

Abstract
CYP73A1 catalyzes cinnamic acid hydroxylation, a reaction essential for the synthesis of lignin monomers and most phenolic compounds in higher plants. The native CYP73A1, initially isolated from Jerusalem artichoke (Helianthus tuberosus), was engineered to simplify purification from recombinant yeast and improve solublity and stability in the absence of detergent by replacing the hydrophobic N terminus with the peptitergent amphipathic sequence PD1. Optimized expression and purification procedures yielded 4 mg engineered CYP73A1 L–1 yeast culture. This water-soluble enzyme was suitable for 1H-nuclear magnetic resonance (NMR) investigation of substrate positioning in the active site. The metabolism and interaction with the enzyme of cinnamate and four analogs were compared by UV-visible and 1H-NMR analysis. It was shown that trans-3-thienylacrylic acid, trans-2-thienylacrylic acid, and 4-vinylbenzoic acid are good ligands and substrates, whereas trans-4-fluorocinnamate is a competitive inhibitor. Paramagnetic relaxation effects of CYP73A1-Fe(III) on the 1H-NMR spectra of cinnamate and analogs indicate that their average initial orientation in the active site is parallel to the heme. Initial orientation and distances of ring protons to the iron do not explain the selective hydroxylation of cinnamate in the 4-position or the formation of single products from the thienyl compounds. Position adjustments are thus likely to occur during the later steps of the catalytic cycle.
Cytochromes P450 of the CYP73 family catalyze the 4-hydroxylation of cinnamic acid, an early and obligatory step in the biosynthesis of most phenolic compounds such as lignin monomers, flavonoids, coumarins, stilbenes, lignans, and tannins (Dixon, 2001). CYP73 proteins are present at high levels in all vascular plants, and up to 20% of the terrestrial plant biomass is processed through their active sites. The CYP73 family appeared early during plant evolution and does not display the multiple gene duplication and divergence that is evident for most other plant P450 families (Paquette et al., 2000). The importance of their physiological function thus seems to have exerted a constraint on CYP73 evolution and on the conservation of active site structure.
Very limited data are available on the structure of the active sites of plant P450s. Most of the extent information has been obtained using site-directed mutagenesis, guided by homology modeling on crystallized bacterial protein (Kahn et al., 2001; Sawada et al., 2002), or based on the results of exchanging domains between closely related enzymes (Schalk and Croteau, 2000). CYP73A1, the first to be highly expressed in a recombinant system (Pierrel et al., 1994), is one of the most extensively studied plant P450 enzymes, can metabolize alternate substrates (Pierrel et al., 1994; Schalk et al., 1997, 1998), and has some unusual residues in the most conserved regions involved in heme binding and oxygen activation (Schalk et al., 1999). The natural substrate of CYP73A1 is trans-cinnamic acid (CA). A systematic search for possible alternate substrates has shown that CYP73A1 has quite high substrate specificity but can accommodate in its active site a diverse set of compounds that are structural analogs of the natural substrate. The structural requirements for such analogs to be CYP73 substrates include a planar, aromatic structure, a small size of approximately two adjacent aromatic rings, and an anionic site opposite (i.e. at about 8.5 Å) to the position of oxidative attack (Schalk et al., 1997, 1998). The protein residues that govern recognition and correct orientation of this substrate pattern have not been defined.
To date, only one membrane-bound P450 protein has been crystallized, and investigation of substrate docking in the active site of mammalian and other eukaryotic enzymes has relied on indirect methods such as homology models, site-directed mutagenesis, and adduct formation. We have built a model of CYP73A1, based on the structures of crystallized P450s (Schoch et al., 2003), to guide site-directed mutagenesis experiments. Identification of residues in the model that would be likely to interact with the substrate would benefit from information on substrate orientation and positioning in the active site, which might vary greatly depending on the substrate and P450 protein. 1H-NMR was chosen to examine on cinnamic acid orientation in the active site of CYP73A1, because paramagnetic relaxation experiments have been used successfully to determine the distances between the protons of bound substrates and the heme iron in several bacterial and mammalian P450 enzymes (Modi et al., 1995, 1996a, 1996b, 1997; Poli-Scaife et al., 1997; Regal and Nelson, 2000). CYP73A1 substrates are water soluble at concentrations in the range suitable for NMR relaxation experiments. 1H relaxation time spectroscopy requires large amounts of highly purified, stable, and soluble enzyme. However, the native enzyme is difficult to purify and completely insoluble in the absence of detergent. A modified form of CYP73A1 with increased stability and water solubility was engineered for use in these experiments. In this report, we describe the enzyme modifications, the purification procedure, and the paramagnetic relaxation rates determined by NMR for the protons of cinnamic acid and some new substrate analogs within the active site. Our data suggest a strategy for the production of soluble enzyme, which would be also suitable for crystallographic analysis. The results indicate that the substrates lie roughly parallel to the heme in the oxidized form of the enzyme.
RESULTS
Engineering of a Soluble and Stable Form of CYP73A1
Previous attempts at CYP73A1 purification (Gabriac et al., 1991) have shown that the protein is too hydrophobic to remain in solution in the absence of detergent and is unstable upon storage, especially in low detergent and glycerol. The native form of the enzyme was thus not suitable for long-lasting NMR experiments that have to be performed in the absence of detergent. The poor solubility of the native enzyme also limits effective purification and suitability for crystallization. To improve CYP73A1 properties, several engineered forms of the enzyme were generated by directed mutagenesis (Fig. 1). For easy purification, a tag of four His residues was added to the C-terminal end of the protein (Schalk et al., 1999), and solubility was increased by replacing the first 20 amino acids of the membrane-anchoring segment at the N terminus with the peptitergent sequence PD1. PD1 is an amphipathic helix of 25 amino acids initially designed to replace detergents for the solubilization of intrinsic membrane proteins (Schafmeister et al., 1993). Its interaction with transmembrane domains was shown to maintain bacteriorhodopsin in solution for several days. Replacement of the N-terminal sequence of a mammalian P450 with PD1 (Sueyoshi et al., 1995) enhanced expression in Escherichia coli of the mouse PD1-CYP2A4 compared with the wild type, and produced an enzyme that was soluble in the absence of detergent and showed increased stability. The expression, subcellular localization, and stability of this type of construct in yeast have not been reported. However, yeast is better suited than E. coli to assay P450 activity that requires the presence of P450 reductase, and it has been shown to be an efficient system for production of large amounts of native CYP73A1 (Urban et al., 1994). Therefore, three constructs were generated from CYP73A1: the first with PD1 at the N terminus and 4-His tag at the C terminus, the two others with single modifications, to check for the impact of the N- and C-terminal modifications on the catalytic properties and stability of the enzyme. The three constructs were expressed in yeast W(R).
Figure 1.

Engineering of CYP73A1 for improved purification and water solubility.
Analysis of total P450 content in recombinant yeast microsomes (Table I) shows that the N-terminal exchange for PD1 does neither significantly alter the membrane localization of CYP73A1 nor its level of expression in yeast. There is no PD1-P450 accumulation in the soluble fraction compared with the wild type. Introduction of the 4-His-tag however leads to a 20% to 40% decrease in protein expression.
Table I.
