

Abstract
Progression through mitosis and cytokinesis requires the sequential proteolysis of several cell-cycle regulators. This proteolysis is mediated by the ubiquitin-proteasome system, with the E3 ligase being the anaphase-promoting complex, also known as the cyclosome (APC/C). The APC/C is regulated by two activators, namely Cdc20 and Cdh1. The current view is that prior to anaphase, the APC/C is activated by Cdc20, but that following anaphase, APC/C switches to Cdh1-dependent activation. However, here we present an analysis of the kinetochore protein Cenp-F that is inconsistent with this notion. Although it has long been appreciated that Cenp-F is degraded sometime during or after mitosis, exactly when and how has not been clear. Here we show that degradation of Cenp-F initiates about six minutes after anaphase, and that this is dependent on a C-terminal KEN-box. Although these two observations are consistent with Cenp-F being a substrate of Cdh1-activated APC/C, Cenp-F is degraded normally in Cdh1-null cells. By contrast, RNAi-mediated repression of APC/C subunits or Cdc20 does inhibit Cenp-F degradation. These findings therefore suggest that the APC/C does not simply ‘switch’ upon anaphase onset; rather, our observations indicate that Cdc20 also contributes to post-anaphase activation of the APC/C. We also show that the post-anaphase, KEN-box-dependent degradation of Cenp-F requires it to be farnesylated, a post-translational modification usually linked to membrane association. Because so many of the behaviours of Cenp-F are farnesylation-dependent, we suggest that this modification plays a more global role in Cenp-F function.
Keywords: Cdc20, Cenp-F, Cenp-E, APC/C, Farnesylation, Kinetochore
Introduction
Successful completion of chromosome segregation and cytokinesis requires the coordinated activation and inactivation of numerous mitotic regulators. Inactivation is largely brought about by ubiquitin-dependent proteolysis, with the relevant E3 ubiquitin ligase being the anaphase promoting complex, or cyclosome (APC/C) (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995). During the early stages of mitosis, the APC/C targets cyclin A and Nek2A for destruction (den Elzen and Pines, 2001; Di Fiore and Pines, 2007; Hames et al., 2001; Pines and Hunter, 1991). Following chromosome alignment, securin and cyclin B1 are then targeted for degradation, thereby triggering anaphase onset and mitotic exit, respectively (Clute and Pines, 1999; Hagting et al., 2002). After anaphase onset, a number of other APC/C substrates are destroyed including the Aurora kinases, Plk1, annilin, geminin, Skp2 and Tome-1 (Peters, 2006).
The APC/C is activated by two cofactors, Cdc20 and Cdh1. Destruction of pre-anaphase substrates is Cdc20-dependent. Cdc20 is itself an APC/C substrate that is turned over during mitosis when the spindle checkpoint is active (Nilsson et al., 2008). However, once the spindle checkpoint has become satisfied and anaphase initiated, APC/C activation switches from Cdc20 to Cdh1 (Peters, 2006). This switch occurs because prior to anaphase, Cdh1 is inhibited by Cdk1-dependent phosphorylation; following cyclin B1 destruction, Cdk1 activity declines thereby alleviating inhibition of Cdh1 by promoting its association with the APC/C (Kramer et al., 2000; Visintin et al., 1998; Zachariae et al., 1998). This not only initiates Cdh1-activated APC/C (APC/CCdh1)-mediated degradation but, because Cdc20 is itself a APC/CCdh1 substrate, it also inhibits Cdc20-dependent degradation (Huang et al., 2001).
Degradation of Cdc20 substrates is essential for mitotic progression; however, the significance of Cdh1-dependent proteolysis is less clear. Although degradation of Aurora A, Aurora B and Plk1 might facilitate efficient spindle reorganisation and timely cytokinesis (Floyd et al., 2008; Lindon and Pines, 2004), it is striking that, following rescue of placental defects, Cdh1-null mouse embryos develop relatively normally and survive to birth (Garcia-Higuera et al., 2008; Li et al., 2008). Indeed, it was recently shown that despite extensive RNAi-mediated repression of Cdh1, several presumed Cdh1 substrates (including Plk1, Cdc20 and survivin) were degraded normally (Floyd et al., 2008); Aurora A and B were the only substrates stabilised following Cdh1 RNAi.
