stellate genes (Livak 1984; Lyckegaard and Clark 1989)]. It has been suggested that acquisition and amplification of genetic material is a general feature of the Y chromosome (Gvozdev et al. 2005), and two explanations for the multiple copies of Mst77Y can be advanced. The large number of copies may be just an accident (i.e., it would be selectively neutral): heterochromatic regions are prone to rearrangements, at least in part due to their richness in transposable elements. For example, some exons of the rolled gene (heterochromatin of chromosome 2) are duplicated in tandem and the same happens with exons of several Y-linked genes (Hoskins et al. 2002; Kopp et al. 2006). Alternatively, the large number of copies might have been favored by natural selection, perhaps as a mechanism of gene amplification. Two lines of evidence argue against the “selection-driven gene amplification hypothesis.” First, the Y-linked copies of Mst77F added a modest amount of mRNA (20% of the total), although it is possible that this value was much higher in the past. Second, some Mst77Y genes were duplicated after they acquired disruptive mutations (e.g., Mst77Y-10 and Mst77Y-14; Figure 1), and their fixation could not have been driven by selection. Whatever the true explanation for the multicopy state of Mst77Y, it has several important consequences: as in other multicopy genes, selection at individual genes is reduced, and turnover of genes may occur through duplications and losses.
Evolutionary fate of the Mst77Y genes:
We detected purifying selection on several Mst77Y genes, but it is unclear whether it is strong enough to prevent their pseudogenization. After a gene duplication event both copies may remain undifferentiated for a long time or perhaps indefinitely (Tanaka et al. 2009; Xue and Fu 2009). However, the duplication results in relaxed selective constraints, which may lead to the fixation in one of the copies of deleterious mutations that abolish gene function (nonfunctionalization or pseudogenization) or of advantageous mutations that generate novel gene functions (neofunctionalization) (Lynch and Katju 2004). It is interesting to examine the Mst77Y genes in the context of these three possible fates (neofunctionalization, pseudogenization, and no differentiation). The timescale of these events is an important issue here, but unfortunately it is poorly known. Depending on which estimate of the half-life of Drosophila duplicate genes we use [0.66 MY (Rogers et al. 2009) or 3.2 MY (Lynch and Conery 2003)], Mst77Y genes (which are 4.3 MY old) would seem to have already been stably incorporated in the genome or to still have an undefined fate. Ten Mst77Y genes have clear signs of pseudogenization such as presence of frameshift or nonsense mutations. Among the remaining 8 Mst77Y genes, while we cannot exclude the possibility of neofunctionalization, the observation that they are expressed in the same organ (testis) suggests that they have the same function of the parental gene Mst77F.
What will happen with the 8 functional Mst77Y genes? While this type of question cannot be precisely answered, a comparison with the other D. melanogaster Y-linked genes can give some clues. All 12 known D. melanogaster Y-linked genes originated from a duplication of autosomal genes, and in some cases it seems that both copies were retained, whereas in other cases the autosomal copy disappeared so the gene was effectively transposed to the Y chromosome (Koerich et al. 2008; our unpublished results). More specifically, there are two cases of retention of both copies (CG13125/PPr-Y and FDY/CG11844) and seven cases where it was completely lost (kl-3, kl-5, PP1-Y1, PP1-Y2, ARY, ORY, and PRY). Finally, in three cases a small relic gene was left at the original autosomal position (CG34164/WDY, CG13161/CCY, and probably CG9068/kl-2); this relic gene most likely cannot perform all the original functions, which presumably are now being carried by the full-size, Y-linked copy. Although there is some uncertainty in these numbers (we would need the sequences of more Diptera genomes to be sure), the bottom line is that in most cases (10 of 12) the autosomal copy disappeared or became a relic gene. The Mst77Y genes are young and may be in the initial stages of their establishment, so it is entirely possible that they will degenerate. If they attain stability in the genome, the examples of the other Y-linked genes suggest that it is likely that the autosomal Mst77F gene will eventually degenerate. This would add one more essential gene to the D. melanogaster Y chromosome.