Yeast expression, substrate binding, and activity of the engineered forms of CYP73A1
C4H activity was measured at saturating substrate concentration (100 μm). CYP73A1 expression was evaluated from CO-reduced versus reduced difference spectra. Dissociation constants for the enzyme-substrate complex and εΔA(peak-trough) were calculated from the saturation curves of difference spectra recorded upon cinnamate binding to the oxidized yeast microsomes. P450 and C4H activity were not detected in W(R) transformed with a void plasmid.
| Protein | C4H Activity | Relative Expression | Cinnamate Binding | |
|---|---|---|---|---|
| KD | εΔ(390-420nm) | |||
| min-1 | μm | mm-1 cm-1 | ||
| CYP73A1 | 20 ± 3 | 100 ± 3.6 | 8.8 ± 0.3 | 125 |
| 73His | 22 ± 2.6 | 61 ± 3 | 4.2 ± 0.1 | 121 |
| PD73 | 7.9 ± 0.9 | 98 ± 3.5 | 6.4 ± 0.3 | 113 |
| PD73His | 13 ± 0.3 | 77 ± 4.8 | 2.7 ± 0.1 | 119 |
Interaction spectra with cinnamic acid indicate that the structure of the substrate-binding site is essentially preserved in all constructs (Table I). The ΔA(390–420 nm) at saturating substrate concentrations indicates that heme iron is low spin (S = 1/2) in all the native Fe(III) construct proteins and completely converted to high spin (S = 5/2) upon binding of cinnamate. The minor increase in affinity for cinnamate observed for the 4-His-tagged protein is a possible consequence of the positive charges of the histidines at the surface of the protein that might favor substrate access to the active site. Probably related to this increase in affinity is a small increase in enzyme turnover. The presence of PD1 at the N terminus does not significantly alter the substrate-binding constant, but significantly decreases catalytic activity. This is not unexpected because PD1 exchange of the membrane-anchoring segment is meant to decrease the hydrophobic interactions of CYP73A1 with the membrane. Altered P450-membrane interaction is likely to have an adverse impact on PD1-CYP73A1 coupling with the P450 reductase.
No conversion of P450 into P420 was detected with the constructs under standard conditions (i.e. in 10% [v/v] glycerol and at 20°C) for several hours. The stability of the PD1-CYP73A1–4His (PD73His) mutant was thus further investigated at low concentration of glycerol (3% [v/v]) and high concentration of dithionite (10 mg mL–1) at 30°C for 2 h. The half-life of the P450-CO complex in such drastic conditions was 95.1 ± 8 min for PD73His instead of 45 ± 7 min for CYP73A1.
PD73His is expressed at high levels, has conserved substrate-binding properties, and shows an increased stability. Its behavior upon purification in the absence of detergent was thus investigated.
Optimization of the Yeast Expression System
The levels of expression and stability of plant P450 enzymes in yeast were previously shown to be dependent on the co-expressed P450-reductase (Cabello-Hurtado et al., 1998; Robineau et al., 1998). PD73His expression was thus tested in different yeast strains, overexpressing different P450 reductases from yeast (strain W(R)), or plants, Arabidopsis (WAT11), Jerusalem artichoke (Helianthus tuberosus; WHT1), or common vetch (Vicia sativa; WVS1).
The highest specific expression and activity was obtained in WAT11, which also displays the highest cytochrome c reductase activity (Table II). As previously reported (Cabello-Hurtado et al., 1998; Robineau et al., 1998), WAT11 seems to be an optimal system to obtain high levels of expression of functional P450 in recombinant yeast microsomes. However, around 30% higher yields of PD73His protein are obtained in W(R). Lower activity of this protein compared with the WAT11-expressed enzyme seems to be correlated to the lower expression of P450 reductase, because no trace of conversion of P450 into P420 is detected when the enzyme is reduced and bound to carbon monoxide. However, traces of P420, which are supposed to reflect disruption of the heme thiolate bound in the active center, are detected in the other yeast strains. W(R) was thus selected for expressing PD73His for large-scale purifications.
Table II.
Compared levels of expression of PD73His in the microsomes from yeast strains overexpressing different P450 reductases
Levels of PD73His expression are estimated from the CO-reduced versus reduced difference spectra recorded with the recombinant yeast microsomes. W(R) overexpresses the yeast P450 reductase. WAT11 overexpresses ATR1 from Arabidopsis, WHT1 HTR1 from Jerusalem artichoke, and WVS1 VSR1 from common vetch. All reductases are under the control of the same galactose-inducible promoter (GAL10-CYC1) as PD73His.
| Strain | C4H Activity | Level of PD73His Expression | Cyt c Reductase Activity | |
|---|---|---|---|---|
| pkat mg-1 protein | pmol mg-1 protein | nmol l-1 growth medium | pkat mg-1 protein | |
| W(R) | 54.1 ± 1.3 | 308 ± 6.0 | 46.8 ± 0.9 | 742 ± 89 |
| WAT11 | 157 ± 15.3 | 369 ± 6.2 | 37.1 ± 0.6 | 2,166 ± 28 |
| WHT1 | 70.1 ± 11.2 | 313 ± 6.8 | 26.1 ± 0.5 | 2,049 ± 135 |
| WVS1 | 28.6 ± 1.6 | 179 ± 26 | 24.3 ± 3.6 | 1,394 ± 33 |
Purification of Recombinant PD73His
The purification protocol was derived from the method described by Schalk et al. (1999), taking advantage of the 4-His tag for affinity chromatography on a Ni-chelating matrix. This method provided high yields (35%) of very pure protein, with a heme to protein molar ratio of 1 (17 μmol g–1), which was apparently devoid of contamination detectable by SDS-gel analysis (Fig. 2). This affinity purification procedure was scaled-up by inclusion of an anion-exchange step to exploit the very high pI of the protein (pI calculated from the amino acid sequence = 9.78), so as to obtain up to 4 mg of pure PD73His protein from 1.2 L of culture in a single experiment. The purified protein was immediately dialyzed to remove free His, concentrated to 60 μm, and frozen in 10% (v/v) glycerol and 0.5 m NaCl.
Figure 2.

SDS-PAGE analysis of the purified PD73His fractions. Proteins are revealed by silver staining. S, Emulgen 911-solubilized fraction. Ni-affinity chromatography, S was loaded onto a Hitrap Ni-chelating column (Pharmacia). The column was washed with increasing imidazole concentrations: 10 mm (L1) and 60 mm (L2). The protein was eluted with 50 mm His (E1).
Mass spectrometry analysis of the purified enzyme confirmed the absence of protein contaminants and detected no residual detergent. The determined mass of 59,263 Da was that expected for a protein devoid of posttranslational modification. Cinnamate saturation experiments performed with the purified enzyme led to the determination of a KD of 5 μm and to complete low- to high-spin conversion of the heme-iron (εΔA(390–420 nm) = 125 mm–1 cm–1), showing that the absence of membrane has no impact on the binding of the substrate in the active site and that the enzyme-substrate complex can be considered to be 100% high spin in 1H-NMR experiments.
It was possible to elute and concentrate PD73His in only 10% (v/v) glycerol and in the complete absence of detergent. Provided that the protein was kept at high ionic strength, and in contrast to the wild-type enzyme, no precipitation or enzyme loss by adsorption on the tube walls was observed upon storage. Exchange of the wild-type membrane anchor for PD1 thus results in a very significant increase in water solubility and stability of the purified protein.
Titration of Relaxation Times of Cinnamate Protons with CYP73A1
The 1H-NMR spectrum of cinnamate is not well resolved. Thus the chemical displacement for the ring ortho-protons, and meta- and para-protons give a common signal. The plot of the observed relaxation rates as a function of the substrate-bound fraction for all the resonances of cinnamate (Fig. 3) did not allow independent determination of T1M for the aromatic protons. Thus the iron-proton distances calculated for H3/H4/H5 (Table III) are averaged for their presence probability, which cannot be estimated; the value found, 5.8 Å for these protons, put them closer to the heme than the ethylenic protons. However, H4, which is at the position of cinnamate attack, should be located closer to the iron than H3/H5. To test this hypothesis, substrate analogs with proton signals better differentiated at positions equivalent to C4, C3, and C5 of cinnamate were analyzed.