Centromere protein F (Cenp-F) is a kinetochore protein required for chromosome segregation (Bomont et al., 2005; Holt et al., 2005; Laoukili et al., 2005; Yang et al., 2005). Pulse-chase experiments conducted over 10 years ago showed that Cenp-F is degraded at some time during or after mitosis (Liao et al., 1995). However, exactly when Cenp-F is degraded, the underlying mechanism, and the functional significance of its proteolysis are all unresolved issues. Overexpression of Cdh1 suppresses Cenp-F expression (Sorensenet al., 2000), suggesting that Cenp-F might be an APC/CCdh1 substrate. Consistently, Cenp-F has several KEN-boxes, motifs believed to be Cdh1 degrons (Pfleger and Kirschner, 2000). Surprisingly, Cenp-F is farnesylated (Ashar et al., 2000) and this post-translational modification is required for its ability to efficiently target kinetochores (Hussein and Taylor, 2002). More surprisingly, cells treated with farnesyl transferase inhibitors (FTI) accumulate Cenp-F, suggesting that farnesylation is also required for its turnover (Hussein and Taylor, 2002).
Based on these observations, we hypothesised that Cenp-F is an APC/CCdh1 substrate that needs to be farnesylated to be degraded. This hypothesis makes several predictions: first, Cenp-F degradation should initiate after anaphase; second, degradation should be dependent on one or more KEN-boxes and the CAAX farnesylation motif; and third, inhibition of Cdh1 or farnesyl transferase activity should block Cenp-F degradation specifically after anaphase onset. Here, we directly test these predictions by analysing GFP-tagged fusion proteins in living cells as they progress through mitosis, an approach used previously to great effect when studying, for example, mitotic cyclins, securin and Plk1 (Clute and Pines, 1999; Hagting et al., 2002; Lindon and Pines, 2004; Wolthuis et al., 2008).
Results
A time-lapse assay to differentiate pre- and post-anaphase degradation
To determine when degradation of Cenp-F initiates, we first had to establish an experimental system with sufficient temporal resolution to distinguish between pre- and post-anaphase initiation events. To do this we used securin as a model protein. Degradation of securin initiates before anaphase, consistent with it being a Cdc20 substrate. However, deleting the D-box from securin converts it to a Cdh1 substrate; degradation now initiates after anaphase in a KEN-box-dependent manner (Hagting et al., 2002).
We generated DLD-1-derived stable cell lines harbouring C-terminally GFP-tagged securin transgenes. These transgenes were also Myc-tagged to enable us to check expression by immunoblotting and immunofluorescence. The transgenes were integrated at a pre-defined genomic locus using FRT-Flp-mediated recombination, facilitating direct comparison of different fusions (O'Gorman et al., 1991). Transgene expression was under tetracycline control, allowing us to study the first mitosis following induction. The parental DLD-1 line was also engineered to constitutively express a dsRed-tagged histone H2B, allowing us to monitor chromosome alignment and segregation.
To determine degradation kinetics, stably transfected cells were synchronised at G1-S using a single thymidine block (Fig. 1A). The cells were then released from the block and tetracycline (Tet) added to induce transgene expression. Eight hours later, tetracycline was removed and the cells analysed by time-lapse microscopy. GFP fluorescence was measured every 2 minutes as cells progressed through mitosis. Degradation curves were then generated by plotting the GFP-fluorescence intensity as a function of time, with T0 representing anaphase onset. Fluorescence intensities were normalised, with 100% representing the maximum value prior to or around anaphase, allowing us to generate degradation curves derived from multiple cells.
Fig. 1.

GFP-tagged SecurinΔD and Aurora A are degraded after anaphase. (A) Timeline showing experimental strategy. (B) Line graphs showing the mean degradation over time of securin-GFP (WT, light grey circles, n=15 cells from four experiments), securinΔD-GFP (ΔD, dark grey circles, n=14 from four experiments) and Aurora-A—GFP (dark grey circles, n=7 from one experiment). Immunoblots show the expression of the GFP-fusion proteins. BubR1 was used as a loading control. Note that the lower mobility forms are more apparent in the securinΔD-expressing line. This is because securin is also degraded in interphase by a phosphorylation-mediated SCF-dependent mechanism (Gil-Bernabe, 2006). The ΔD mutant seems refractory to SCF-dependent degradation, resulting in the accumulation of the hyper-phosphorylated forms.