Acknowledgments
We thank L. Koerich, E. Dupim, R. Hassan, D. Guiretti, H. Seuanez, A. Villasante, A. Clark, H. Araujo, K. Carneiro, G. Sutton, R. Hoskins, B. Ahsan, and S. Morishita for help during the work and for valuable comments on the manuscript. This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico-(CMPq), by Coordenação de Aperfeiçoamento do Pessoal de Ensino Superior-CAPES, by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), by National Institutes of Health (NIH) grant GM64590, and by Fogarty International Center–NIH grant TW007604.
Sequence data from this article have been deposited with the EMBL/GenBank data libraries under accession nos. GQ868243–GQ868260.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.107516/DC1.
References
- Abad, J. P., B. de Pablos, M. Agudo, I. Molina, G. Giovinazzo et al., 2004. Genomic and cytological analysis of the Y chromosome of Drosophila melanogaster: telomere-derived sequences at internal regions. Chromosoma 113 295–304. [DOI] [PubMed] [Google Scholar]
- Adams, M. D., S. E. Celniker, R. A. Holt, C. A. Evans, J. D. Gocayne et al., 2000. The genome sequence of Drosophila melanogaster. Science 287 2185–2195. [DOI] [PubMed] [Google Scholar]
- Ahsan, B., T. L. Saito, S.-i. Hashimoto, K. Muramatsu, M. Tsuda et al., 2009. MachiBase: a Drosophila melanogaster 5′-end mRNA transcription database. Nucleic Acids Res. 37 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, J. A., Z. Gu, R. A. Clark, K. Reinert, R. V. Samonte et al., 2002. Recent segmental duplications in the human genome. Science 297 1003–1007. [DOI] [PubMed] [Google Scholar]
- Berglund, J., K. S. Pollard and W. T. Webster, 2009. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 7 e1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges, C. B., 1916. Non-disjunction as proof of the chromosome theory of heredity. Genetics 1 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, A. B., 2002. Origin and evolution of the Drosophila Y chromosome. Curr. Opin. Genet. Dev. 12 664–668. [DOI] [PubMed] [Google Scholar]
- Carvalho, A. B., and A. G. Clark, 2003. Birth of a new gene on the Drosophila Y chromosome. The 44th Annual Drosophila Research Conference, Chicago, Illinois, USA. Abstract 318C, p.113. The Genetics Society of America, March 2003.
- Carvalho, A. B., B. P. Lazzaro and A. G. Clark, 2000. Y chromosomal fertility factors kl-2 and kl-3 of Drosophila melanogaster encode dynein heavy chain polypeptides. Proc. Natl. Acad. Sci. USA 97 13239–13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, A. B., B. A. Dobo, M. D. Vibranovski and A. G. Clark, 2001. Identification of five new genes on the Y chromosome of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 98 13225–13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, A. B., M. D. Vibranovski, J. W. Carlson, S. E. Celniker, R. A. Hoskins et al., 2003. Y chromosome and other heterochromatic sequences of the Drosophila melanogaster genome: How far can we go? Genetica 117 227–237. [DOI] [PubMed] [Google Scholar]
- Carvalho, A. B., L. B. Koerich and A. G. Clark, 2009. Origin and evolution of Y chromosomes: Drosophila tales. Trends Genet. 25 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., and D. Charlesworth, 2000. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355 1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crumpacker, D. W., and J. S. Williams, 1973. Density, dispersion, and population structure in Drosophila pseudoobscura. Ecol. Monogr. 43 499–538. [Google Scholar]
- Degroot, E. J., and M. J. Schervish, 2002. Probability and Statistics. Addison-Wesley Longman, Reading, MA/Menlo Park, CA.
- Dobzhansky, T., and S. Wright, 1943. Genetics of natural populations. X. Dispersion rates in Drosophila pseudoobscura. Genetics 28 304–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorus, S., S. A. Busby, U. Gerike, J. Shabanowitz, D. F. Hunt et al., 2006. Genomic and functional evolution of the Drosophila melanogaster sperm proteome. Nat. Genet. 38 1440–1445. [DOI] [PubMed] [Google Scholar]
- Fuller, M. T., 1986. Genetic analysis of spermatogenesis in Drosophila: the role of the testis-specific beta-tubulin and interacting genes in cellular morphogenesis, pp. 19–41 in Gametogenesis and the Early Embryo, 44th Symposium of the Society of Developmental Biology edited by J. G. Gall. Alan R. Liss, New York.