Figure 3.

Effect of PD73His on the longitudinal relaxation rates of the protons of cinnamic acid. T1obs values were measured after addition of increasing amounts of purified PD73His (E0) from 0 to 6 μm to a solution of cinnamate (S0 = 40 mm) in D2O buffer. Assuming fast exchange of the substrate, 1/T1P = E0/(KD + nS0)×1/T1M where n is the number of exchangeable protons (Mildvan and Gupta, 1978; Poli-Scaife et al., 1997). Slopes provide the values for paramagnetic relaxation times of protons, 1/T1M, which are directly related to the distances to heme iron (Table III).
Table III.
Paramagnetic relaxation times of the protons of ligands bound to PD73His and corresponding calculated iron-proton distances
For each ligand, a complete titration was performed three times.
| Ligand | Parameter | Proton | ||||
|---|---|---|---|---|---|---|
| CA | H5/H4/H3 | H6/H2 | H3′ | H2′ | ||
| T1M(μs) | 239 ± 8 | 311 ± 31 | 796 ± 6 | 737 ± 11 | ||
| r(Å) | 5.76 ± 0.2 | 6.02 ± 0.6 | 7.04 ± 0.05 | 6.95 ± 0.1 | ||
| 3TA | H5 | H2 | H4 | H3′ | H2′ | |
| T1M(μs) | 949 ± 144 | 1,145 ± 138 | 560 ± 34 | 1,145 ± 107 | 997 ± 82 | |
| r(Å) | 7.25 ± 1.1 | 7.48 ± 0.9 | 6.64 ± 0.4 | 7.48 ± 0.7 | 7.31 ± 0.6 | |
| 2TA | H5 | H4 | H3 | H3′ | H2′ | |
| T1M(μs) | 1,127 ± 60 | 1,082 ± 117 | 941 ± 39 | 1,250 ± 49 | 1,310 ± 137 | |
| r(Å) | 7.46 ± 0.4 | 7.41 ± 0.8 | 7.24 ± 0.3 | 7.59 ± 0.3 | 7.65 ± 0.8 | |
| 4VB | Hb | Hc | Ha | H3/H5 | H2/H6 | |
| T1M(μs) | 663 ± 29 | 1,427 ± 257 | 699 ± 20 | 475 ± 29 | 420 ± 27 | |
| r(Å) | 6.83 ± 0.3 | 7.76 ± 1.4 | 6.89 ± 0.2 | 6.46 ± 0.4 | 6.33 ± 0.4 | |
| 4FC | H5/H3 | H6/H2 | H3′ | H2′ | ||
| T1M(μs) | 213 ± 15 | 217 ± 11 | 424 ± 33 | 493 ± 23 | ||
| r(Å) | 5.65 ± 0.4 | 5.67 ± 0.3 | 6.34 ± 0.5 | 6.50 ± 0.3 |
Characterization of Alternate Ligands of CYP73A1
The four structural analogs of cinnamate likely to present better resolved 1H-NMR spectra were selected (Fig. 4), submitted to preliminary 1H-NMR analysis, and investigated for their binding properties to recombinant PD73His. The proton signals of all four molecules were distinct from those of glycerol. The thiophene ring protons of trans-2-thienylacrylic acid (2TA) and trans-3-thienylacrylic acid (3TA) gave independent signals. 4-Fluorocinnamate has no proton at position 4 and gives well separated signals for its ortho- and meta-protons. The 4-vinyl benzoate has two distinct vinylic protons.
Figure 4.

Cinnamate analogs tested as ligands of PD73His.
Binding properties determined from UV-visible difference spectra of recombinant yeast microsomes (Table IV) show that the dissociation constants of the four enzyme-ligand complexes are very similar to those measured for cinnamate and that the values for maximal spectral changes determined with 3TA, 2TA, and 4-vinylbenzoic acid (4VB) are indicative of complete or almost complete (i.e. 87%–100%) low- to high-spin conversion of the enzyme. In agreement with these binding data, 3TA, 2TA, and 4VB were found to be both substrates of CYP73A1 and strong competitive inhibitors of its cinnamate 4-hydroxylase (C4H) activity (Table II; Fig. 5). However, neither 3TA nor 2TA, which are analogs of well-documented metabolic inactivators of mammalian drug-metabolizing enzymes (Minoletti et al., 1999), inactivated CYP73A1. This suggests either that oxygenation does not occur at the sulfur atom or that no nucleophilic residue is located close to the position of attack in the active site of CYP73A1. Metabolism of 2TA and 3TA leads to the formation of a single product. Similarly, only one metabolite is formed upon 4VB oxygenation. Attempts to characterize the 2TA and 3TA metabolites generated by CYP73A1 were impaired by the instability of the products. However, the extent of metabolism indicated that 3TA, 2TA, and 4VB would be good probes for further NMR investigations.
Table IV.
Binding characteristics and metabolism of the alternate ligands of CYP73A1
Measurements were performed at 27°C with recombinant yeast microsomes. Dissociation constant for the enzyme-ligand complex and spin change were calculated from the saturation curves of difference spectra recorded upon ligand binding to the oxidized microsomes. Activities were measured at saturating substrate concentrations (150 and 400 μm). Extracted products were analyzed and quantified by HPLC and diode array detection. Activities were related to that measured with cinnamate as substate: 54 pkat.mg-1 protein. Inhibition of C4H activity was competitive. n.d. refers to not determined.
| Ligand | Binding Characteristics | Relative Activity | KI | |
|---|---|---|---|---|
| KD | Spin Change | |||
| CA | μM | % | % | μM |
| 8.8 ± 0.3 | 100 ± 1.4 | 100 ± 0.6 | - | |
| 3TA | 7.7 ± 0.6 | 101 ± 0.8 | 29 ± 0.7 | 2.1 ± 0.1 |
| 2TA | 6.3 ± 0.4 | 100 ± 1.5 | 41 ± 1.1 | 2.3 ± 0.2 |
| 4VB | 12 ± 0.9 | 87 ± 2.5 | 8.1 ± 0.4 | 5.6 ± 0.7 |
| 4FC | 9 ± 0.9 | 57 ± 1.7 | no metabolism | n.d. |
Figure 5.

Inhibition of CYP73A1 C4H activity by 2TA. Lineweaver-Burk representation of the inhibition of CYP73A1-dependent metabolism of cinnamic acid in recombinant yeast microsomes: P450 (3 nm in the assay) was incubated in the presence of 2TA (0, 1, or 3 μm). Data are means of duplicate experiments. Competitive inhibition was also observed with 3TA incubated under the same conditions and with 4VB (0, 20, or 30 μm in the assays). No time-dependent inactivation of the C4H activity was detected with these compounds. C4H activity is given in nanokatals per milligram of microsomal protein.
1H-NMR Relaxation Titration of PD73His Complexes with Alternate Substrates
Titration of saturating amounts of the above substrates with increasing concentrations of soluble CYP73A1 allowed the calculation of T1m for each proton of these molecules.