Consistent with previous observations (Hagting et al., 2002; Holland and Taylor, 2006), degradation of securin-GFP initiated around 25 minutes prior to anaphase onset and degradation of securin-ΔD-GFP initiated at anaphase (Fig. 1B). Furthermore, degradation of an Aurora-A—GFP fusion — a bone fide Cdh1 substrate — initiated at around 10 minutes after anaphase onset (Fig. 1B). Thus, the time-lapse assay does indeed have sufficient temporal resolution to distinguish between degradation events initiated before and after anaphase onset.
Degradation of Cenp-F initiates after anaphase and is KEN-box-dependent
Having established an assay capable of resolving pre- and post-anaphase degradation events, we turned our attention to Cenp-F, initially focusing on the C-terminal 630 amino acids (C630, Fig. 2A). Importantly, we previously showed that this domain is cell-cycle regulated, akin to the endogenous protein (Hussein and Taylor, 2002). Following tetracycline-induction, GFP-C630 was detectable at kinetochores in prometaphase (Fig. 2B,D), consistent with prior observations (Hussein and Taylor, 2002). Analysis of the GFP-C630 degradation curves showed that proteolysis initiated ~6 minutes after anaphase (Fig. 2F), consistent with our hypothesis that Cenp-F is an APC/CCdh1 substrate.
Fig. 2.

GFP-tagged Cenp-F is degraded after anaphase in a KEN-dependent manner. (A) Schematic of Cenp-F showing the domain encoding C630. (B) Immunoblots showing the tetracycline-induced expression of GFP-tagged C630 (WT), C630 ΔKEN6 and C630 ΔKEN7 mutants. Bub3 was used as a loading control. (C) Immunoblot showing the tetracycline-induced expression of GFP-tagged Cenp-F (WT) and Cenp-F ΔKEN7. BubR1 was used as a loading control. (D) Immunofluorescence of mitotic cells expressing GFP-tagged C630 and C630 ΔKEN7, stained as indicated. Enlargements of highlighted (boxed) areas show kinetochore localisation as judged by Bub1. Scale bars: 5 μm. (E) Immunofluorescence of mitotic cells expressing GFP-tagged Cenp-F and Cenp-F ΔKEN7, stained as indicated. Enlargements of highlighted (boxed) areas show kinetochore localisation as judged by Bub1. Scale bars: 5 μm. (F) Line graphs showing the mean degradation over time of GFP-C630 (WT, green circles, n=27 from five experiments), GFP-C630 ΔKEN6 (yellow circles, n=12 from four experiments), GFP-C630 ΔKEN7 (red circles, n=14 from four experiments), GFP—Cenp-F (WT, green circles, n=8 from two experiments) and GFP—Cenp-F ΔKEN7 (red circles, n=14 from four experiments).
Cenp-F has seven KEN boxes and two of these, KEN6 and KEN7, are present in C630 (Fig. 2A). To determine whether these motifs are required for proteolysis of C630, we generated cell lines expressing GFP-tagged C630 fusions in which either KEN6 or KEN7 were mutated to AAA (Fig. 2B). Both ΔKEN mutants localised to kinetochores in prophase (Fig. 2D and not shown). However, whereas the ΔKEN6 mutant was degraded in a manner similar to GFP-C630, the ΔKEN7 mutant was completely stabilised (Fig. 2F). Thus, degradation of GFP-C630 requires a specific KEN box, again consistent with Cenp-F being an APC/CCdh1 substrate. So far, we have not been able to determine why KEN7 is required for degradation of Cenp-F and the other six KEN motifs are not. Interestingly, mouse Cenp-F has only two KEN sequences, both of which are conserved in the human protein, namely KEN2 and KEN7.
To confirm that lessons learnt from C630 are relevant to the full-length protein, we generated cell lines expressing GFP-tagged full-length Cenp-F fusions (Fig. 2C). The exogenous GFP-tagged Cenp-F appeared to behave like endogenous Cenp-F, localising to the nuclear envelope in late G2 before then targeting kinetochores in late prophase (supplementary material Fig. S1). Importantly, both GFP—Cenp-F and the ΔKEN7 mutant localised to kinetochores in prophase (Fig. 2E). Like GFP-C630, degradation of GFP—Cenp-F initiated after anaphase and was dependent on KEN7 (Fig. 2F). Thus, taken together, these observations suggest that Cenp-F is degraded after anaphase in a KEN-dependent manner, arguing that it is indeed an APC/CCdh1 substrate.