- Galtier, N., L. Duret, S. Glémin and V. Ranwez, 2009. GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet. 25 1–5. [DOI] [PubMed] [Google Scholar]
- Gatti, M., and S. Pimpinelli, 1983. Cytological and genetic analysis of the Y chromosome of Drosophila melanogaster. Chromosoma 88 349–373. [Google Scholar]
- Gepner, J., and T. S. Hays, 1993. A fertility region on the Y chromosome of Drosophila melanogaster encodes a dynein microtubule motor. Proc. Natl. Acad. Sci. USA 90 11132–11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gvozdev, V. A., G. L. Kogan and L. A. Usakin, 2005. The Y chromosome as a target for acquired and amplified genetic material in evolution. BioEssays 27 1256–1262. [DOI] [PubMed] [Google Scholar]
- Hazelrigg, T., P. Fornili and T. C. Kaufman, 1982. A cytogenetic analysis of X- ray induced male steriles on the Y chromosome of Drosophila melanogaster. Chromosoma 87 535–559. [Google Scholar]
- Hogrefe, H. H., and M. C. Borns, 2003. High-fidelity PCR enzymes, p. 520 in PCR Primer: A Laboratory Manual, edited by C. W. Dieffenbach and G. S. Dveksler. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Hoskins, R., C. Smith, J. Carlson, A. B. Carvalho, A. Halpern et al., 2002. Heterochromatic sequences in a Drosophila whole-genome shotgun assembly. Genome Biol. 3 Research0085.0081–0085.0016. [DOI] [PMC free article] [PubMed]
- Hoskins, R. A., J. W. Carlson, C. Kennedy, D. Acevedo, M. Evans-Holm et al., 2007. Sequence finishing and mapping of Drosophila melanogaster heterochromatin. Science 316 1625–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic, K., T. Berleth and N. J. Provart, 2004. BlastDigester—a web-based program for efficient CAPS marker design. Trends Genet. 20 280–283. [DOI] [PubMed] [Google Scholar]
- Kalderon, D., and G. M. Rubin, 1988. Isolation and characterization of Drosophila cAMP-dependent protein kinase genes. Genes Dev. 2 1539–1556. [DOI] [PubMed] [Google Scholar]
- Kennison, J. A., 1981. The genetic and cytological organization of the Y chromosome of Drosophila melanogaster. Genetics 98 529–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerich, L. B., X. Wang, A. G. Clark and A. B. Carvalho, 2008. Low conservation of gene content in the Drosophila Y chromosome. Nature 456 949–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp, A., A. K. Frank and O. Barmina, 2006. Interspecific divergence, intrachromosomal recombination, and phylogenetic utility of Y-chromosomal genes in Drosophila. Mol. Phylogenet. Evol. 38 731–741. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond, S. L., S. D. W. Frost and S. V. Muse, 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21 676–679. [DOI] [PubMed] [Google Scholar]
- Lahn, B. T., and D. C. Page, 1999. Four evolutionary strata on the human X chromosome. Science 286 964–967. [DOI] [PubMed] [Google Scholar]
- Livak, K. J., 1984. Organization and mapping of a sequence on the Drosophila melanogaster X and Y chromosomes that is transcribed during spermatogenesis. Genetics 107 611–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyckegaard, E. M. S., and A. G. Clark, 1989. Ribosomal DNA and Stellate gene copy number variation on the Y chromosome of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 86 1944–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and J. S. Conery, 2003. The evolutionary demography of duplicate genes. J. Struct. Funct. Genomics 3 35–44. [PubMed] [Google Scholar]
- Lynch, M., and V. Katju, 2004. The altered evolutionary trajectories of gene duplicates. Trends Genet. 20 544–549. [DOI] [PubMed] [Google Scholar]
- Mendez-Lago, M., J. Wild, S. L. Whitehead, A. Tracey, B. de Pablos et al., 2009. Novel sequencing strategy for repetitive DNA in a Drosophila BAC clone reveals that the centromeric region of the Y chromosome evolved from a telomere. Nucleic Acids Res. 37 2264–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse, S. V., and B. S. Gaut, 1994. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 11 715–724. [DOI] [PubMed] [Google Scholar]