The T1m values and calculated iron-proton distances for the different CYP73A1 ligands are summarized in Table III. As predicted from the large iron spin transitions induced by the binding of the ligands (Table IV), they all bind close to the heme. The most consistent results were obtained with the strongest ligands of CYP73A1, CA, 4FC, 2TA, and 3TA. Information provided by all of the 1H-NMR analyses coincided to show that the differences in the distance to the iron of all protons of each ligand were small. This indicates that the ligands do not stand upright above the iron, but that their average position is roughly parallel to the heme (or that they move fast enough at a similar average distance). Protons located closest to the carboxylate suspected of anchoring the substrate on the protein are, as anticipated, the most distant to the iron. The expected positions of hydroxylation on each substrate are within 5.5 to 7.5 Å from the iron. Surprisingly, the aromatic or thiophene rings do not seem to be positioned so as to favor the oxidative attack at the 4 position of cinnamate or its equivalent in 2TA or 3TA. All ring protons appear nearly equidistant to the iron.
Position of the Substrate Relative to CYP73A1 Heme Iron
A three-dimensional model consisting of each substrate plus iron and incorporating the iron-proton distances in Table III was built. The starting conformations of the substrates used for constructing the model were drawn from x-ray crystallographic data or deduced from energy minimization calculations. Individual sets were then superimposed with constraints on the superimposition of the iron atoms, the carboxylate oxygens, and when possible, the geometrical centroid of the rings. In the best final conformation, obtained by performing superimposition based just on carboxylate and iron atoms, all of the potential hydroxylation sites were close, and carbon 4 of the cinnamic acid analogs was located from 5.5 to 7 Å from the iron (Fig. 6). The model was built assuming a stable orientation and position of the substrate in the active site. It must however be kept in mind that the substrate might be somehow mobile at this stage in its interaction with the protein and that the measured distances between the iron and the protons only reflect an average relative position. Incorporation of this predicted substrate orientation in a three-dimensional model of the CYP73A1 protein and directed mutagenesis confirmation of this orientation are described elsewhere (Schoch et al., 2003).
Figure 6.

Orientation of the substrates relative to the heme of CYP73A1 based on NMR-derived T1M measurements. Ligands are superposed relatively to the heme-iron in red (top). Distances of 2TA protons to heme iron are indicated in Å (bottom)
DISCUSSION
High-yield purification of a plant P450 enzyme water soluble and stable enough for allowing NMR or crystallographic studies has not been reported. The first objective of this work was to modify the native form of CYP73A1 to engineer a protein with an intact catalytic center and substrate-binding properties that would be easy to purify and be stable in the absence of detergent. Exchange of the N-terminal membrane-anchoring segment of CYP73A1 for the amphipathic helix PD1 led to a highly expressed protein in yeast that remained bound to the endoplasmic reticulum membrane. Contrary to the previous report on the PD1-engineered CYP2A4 expressed in E. coli (Sueyoshi et al., 1995), there was no increase in protein expression nor obvious change in protein intracellular location, although the high expression of wild-type CYP73A1 in yeast microsomal fraction was conserved in the PD73His construct. The substrate-binding constants of the engineered protein and complete low- to high-spin conversion upon addition of substrate were also conserved. Structure and accessibility of the active site of the whole population of P450 protein was preserved. The partial activity loss could reflect an altered binding and orientation on the membrane, which results in nonoptimal contact with the P450 reductase and impaired electron transfer. A similar loss in activity was recorded for the modified CYP2A4 when reconstituted in an artificial liposome system. However, contrary to E. coli-expressed CYP2A4, active PD73His could not be solubilized with Na2CO3. Detergent was needed for initial protein solubilization, but could be completely omitted in latter stages of purification. The enzyme was stable upon storage in the absence of detergent and in glycerol concentrations as low as 10%.
PD1 was designed to form an amphipathic α-helix with a flat hydrophobic surface (Schafmeister et al., 1993). It also contains a large proportion of charged or hydrophilic amino acids. This helix was meant to sequester hydrophobic groups from the solvent and is unlikely to be inserted into the membrane. Its fate upon cotranslational insertion and binding of PD73His to the membrane is difficult to predict or to deduce from our data. Upon protein solubilization and purification, it could interact with the F-G loop or any other hydrophobic residues at the membrane-protein interface responsible for the residual binding to the endoplasmic reticulum (Williams et al., 2000) and shield these residues from the solvent. Such an occurrence may explain both the increases in water solubility and stability observed in these experiments. Other hydrophobic interactions might also occur between the purified protein monomers. It was previously shown that purified PD1-linked CYP2A4 in detergent-free buffer forms octamers via hydrophobic interactions (Sueyoshi et al., 1995). The oligomerization state of purified PD73His was not investigated, but if some protein association did occur, it did not seem to have any impact on the accessibility of substrates to the active site. Ni-affinity purification of the engineered CYP73A1 led to milligram amounts of detergent-free protein that was further analyzed for substrate positioning by 1H-NMR.
Other strategies have been used to engineer soluble P450s expressed in E. coli, usually based on truncation of N-terminal transmembrane anchor before or after membrane insertion. Only one, so far, led to successful protein crystallization (Cosme and Johnson, 2000). The alterations involved both deletion of the N-terminal membrane-spanning domain and modifications in the hydrophobic region corresponding to the F-G loop that is expected to contribute to membrane association of the protein. Peptitergent exchange of the N terminus might thus represent an interesting alternative for the production of a protein amenable to crystallization. The use of such amphipathic peptide could be designed to bind the hydrophobic face of the protein to restore the lack of membrane interactions and improve stability to the protein in solution or in the crystal.
Several aromatic protons of cinnamic acid, the natural substrate of CYP73A1, give equivalent signals on 1H-NMR spectra. Cinnamate analogs with substituted aromatic or heterocyclic rings were therefore tested as ligands and substrates of CYP73A1 to refine the NMR analysis and obtain more precise information on the positioning of the substrate in the active site. This led to the demonstration that 4-fluorocinnamate is a strong competitive inhibitor of the C4H, and that two thiophene propenoic acid analogs of cinnamate are good competitive inhibitors and substrates of the enzyme. Another analog, 4VB, behaved as a weaker ligand and poorer substrate of CYP73A1. All four ligands were included in the NMR analysis. The clearest information provided by this analysis is that the average initial orientation of the ligand in the active site of the resting Fe(III) protein is roughly parallel to the heme. The calculated distances of the different ring protons to the iron are very similar and in a 5.6 to 7.5 Å range. This is similar to the distances between hydroxylation sites and the heme iron reported for other P450 enzymes (Modi et al., 1995, 1996b; Li and Poulos, 1997; Poli-Scaife et al., 1997; Regal and Nelson, 2000). In particular in the case of the drug diclofenac in the active site of human CYP2C9, the 4′ carbon, which is the hydroxylation site, was found at 5 ± 0.5 Å of the iron. A similar value has now been found in an x-ray structure of the CYP2C5-diclofenac complex (Wester et al., 2003).