Previously, we showed that ectopic expression of C630 delayed the cell cycle some time during G2 and/or mitosis (Hussein and Taylor, 2002). We also showed that this delay was alleviated when the CAAX motif of C630 was mutated. Consistent with these observations, our time-lapse analysis showed that GFP-C630 delayed mitotic progression (see later). Whereas cells expressing GFP took ~49 minutes to proceed from nuclear envelope breakdown to anaphase, cells expressing GFP-C630 took ~73 minutes, largely due to delay in chromosome alignment. Mutating the CAAX motif alleviated this delay; cells expressing GFP-C630 C:S (whereby the cysteine in the CAAX motif is substituted with a serine residue) took ~59 minutes to complete mitosis. This affect does not seem to be due to the CAAX mutant being non-degradable; expression of GFP-C630 ΔKEN7 also delayed chromosome alignment. In contrast to GFP-C630, expression of GFP-tagged full-length Cenp-F did not delay mitosis (data not show). Importantly, C630 lacks the binding site for Ndel1 and Nde1 (Vergnolle and Taylor, 2007). One possibility therefore is that by displacing endogenous Cenp-F from kinetochores (see Hussein and Taylor, 2002), expression of C630 reduces the levels of kinetochore-bound Ndel1 and Nde1, which in turn compromises chromosome alignment.
Post-anaphase degradation of Cenp-F is dependent on farnesylation
As mentioned above, in a previous study we showed that C630 is cell-cycle regulated, much like endogenous Cenp-F. More specifically, when C630 is constitutively expressed from a CMV promoter, it accumulates in G2 cells but is largely absent from G1 cells (Hussein and Taylor, 2002). Interestingly, when we mutated the CAAX farnesylation motif of C630, it accumulated in G1 cells. In addition, when we treated HeLa cells with an FTI, endogenous Cenp-F accumulated in G1 cells (Hussein and Taylor, 2002). On the basis of these indirect observations, we speculated that the degradation of Cenp-F is dependent on farnesylation. The new data shown here is consistent with the notion that Cenp-F is an APC/CCdh1 substrate. However, there is no evidence to suggest that degradation of other APC/CCdh1 substrates requires them to be farnesylated. Therefore, we used the time-lapse assay to determine more directly whether farnesylation is required for the initiation of Cenp-F degradation after anaphase.
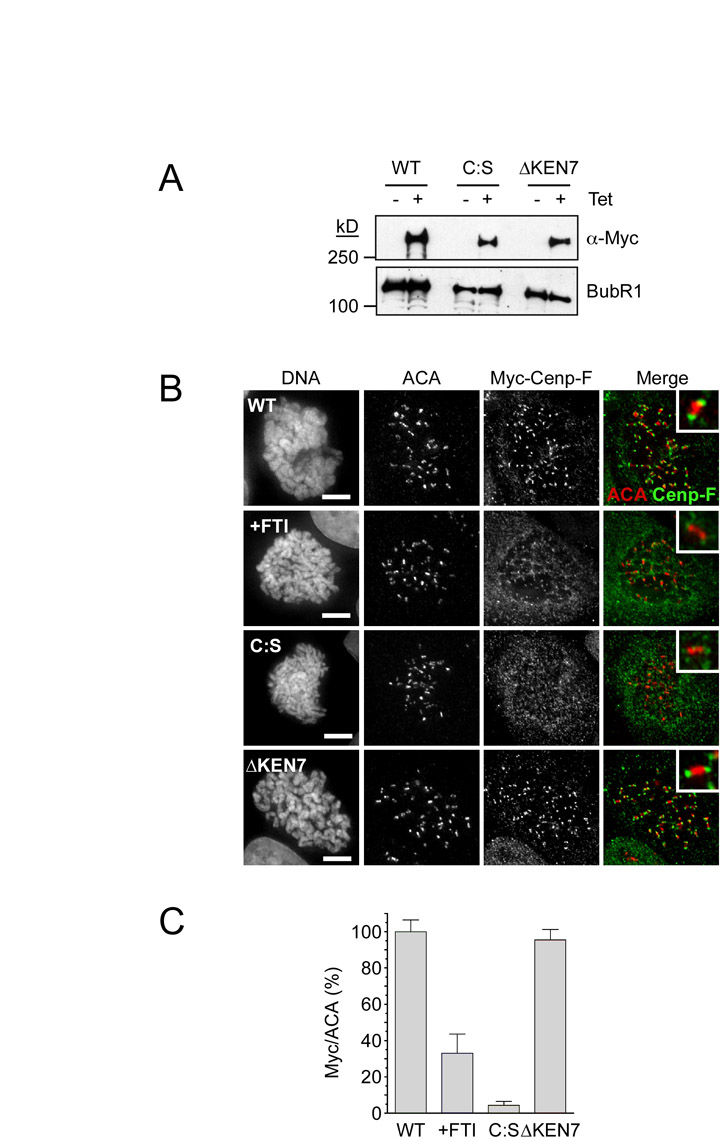
To test whether the CAAX motif of Cenp-F is required for its degradation, we generated cell lines expressing GFP-tagged C630 and full-length Cenp-F fusions harbouring C:S mutations (Fig. 3A,B). Consistent with our previous data, the C630 C:S mutant localised poorly to kinetochores (Fig. 3D). Surprisingly, the full-length C:S mutant was more readily detectable at some kinetochores, suggesting that in the absence of farnesylation, N-terminal sequences not present in C630 might stabilise Cenp-F at the kinetochore. However, quantification of Myc-tagged Cenp-F C:S localisation showed that it was dramatically reduced compared to wild type (supplementary material Fig. S2).