- Myers, E. W., G. G. Sutton, A. L. Delcher, I. M. Dew, D. P. Fasulo et al., 2000. A whole-genome assembly of Drosophila. Science 287 2196–2204. [DOI] [PubMed] [Google Scholar]
- R Development Core Team, 2007. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
- Raja, S. J., and R. Renkawitz-Pohl, 2005. Replacement by Drosophila melanogaster protamines and Mst77F of histones during chromatin condensation in late spermatids and role of sesame in the removal of these proteins from the male pronucleus. Mol. Cell. Biol. 25 6165–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala, B., and Z. Yang, 2007. Inferring speciation times under an episodic molecular clock. Syst. Biol. 56 453–466. [DOI] [PubMed] [Google Scholar]
- Rice, W. R., 1996. Evolution of the Y sex chromosome in animals. Bioscience 46 331–343. [Google Scholar]
- Rogers, R. L., T. Bedford and D. L. Hartl, 2009. Formation and longevity of chimeric and duplicate genes in Drosophila melanogaster. Genetics 181 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, M. T., D. V. Grafham, A. J. Coffey, S. Scherer, K. McLay et al., 2005. The DNA sequence of the human X chromosome. Nature 434 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, S. R. H., and K. Kaiser, 1993. Drosophila melanogaster male germ line-specific transcripts with autosomal and Y-linked genes. Genetics 134 293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletsky, H., T. Kuroda-Kawaguchi, P. J. Minx, H. S. Cordum, L. Hillier et al., 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423 825–837. [DOI] [PubMed] [Google Scholar]
- Smith, C. D., S. Q. Shu, C. J. Mungall and G. H. Karpen, 2007. The Release 5.1 annotation of Drosophila melanogaster heterochromatin. Science 316 1586–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemori, N., and M. T. Yamamoto, 2009. Proteome mapping of the Drosophila melanogaster male reproductive system. Proteomics 9 2484–2493. [DOI] [PubMed] [Google Scholar]
- Tamura, K., S. Subramanian and S. Kumar, 2004. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol. Biol. Evol. 21 36–44. [DOI] [PubMed] [Google Scholar]
- Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA 4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24 1596–1599. [DOI] [PubMed] [Google Scholar]
- Tanaka, K. M., K. R. Takahasi and T. Takano-Shimizu, 2009. Enhanced fixation and preservation of a newly arisen duplicate gene by masking deleterious loss-of-function mutations. Genet. Res. 91 267–280. [DOI] [PubMed] [Google Scholar]
- Vibranovski, M. D., L. B. Koerich and A. B. Carvalho, 2008. Two new Y-linked genes in Drosophila melanogaster. Genetics 179 2325–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, P. S., H. A. Erlich and R. Higuchi, 1992. Preferential PCR amplification of alleles: mechanisms and solutions. Genome Res. 1 241–250. [DOI] [PubMed] [Google Scholar]
- Xue, C., and Y. Fu, 2009. Preservation of duplicate genes by originalization. Genetica 136 69–78. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 2007. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang, Z., and J. P. Bielawski, 2000. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara, J. C., C. H. DeCrease and B. T. Wakimoto, 2005. Evolution of heterochromatic genes of Drosophila. Proc. Natl. Acad. Sci. USA 102 10958–10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurovcova, M., and W. F. Eanes, 1999. Lack of nucleotide polymorphism in the Y-linked sperm flagellar dynein gene Dhc-Yh3 of Drosophila melanogaster and D. simulans. Genetics 153 1709–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylstra, P., H. S. Rothenfluh, G. F. Weiller, R. V. Blanden and E. J. Steele, 1998. PCR amplification of murine immunoglobulin germline V genes: strategies for minimization of recombination artefacts. Immunol. Cell. Biol. 76 395–405. [DOI] [PubMed] [Google Scholar]