It is possible that a bias leading to an overestimation of the distances was introduced if the exchange between the free and bound ligands is not fast enough to neglect the residence time for the protons near the paramagnetic site in the calculations. Most ring protons appear almost equidistant to the iron, whereas the enzyme exclusively attacks the 4-position of cinnamic acid. This rather suggests that further position adjustments occur during the next steps of the catalytic cycle. Evidence of such substrate movement has been reported upon interaction with the reductase (Modi et al., 1997), after entry of the first electron (Crull et al., 1989; Modi et al., 1996b; Schlichting et al., 2000) or formation of oxygenated intermediates (Paulsen and Ornstein, 1993; Schlichting et al., 2000). It is quite conceivable that the ring carbon 4 is shifted a few angstroms nearer the active center by a conformational change upon reduction of the protein or by the steric constraints imposed by the heme-bound oxygen. The movement of the C10 and C11 protons of laurate in the active site of P450BM3 upon transfer of the first electron was shown to be as large as 6 Å, bringing them to a correct position and orientation for hydroxylation (Modi et al., 1996b; Li and Poulos, 1997). The substrate position in the initial Fe(III) P450-ligand complex shown in Figure 6 seems to be confirmed by site-directed mutagenesis based on substrate docking in the modeled CYP73A1 protein (Schoch et al., 2003), which suggests that the carboxylate of CA is most likely anchored on N302 in the I helix (SRS4) and that the 4 proton of its aromatic ring is stabilized by I371 in SRS5. Movement of the substrate in the active site during the catalytic cycle is thus very likely to be needed not only to allow cinnamate hydroxylation, but also to explain the exclusive attack on C4 of the aromatic ring.
MATERIALS AND METHODS
Chemicals
CA, 2TA, 4VB, and NADPH were from Sigma (l'Isle d'Abeau Chesnes, France); trans-4-fluorocinnamate (4FC) was from Lancaster (Morecambe, UK); 3TA was from Maybridge (Cornwall, UK); and trans-[3-14C]cinnamate was from Isotopchim (Ganagobie, France).
Construction of Peptitergent and His-Tagged Forms of CYP73A1
The modified CYP73A1 cDNAs were generated by PCR, using as a template the double-stranded wild-type CYP73A1 coding sequence subcloned as an EcoRI-BamHI fragment into the pBluescript SK phagemid and the following primers: for exchanging the membrane-anchoring segment for a peptitergent sequence, sense (1) 5′-CAG GAT CCA TGG AAG AAT TAT TAA AAC AAG CTT TAC AAC AAG CTC AAC AAT TAT TAC AAC AAG CTC AAG AAT TAG CTA AAA AAA TAC TAA TCT CCA AAC TCC GCG G-3′ and antisense (2) 5′-GGG AAT TCC CTT AAA ATG ACC TAG GTT TAG CTA CG-3′; for generation of a C-terminally 4-His-tagged protein, sense (3) 5′-GGG ATC CCA TGG ACC TCC TCC TCA TAG AAA AAA CCC TCG TCG-3′ and antisense (4) 5′-ATC GGA ATT CCC TTA ATG ATG ATG ATG AAA TGA CCT AGG TTT AGC TAC G-3′. For generation of the double mutant with a peptitergent at the N terminus and 4-His at the C terminus, amplification was performed using the primers 1 and 4.
PCR mixtures (50 μL) contained 150 μm of each dNTP, 1 μm of each primer, 50 ng of template, 1.25 units of Pfu DNA polymerase (Stratagene, La Jolla, CA), 20 mm Tris-HCl, pH 8.75, 10 mm KCl, 100 mm (NH4)2SO4, 2 mm MgSO4, 0.1% (w/v) Triton X-100, and 0.1 g mL–1 bovine serum albumin. The polymerase was added after 5 min of preheating at 92°C. Thirty cycles of amplification (1 min at 92°C; 2 min at 52°C; and 2 min at 72°C) were completed by 10-min extension at 72°C. PCR provided full-length cDNAs with flanking EcoRI and BamHI sites that were purified on agarose gel, digested, and ligated into the shuttle vector pYeDP60 (Urban et al., 1990). Clones of Escherichia coli transformed with this vector were first checked for correct size of the inserts. Inserts of correct size were sequenced, and plasmids with no additional mutations in the cDNA were selected for expression in yeast.
Yeast Expression and Microsome Preparation
Several modified strains of Brewer's yeast (Saccharomyces cerevisiae) were tested to optimize expression of the mutant proteins: W(R) (Truan et al., 1993) overexpressing the yeast P450 reductase, WAT11 (Pompon et al., 1996) overexpressing the P450 reductase ATR1 from Arabidopsis, and WVS1 and WHT1 (Latunde-Dada et al., 2001) overexpressing P450-reductases from common vetch (Vicia sativa) and Jerusalem artichoke (Helianthus tuberosus).
Yeast transformation and preparation of microsomes (high-density procedure) were described by Urban et al. (1994) and Pompon et al. (1996), respectively. To achieve optimal expression, a yeast colony grown on an SGI (1 g L–1 bactocasamino acids, 7 g L–1 yeast nitrogen base, 20 g L–1 Glc, and 20 mg L–1 Trp) plate was tooth-picked into 50 mL of SGI and grown for 18 h at 30°C to a density of 6 × 107 cells mL–1. This preculture was diluted in YPGE (5 g L–1 glucose, 10 g L–1 yeast extract, 10 g L–1 bactopeptone, 3% [v/v] ethanol) to a density of 2 × 105 cells mL–1, and grown for 30 to 31 h until it reached a density of 8 × 107 cells mL–1, and then protein expression was induced by addition of 10% of a Gal aqueous solution at 200 g L–1. Final density after 17 h of induction at 28°C was routinely around 2 × 108 cells mL–1. Microsomal membranes were isolated after mechanical disruption of yeast cells with glass beads (Pompon et al., 1996). Microsomes from yeasts transformed with void pYeDP60 were used as negative controls.
Spectrophotometric Measurements and Catalytic Activity
P450 content was calculated from CO-reduced versus reduced difference spectra (Omura and Sato, 1964). Low- to high-spin conversion and dissociation constants of enzyme-ligand complexes were evaluated from type I ligand-binding spectra using the εpeak-trough = 125 mm–1 cm–1 (Schalk et al., 1997). NADPH oxidation was quantified as by Werck-Reichhart et al. (1988).
CA hydroxylation was assayed using radiolabeled trans-[3-14C]cinnamic acid and thin-layer chromatography analysis of the metabolites (Reichhart et al., 1980). For determination of the kinetic constants, data were fitted using the nonlinear regression program DNRPEASY derived from DNRP53 (Duggleby, 1984). Cytochrome c reductase activity of the P450 reductase was assayed as by Benveniste et al. (1986).
Metabolism of alternate subtrates was analyzed by HPLC. The assay contained, in a total volume of 200 μL, 100 mm sodium phosphate, pH 7.4, 80 μg of yeast microsomal protein, 150 to 400 μm of substrate, and 600 μm of NADPH. The reaction was incubated at 27°C for 20 to 60 min and was quenched by the addition of 20 μL of HCl 4 n. The products were extracted three times with 2 volumes of ether:petroleum ether (50:50, v/v), the organic phases were pooled and evaporated under argon, and the residue was dissolved in the initial mobile phase (acetonitrile:water:acetic acid (10:90:0.2, v/v). Reverse-phase HPLC analysis was performed on a LiChrosorb RP-18 column (Merck, 4 × 125 mm, 5 μm) at a flow rate of 1 mL min–1, with elution 5 min isocratic and then a 20-min linear gradient from 10% to 52% (v/v) acetonitrile. Microsomes of yeasts transformed with a void plasmid or incubations without NADPH were used as controls to check for the absence of CYP73-independent metabolism and to calculate yields of organic extraction. Wavelength of detection and retention times for substrate and product are, respectively: 2TA (300 nm), 10.8 and 14 min; 3TA (280 nm), 11.7 and 14 min; 4VB (268 nm), 12.1 and 15.7 min; and cinnamate (275 nm and radio-detection), 7.5 and 12 min.
Protein Solubilization and Purification
Yeast microsomes resuspended in 20 mm Tris-HCl, pH 8, containing 30% (v/v) glycerol were solubilized by adding dropwise 1.25% (w/v) Emulgen 911 (Kao Atlas, Tokyo) and were stirred on ice for 15 min (Schalk et al., 1999).