Fig. 3.

Degradation of Cenp-F is farnesylation-dependent. (A,B) Immunoblots showing the tetracycline-induced expression of GFP-tagged C630 C:S and GFP-tagged Cenp-F C:S. Bub3 and BubR1 were used as loading controls. (C) Anti-Myc immunoprecipitates of GFP-C630 (WT), GFP-C630 treated with FTI, GFP-C630 C:S and GFP-C630 ΔKEN7, from cells treated with AGOH. The precipitates were immunoblotted with anti-Myc and anti-AGOH, as indicated. (D) Immunofluorescence of mitotic cells expressing GFP-C630 C:S and GFP—Cenp-F C:S, stained as indicated. Enlargements of highlighted (boxed) areas show kinetochore localisation as judged by Bub1. Scale bars: 5 μm. (E) Line graphs showing the mean degradation over time of GFP-C630 treated with FTI (yellow circles, n=16 from four experiments), GFP-C630 C:S (red circles, n=30 from three experiments), GFP—Cenp-F treated with FTI (yellow circles, n=9 from five experiments), and GFP—Cenp-F C:S (red circles, n=13 from four experiments). Note that WT C630 and WT Cenp-F from Fig. 2 are shown as green lines merely for comparison.
First, we wanted to confirm that Cenp-F is indeed farnesylated and that this could be inhibited both by mutation of the CAAX motif and by exposure to FTIs. To do this, we cultured cells expressing GFP-tagged C630 in the alternative farnesyl transferase substrate, anilinogeraniol (AGOH) (Troutman et al., 2005). AGOH is a prodrug that is converted to 8-anilinogeranyl diphosphate (AGPP) in cells. This in turn can be used by farnesyl transferases as a substrate to modify CAAX motifs, thereby adding the AG moiety instead of a natural farnesyl group. The advantage of this approach is that an anti-AG antibody can then be used to detect the modified protein. GFP-tagged C630 was then immunoprecipitated and the immune complexes blotted with an antibody that recognises the AG moiety. The anti-AG antibody recognised GFP-tagged C630 (Fig. 3C), confirming that this proteins is indeed farnesylated in cells, and that this is unaffected by mutation of KEN7. However, mutating the cysteine residue in the CAAX motif to serine, or treating the cells with an FTI, abolished anti-AG reactivity, confirming that farnesylation was blocked.
Analysis of the degradation curves showed that mutating the CAAX farnesylation motif stabilised both GFP-C630 and full-length Cenp-F (Fig. 3E). Similarly, culturing the cells with an FTI blocked their post-anaphase degradation. Note that the FTI did not affect the degradation of securin, securinΔD or Aurora A (not shown). Clearly therefore, this more direct analysis argues that farnesylation of Cenp-F is indeed required for its post-anaphase degradation.
Cenp-E tail turnover is dependent on farnesylation
As far as we are aware, the role of farnesylation in a cell-cycle-regulated degradation process is unprecedented. This in itself raises numerous questions, one of which is whether this phenomenon is restricted to Cenp-F. Interestingly, the kinetochore-associated kinesin Cenp-E is also farnesylated (Ashar et al., 2000). Furthermore, Cenp-E is degraded sometime shortly after mitosis (Brown et al., 1994). Therefore, we asked whether farnesylation of Cenp-E is required for its post-mitotic degradation. To do this, by analogy with Cenp-F, we focused on the C-terminal region of Cenp-E, referred to as the Cenp-E tail (Fig. 4A). We generated cell lines expressing the Cenp-E tail as a GFP-tagged fusion (Fig. 4 B). The GFP-tagged Cenp-E tail was recruited to kinetochores in prometaphase and was then depleted from kinetochores following chromosome alignment, suggesting that it is useful proxy for the full-length protein (supplementary material Fig. S4). Importantly, when cells were cultured in AGOH and the Cenp-E tail immunopurified, the fusion protein was detected by the anti-AG antibody (Fig. 4C), confirming that the fusion protein can be farnesylated. Moreover, mutation of the CAAX motif or exposure of the cells to an FTI blocked this modification (Fig. 4C).
Fig. 4.

Turnover of the Cenp-E tail is farnesylation-dependent. (A) Schematic of Cenp-E showing the tail domain. (B) Immunoblot showing the tetracycline-induced expression of GFP-tagged Cenp-E tail and tail C:S in DLD1 cells. Coomassie blue staining is shown as a loading control. (C) Anti-Myc immunoprecipitates of GFP—Cenp-E tail, GFP—Cenp-E tail treated with FTI and GFP—Cenp-E tail C:S from cells treated with AGOH. The precipitates were then immunoblotted with anti-Myc and anti-AGOH, as indicated. Arrow indicates Myc-tail protein. Arrowhead indicates an unknown farnesylated endogenous protein. M, Myc IP; C, control IP. (D) DNA content of asynchronous DLD1 cultures showing either whole cell populations (red) or only the GFP-positive cells. (E) Immunofluorescence of asynchronous cells expressing GFP—Cenp-E tail, GFP—Cenp-E tail C:S and GFP—Cenp-E tail treated with FTI, then imaged to detect GFP and cyclin B1 (red). Scale bars: 10 μm.
Note that ectopic expression of the Cenp-E tail induced a prolonged mitotic delay (supplementary material Fig. S4). This has thus far hindered our attempts to analyse the degradation of the Cenp-E tail by time-lapse GFP imaging. Therefore, to test the role of farnesylation in Cenp-E turnover, we turned to a less direct but nevertheless informative approach. We first induced expression of the GFP-tagged Cenp-E tail, then fixed the cells and stained them with propidium iodide to determine the DNA content. We then compared the DNA-content profiles of the whole population and the GFP-positive subpopulation. In cells expressing the tail, virtually all the GFP-positive cells had 4N DNA contents, consistent with the Cenp-E tail only being present in G2 and M-phase cells (Fig. 4D). However, when the CAAX motif was mutated or when the cells were cultured with an FTI, cells with 2N DNA contents were now GFP-positive, indicating the accumulation of the Cenp-E tail in G1 cells. To confirm this, we analysed cells by immunofluorescence, co-staining for cyclin B1, which is absent from G1 cells. In cells expressing the Cenp-E tail, GFP fluorescence was very low but, when detectable, it was only present in mitotic cells staining positive for cyclin B1 (Fig. 4E). By contrast, the C:S mutant was readily detectable in most interphase cells, including those negative for cyclin B1, again indicating its accumulation in G1 cells. The Cenp-E tail was detectable in cyclin-B1-negative cells treated with an FTI (Fig. 4E). These observations therefore are entirely consistent with the notion that the Cenp-E tail also needs to be farnesylated to be degraded.