For purification of His-tagged CYP73A1 by affinity chromatography, 200 mg of Emulgen 911 solubilized protein (i.e. 40 nmol of P450) was diluted with 1 volume of 20 mm Tris-HCl, pH 8, containing 1 m NaCl and centrifuged at 100,000g for 45 min. The supernatant was applied at a flow rate of 0.1 mL min–1 to a 5-mL HiTrap-chelating column (Pharmacia, Uppsala) complexed with Ni2+ using the procedure recommended by the manufacturer and equilibrated in 20 mm Tris-HCl, pH 8, containing 0.5 m NaCl, 10 mm imidazole, 0.5% (w/v) Emulgen 911, and 10% (v/v) glycerol (buffer A). The column was then washed at a flow rate of 1 mL min–1, successively with 60 mL of buffer A, 40 mL of buffer A with 0.1% (w/v) Emulgen 911 and 20 mm imidazole, and 30 mL of 20 mm Tris-HCl, pH 8, 0.5 m NaCl, 10% (v/v) glycerol, and 60 mm imidazole. The protein was then eluted with 20 mL of 20 mm Tris-HCl, pH 8, containing 0.5 m NaCl, 10% (v/v) glycerol, and 50 mm His.
For large-scale purification (starting from 150 nmol of P450), another protocol was developed, combining the Mono Q (5 mL) and chelating columns (1 mL), to avoid saturation of the Ni2+ column. Acidic proteins were bound to the MonoQ, but not the engineered CYP73A1 (pI calculated from the amino acid sequence = 9.78). Differences with the affinity chromatography protocol were in the dilution of the solubilized protein (5-fold) and in the first wash that was performed without NaCl. Before using NaCl buffer, the Mono Q column was discarded. NaCl was always needed to keep the protein in solution and to avoid precipitation in the purified fractions in low glycerol and in the absence of detergent.
Analysis of Purified Fractions
SDS-PAGE analysis (Laemmli, 1970) was performed on 10% (w/v) acrylamide and 0.27% (v/v) bisacrylamide gels. Proteins were stained with AgNO3 according to Morrissey (1981).
Purified protein were quantified using the Pierce bicinchoninic acid assay. Heme content was calculated from the Soret band of the electronic absorption spectrum of the oxidized protein using the ε424 nm = 180 mm–1 cm–1 (Omura and Sato, 1964). Eluted fractions containing a protein apparently devoid of contamination as determined on SDS gels were pooled, concentrated, and dialyzed in deuterated NMR buffer in an Ultrafree centrifugal concentrator (Millipore, Bedford, MA).
Mass spectroscopy analyses were performed (with 0.3 nmol of purified P450) by MALDI-TOF with a Reflex III (Bruker, Wissembourg, France) and by Electrospray Ionization, Time of flight with a LCT (Micromass) coupled to a HPLC apparatus SMART (Pharmacia).
NMR Spectroscopy
Proton NMR measurements were made on a Bruker AMX250 spectrometer operating at 250.13 MHz, internally locked on the 2H signal of the solvent in a standard 5-mm tube; signals were again referenced to HDO at 4.75 ppm. Temperature was kept at 300° K. Substrates were solubilized at 40 mm in deuterated 100 mm sodium phosphate buffer containing 10% (v/v) glycerol and 0.5 m NaCl; pH was adjusted to 7.4 with NaOD. Cinnamate was completely soluble in these conditions and was assumed to be freely exchangeable at the sodium phosphate and NaCl concentrations used for the NMR relaxation experiments.
The 1H-NMR spectra (250 MHz, D2O, δ [ppm]) are for VB: 7.88 (d, J2,3 = 8.2 Hz, 2H, H2), 7.59 (d, J2,3 = 8.2 Hz, 2H, H3), 6.88 (dd, Ja,b = 11.0 Hz, Ja,c = 17.8 Hz, 1H, Ha), 6.69 (d, Ja,c = 17.7 Hz, 1H, Hc), and 5.43 (d, Ja,b = 11.0 Hz, 1H, Hb); for 4FC: 7.66 (dd, J2,3 = 8.8 Hz, JH,F = 5.6 Hz, 2H, H3, and H5), 7.41 (d, J2′,3′ = 16.1 Hz, 1H, H3′), 7.21 (t, J2,3 = 8.7 Hz, 2H, H2, and H6), and 6.49 (d, J2′,3′ = 16.2 Hz, 2H, H2′); for 3TA: 7.67 (s, 1H, H2), 7.52 (d, J4,5 = 4.8 Hz, 1H, H5), 7.48 (d, J4,5 = 4.6 z, 1H, H4), 7.44 (d, J2′,3′ = 16.1 Hz, 1H, H3′), and 6.40 (d, J2′,3′ = 15.9 Hz, 1H, H2′); for 2TA: 7.56 (d, J2′,3′ = 15.5 Hz, 1H, H3′), 7.52 (d, J4,5 = 4.0 Hz, 1H, H5), 7.37 (d, J3,4 = 3.3 Hz, 1H, H3), 7.16 (t, J3,4,5 = 4.6 Hz, 1H, H4), and 6.35 (d, J2′,3′ = 15.8 Hz, 1H, H2′); CA is 7.66 (d, J2,3 = 5.2 Hz, 2H, C-2H, H6), 7.47–7.51 (m, 3H, H3, H4 andH5), 7.44 (d, J2′,3′ = 17.2 Hz, 1H, H3′), and 6.57 (d, J2′,3′ = 16.0 Hz, 1H, H2′).
NMR Relaxation Titration Experiments with CYP73
Molecule protons bound near a paramagnetic center (here Fe3+) have their relaxation rates increased by the fluctuating magnetic moment of the electron spin. If ligands rapidly exchange with molecules in bulk solution, the observed longitudinal relaxation time will represent the weighted average of those of the protons from the molecules in bulk solution and bound to the enzyme. For saturating substrate concentrations:
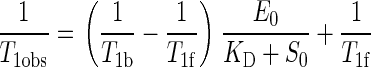 |
(1) |
where T1obs is the observed relaxation time, T1b is the relaxation time of the bound substrate, T1f is the relaxation time for the free substrate, KD is the dissociation constant of the enzyme substrate complex, E0 and S0 are respectively the total enzyme and substrate concentrations.
The slope of a plot of T1obs–1 versus E0/(KD + S0) gives the value of T1b, which is correlated to the paramagnetic relaxation time (T1M) by the following equation
 |
(2) |
where τm is the residence time of the substrate in the enzyme active site. Because τm values greater than 10–4 s are unlikely for type I substrates with a dissociation constant of 5 to 100 μm, Equation 2 can be simplified into
 |
(3) |
The Solomon and Bloembergen (1956) equation correlates paramagnetic relaxation time of protons (T1M) to the distance to the paramagnetic center as discussed in detail by Poli-Scaife et al. (1997).
 |
(4) |
γI and γS are respectively the nuclear and electron giromagnetic ratio, h is the reduced Planck's constant, S is the total electron spin, τc is the correlation time, which describes the process that modulates the electron-nuclear dipolar coupling, and ωI and ωS are respectively the proton and the electron Larmor frequencies. The first term of this equation describes the dipolar interaction; the second one describes the contact interaction, which is always negligible because for the proton A/h < 1 MHz.
The correlation time τc is given by
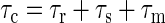 |
(5) |
where τr is the rotational correlation time, τs is the electron spin relaxation time, and τm is the time for the chemical exchange. The fastest process in solution will contribute to τc most significantly. In the case of hemoproteins with high-affinity substrates, we can assume that the substrate is in fast exchange (thus τM ≪ T1M); for an exchangeable proton, we can admit that τS = τc, and we have determined previously that it was approximately 2 × 10–10 s. Thus the term containing τc becomes a constant, and the Solomon-Bloembergen can be rewritten in a simplified form as:
 |
(6) |
where k depends of the instrument used and of the temperature.
The proton longitudinal relaxation times (T1) were measured by standard inversion recovery sequence (180° - τ - 90°). A delay of five times the longest T1 was employed between each sequence, and at least 19 different τ values were used from 20 ms to 10 s in preliminary experiments; in subsequent experiments, 11 appropriate τ values were used, allowing for shorter titrations. T1 value was calculated by nonlinear least squares fitting of the primary data of the peak area as a function of the delay τ, using the correlation time value τc experimentally estimated by Poli-Scaife et al. (1997) using the same instrument and working conditions, which is in good agreement with the values previously reported for other P450s (Modi et al., 1995; Regal and Nelson, 2000).
T1 was calculated in the initial substrate solution, and in the presence of eight to 10 increasing amounts of purified P450 ranging from 0 to 6 μm. After the titration, the P450 was reduced with dithionite and complexed to CO. The conversion to its diamagnetic complex restored T1 values identical to T1 of P450-free substrate, indicating that no significant paramagnetic contribution due to impurities nor contribution of the diamagnetic part of the protein was involved. Total titration time was always less than 4 h. FeIICO versus FeII electronic spectra were also recorded at the end of the experiments to confirm protein integrity. No formation of P420 was detected, however formation of the CO complex was greatly slowed down compared with enzyme in absence of substrate. T1M were calculated as the slope of the curve T1obs–1 versus E0/(KD + nS0) for proton in the NMR spectrum. The proton-iron distances were then calculated as: r = (T1M*k)1/6 using k = 153 Å μs–1, a constant value that we had previously determined on the same instrument in the same experimental conditions (Poli-Scaife et al., 1997).
Computer-Assisted Molecular Modeling
Molecular modeling studies were carried out on a SGI Indy workstation. The geometries of the molecules were taken from the Cambridge Crystallographic Data bank (Allen and Motherwell, 2002; REFCODES = CINMAC, TBTYAC) or, when necessary, were interactively built using the routines of Accelrys InsightII. 4FC was built starting from the chlorine derivative with refcode PCTCIN. Energy minimization was done using Accelrys Discover program (v97.0, force field cvff, minimizer conjugate gradient, maximum derivative 0.001 kcal mol–1). To model a possible relative position of the iron atom to each molecule that would reflect the NMR results, we built sets made of one molecule and one dummy uncharged atom playing the role of the iron atom and then minimized each set under the constraints of NMR distances between the dummy atom (iron) and the substrate protons. The iron protons distances obtained for all sets were within the experimental errors. For superimposing the different sets, we defined three points: the iron atom, the geometrical centroid of the carboxylate oxygens, and the geometrical centroid of the ring. The best fit for superimposing the iron atoms was obtained by performing superimposition based on carboxylate and iron atoms.
Acknowledgments
The skilled technical assistance of Monique Le Ret is greatly appreciated. Critical readings of the manuscript by Keith Griffin are gratefully acknowledged. The W(R) and WAT11 yeast strains and the pYeDP60 expression vector were kindly provided by Drs. Denis Pompon and Philippe Urban.
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.103.020305.
This work was supported by the Centre National de la Recherche Scientifique Programme Chimie-Physique du Vivant and by a fellowship from the French Ministry of Research to G.A.S.
References
- Allen FH, Motherwell WDS (2002) Applications of the Cambridge Structural Database in organic chemistry and crystal chemistry. Acta Crystallogr Sect B 58: 407–422 [DOI] [PubMed] [Google Scholar]
- Benveniste I, Gabriac B, Durst F (1986) Purification and characterization of the NADPH-cytochrome P-450 (cytochrome c) reductase from higher-plant microsomal fraction. Biochem J 235: 365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabello-Hurtado F, Batard Y, Salaün JP, Durst F, Pinot F, Werck-Reichhart D (1998) Cloning, expression in yeast, and functional characterization of CYP81B1, a plant cytochrome P450 that catalyzes in-chain hydroxylation of fatty acids. J Biol Chem 273: 7260–7267 [DOI] [PubMed] [Google Scholar]
- Cosme J, Johnson EF (2000) Engineering microsomal cytochrome P450 2C5 to be a soluble, monomeric enzyme: mutations that alter aggregation, phospholipid dependence of catalysis, and membrane binding. J Biol Chem 275: 2545–2553 [DOI] [PubMed] [Google Scholar]
- Crull GB, Kennington JW, Garber AR, Ellis PD, Dawson JH (1989) F nuclear magnetic resonance as a probe of the spatial relationship between the heme iron of cytochrome P-450 and its substrate. J Biol Chem 264: 2649–2655 [PubMed] [Google Scholar]
- Dixon RA (2001) Natural products and plant disease resistance. Nature 411: 843–847 [DOI] [PubMed] [Google Scholar]
- Duggleby RG (1984) Regression analysis of nonlinear Arrhenius plots: an empirical model and a computer program. Comput Biol Med 14: 447–455 [DOI] [PubMed] [Google Scholar]
- Gabriac B, Werck-Reichhart D, Teutsch H, Durst F (1991) Purification and immunocharacterization of a plant cytochrome P450: the cinnamic acid 4-hydroxylase. Arch Biochem Biophys 288: 302–309 [DOI] [PubMed] [Google Scholar]
- Kahn RA, Le Bouquin R, Pinot F, Benveniste I, Durst F (2001) A conservative amino acid substitution alters the regiospecificity of CYP94A2, a fatty acid hydroxylase from the plant Vicia sativa. Arch Biochem Biophys 391: 180–187 [DOI] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- Latunde-Dada AO, Cabello-Hurtado F, Czittrich N, Didierjean L, Schopfer C, Hertkorn N, Werck-Reichhart D, Ebel J (2001) Flavonoid 6-hydroxylase from soybean (Glycine max L.), a novel plant P-450 monooxygenase. J Biol Chem 276: 1688–1695 [DOI] [PubMed] [Google Scholar]
- Li H, Poulos TL (1997) The structure of the cytochrome P450BM3 heam domain complexed with the fatty acid substrate, palmitoleic acid. Nat Struct Biol 4: 140–146 [DOI] [PubMed] [Google Scholar]
- Mildvan AS, Gupta RK (1978) Binding free energy calculations for P450cam-substrate complexes. Methods Enzymol 49: 322–359651672 [Google Scholar]
- Minoletti C, Dijols S, Dansette PM, Mansuy D (1999) Comparison of the substrate specificities of human liver cytochrome P450s 2C9 and 2C18: application to the design of a specific substrate of CYP 2C18. Biochemistry 38: 7828–7836 [DOI] [PubMed] [Google Scholar]
- Modi S, Gilham DE, Sutcliffe MJ, Lian LY, Primrose WU, Wolf CR, Roberts GC (1997) 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine as a substrate of cytochrome P450 2D6: allosteric effects of NADPH-cytochrome P450 reductase. Biochemistry 36: 4461–4470 [DOI] [PubMed] [Google Scholar]
- Modi S, Paine MJ, Sutcliffe MJ, Lian LY, Primrose WU, Wolf CR, Roberts GC (1996a) A model for human cytochrome P450 2D6 based on homology modeling and NMR studies of substrate binding. Biochemistry 35: 4540–4550 [DOI] [PubMed] [Google Scholar]
- Modi S, Primrose WU, Boyle JM, Gibson CF, Lian LY, Roberts GC (1995) NMR studies of substrate binding to cytochrome P450 BM3: comparisons to cytochrome P450 cam. Biochemistry 34: 8982–8988 [DOI] [PubMed] [Google Scholar]
- Modi S, Sutcliffe MJ, Primrose WU, Lian LY, Roberts GC (1996b) The catalytic mechanism of cytochrome P450 BM3 involves a 6 A movement of the bound substrate on reduction. Nat Struct Biol 3: 414–417 [DOI] [PubMed] [Google Scholar]
- Morrissey JH (1981) Silver stain for proteins in polyacrylamide gels: a modified procedure with enhanced uniform sensitivity. Anal Biochem 117: 307–310 [DOI] [PubMed] [Google Scholar]
- Omura T, Sato R (1964) The carbon monoxide binding pigment of liver microsomes: I. Evidence for its hemoprotein nature. J Biol Chem 239: 2370–2378 [PubMed] [Google Scholar]
- Paquette SM, Bak S, Feyereisen R (2000) Intron-exon organization and phylogeny in a large superfamily, the paralogous cytochrome P450 genes of Arabidopsis thaliana. DNA Cell Biol 19: 307–317 [DOI] [PubMed] [Google Scholar]
- Paulsen MD, Ornstein RL (1993) Substrate mobility in thiocamphor-bound cytochrome P450cam: an explanation of the conflict between the observed product profile and the x-ray structure. Protein Eng 6: 359–365 [DOI] [PubMed] [Google Scholar]
- Pierrel MA, Batard Y, Kazmaier M, Mignotte-Vieux C, Durst F, Werck-Reichhart D (1994) Catalytic properties of the plant cytochrome P450 CYP73 expressed in yeast: substrate specificity of a cinnamate hydroxylase. Eur J Biochem 224: 835–844 [DOI] [PubMed] [Google Scholar]
- Poli-Scaife S, Attias R, Dansette PM, Mansuy D (1997) The substrate binding site of human liver cytochrome P450 2C9: an NMR study. Biochemistry 36: 12672–12682 [DOI] [PubMed] [Google Scholar]
- Pompon D, Louerat B, Bronine A, Urban P (1996) Yeast expression of animal and plant P450s in optimized redox environments. Methods Enzymol 272: 51–64 [DOI] [PubMed] [Google Scholar]
- Reichhart D, Salaün JP, Benveniste I, Durst F (1980) Time course of induction of cytochrome P450, NADPH-cytochrome c reductase, and cinnamic acid hydroxylase by phenobarbital, ethanol, herbicides, and manganese in higher plant microsomes. Plant Physiol 66: 600–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regal KA, Nelson SD (2000) Orientation of caffeine within the active site of human cytochrome P450 1A2 based on NMR longitudinal (T1) relaxation measurements. Arch Biochem Biophys 384: 47–58 [DOI] [PubMed] [Google Scholar]
- Robineau T, Batard Y, Nedelkina S, Cabello-Hurtado F, LeRet M, Sorokine O, Didierjean L, Werck-Reichhart D (1998) The chemically inducible plant cytochrome P450 CYP76B1 actively metabolizes phenylureas and other xenobiotics Plant Physiol 118: 1049–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Kinoshita K, Akashi T, Aoki T, Ayabe S (2002) Key amino acid residues required for aryl migration catalysed by the cytochrome P450 2-hydroxyisoflavanone synthase. Plant J 31: 555–564 [DOI] [PubMed] [Google Scholar]
- Schafmeister CE, Miercke LJ, Stroud RM (1993) Structure at 2.5 A of a designed peptide that maintains solubility of membrane proteins. Science 262: 734–738 [DOI] [PubMed] [Google Scholar]
- Schalk M, Batard Y, Seyer A, Nedelkina S, Durst F, Werck-Reichhart D (1997) Design of fluorescent substrates and potent inhibitors of CYP73As, P450s that catalyze 4-hydroxylation of cinnamic acid in higher plants. Biochemistry 36: 15253–15261 [DOI] [PubMed] [Google Scholar]
- Schalk M, Cabello-Hurtado F, Pierrel MA, Atanossova R, Saindrenan P, Werck-Reichhart D (1998) Piperonylic acid, a selective, mechanism-based inactivator of the trans-cinnamate 4-hydroxylase: a new tool to control the flux of metabolites in the phenylpropanoid pathway. Plant Physiol 118: 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalk M, Croteau R (2000) A single amino acid substitution (F363I) converts the regiochemistry of the spearmint (–)-limonene hydroxylase from a C6- to a C3-hydroxylase. Proc Natl Acad Sci USA 97: 11948–11953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalk M, Nedelkina M, Schoch G, Batard Y, Werck-Reichhart D (1999) Role of unusual amino acid residues in the proximal and distal heme regions of a plant P450, CYP73A1. Biochemistry 38: 6093–6103 [DOI] [PubMed] [Google Scholar]
- Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG (2000) The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287: 1615–1622 [DOI] [PubMed] [Google Scholar]
- Schoch GA, Attias R, Le Ret M, Werck-Reichhart D (2003) Key substrate recognition residues in the active site of a plant cytochrome P450, CYP73A1: homology model guided site-directed mutagenesis. Eur J Biochem 270: 3684–3685 [DOI] [PubMed] [Google Scholar]
- Solomon I, Bloembergen N (1956) Nuclear magnetic interactions in the HF molecule. J Chem Phys 25: 261–266 [Google Scholar]
- Sueyoshi T, Park LJ, Moore R, Juvonen RO, Negishi M (1995) Molecular engineering of microsomal P450 2a-4 to a stable, water-soluble enzyme. Arch Biochem Biophys 322: 265–271 [DOI] [PubMed] [Google Scholar]
- Truan G, Cullin C, Reisdorf P, Urban P, Pompon D (1993) Enhanced in vivo monooxygenase activities of mammalian P450s in engineered yeast cells producing high levels of NADPH-P450 reductase and human cytochrome b5. Gene 125: 49–55 [DOI] [PubMed] [Google Scholar]
- Urban P, Cullin C, Pompon D (1990) Maximizing the expression of mammalian cytochrome P-450 monooxygenase activities in yeast cells. Biochimie 72: 463–472 [DOI] [PubMed] [Google Scholar]
- Urban P, Werck-Reichhart D, Teutsch HG, Durst F, Regnier S, Kazmaier M, Pompon D (1994) Characterization of recombinant plant cinnamate 4-hydroxylase produced in yeast: kinetic and spectral properties of the major plant P450 of the phenylpropanoid pathway. Eur J Biochem 222: 843–850 [DOI] [PubMed] [Google Scholar]
- Werck-Reichhart D, Jones OTG, Durst F (1988) Haem synthesis during cytochrome P-450 induction in higher plants: 5-aminolaevulinic acid synthesis through a five-carbon pathway in Helianthus tuberosus tuber tissues aged in the dark. Biochem J 24: 473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wester MR, Johnson EF, Marques-Soares C, Dijols S, Dansette PM, Mansuy D, Stout CD (2003) The structure of mammalian cytochrome P450 2C5 complexed with diclofenac at 2.1Å resolution: evidence for an induced fit model of substrate binding. Biochemistry 42: 9335–9345 [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Sridhar V, Johnson E, Mc Ree DE (2000) Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell 5: 121–131 [DOI] [PubMed] [Google Scholar]