Cdh1 RNAi only partially inhibits Cenp-F degradation
Our observations showing that degradation of Cenp-F initiates after anaphase in a KEN-box-dependent manner suggests that Cenp-F is an APC/CCdh1 substrate. However, the fact that Cenp-F, along with Cenp-E, also needs to be farnesylated in order to be degraded is a feature not typical of other known APC/CCdh1 substrates. Therefore, we wished to ask more directly whether Cdh1 is required for Cenp-F degradation. To this end, we used siRNA duplexes to repress Cdh1 levels by more than 90% (Fig. 5A). Importantly, this level of repression was sufficient to almost completely block degradation of an Aurora-A—GFP fusion; after 2 hours fluorescence levels had slowly decreased to ~85% (Fig. 5B). The effect of Cdh1 RNAi on C630 and full-length Cenp-F was less straightforward. Following anaphase onset, degradation of both C630 and the full-length protein was initiated with kinetics similar to those in control cells (Fig. 5C). However, whereas the levels of C630 and full-length Cenp-F fell to less than 30% within 120 minutes, their levels only fell to ~60% following Cdh1 RNAi. One possible explanation for this result might be incomplete RNAi-mediated repression; perhaps the residual Cdh1 protein was sufficient to trigger timely degradation of Cenp-F, but not sufficient to push the process to completion. To test this possibility, we turned to mouse embryo fibroblasts (MEFs) that completely lack Cdh1 protein (Garcia-Higuera et al., 2008).
Fig. 5.

Cdh1 RNAi partially suppresses Cenp-F degradation. (A) Immunoblot showing the repression of Cdh1 24 hours after transfection with siRNA duplexes. BubR1 was used as a loading control. (B) Line graphs showing the mean degradation over time of Aurora-A—GFP treated with control (light grey, n=8 from two experiments) and Cdh1 RNAi (dark grey, n=10 from two experiments). (C) Line graphs showing the mean degradation over time of GFP-C630 treated with control (light grey, n=6 from four experiments) and Cdh1 RNAi (dark grey, n=10 from four experiments), and GFP—Cenp-F treated with control (light grey, n=5 from four experiments) and Cdh1 RNAi (dark grey, n=12 from four experiments).
Cenp-F degradation occurs normally in Cdh1-null MEFS
To test the role of Cdh1 more rigorously, we immortalised CDH1+/+ and CDH1−/− mouse embryo fibroblasts (iMEFs) and then transiently transfected them with expression vectors encoding GFP-tagged fusions and analysed them by time-lapse microscopy 48 hours later. First, we wished to determine whether the lessons learnt using human DLD-1 cells also applied to mouse fibroblasts. Importantly, GFP-C630 was degraded in CDH1+/+ iMEFs, with initiation occurring shortly after anaphase (Fig. 6A). In addition, this was blocked when either KEN7 or the CAAX motif was mutated (Fig. 6A). Thus, the mechanisms responsible for Cenp-F degradation appear to be conserved between mouse and human cells. Therefore we then turned to the CDH1−/− iMEFs.
Fig. 6.

Cenp-F is degraded normally in Cdh1-null iMEFS. (A) Line graphs showing the mean degradation over time of GFP-C630 (WT) in wild-type (green circles, n=19 from three experiments) and CDH1−/− iMEFs (green circles, n=13 from four experiments); GFP-C630 C:S in wild-type (yellow circles, n=13 from two experiments) and CDH1−/− iMEFs (yellow circles, n=8 from one experiment); and GFP-C630 ΔKEN7 in wild-type (red circles, n=12 from two experiments) and CDH1−/− iMEFs (red circles, n=10 from three experiments). (B) Line graphs showing the mean degradation over time of: GFP—Aurora-B in wild-type (grey circles, n=11 from two experiments) and CDH1−/− iMEFs (black circles, n=14 from two experiments); GFP-Plk1 in wild-type (grey circles, n=9 from two experiments) and CDH1−/− iMEFs (black circles, n=3 from two experiments).
Immunoblotting confirmed that CDH1−/− iMEFs were indeed Cdh1-deficient (Fig. 6B). Furthermore, a GFP-tagged Aurora B fusion was stabilised in CDH1−/− iMEFs, confirming that these cells were Cdh1-deficient. Strikingly, however, GFP-C630 was degraded in CDH1−/− iMEFs in a manner completely indistinguishable from that observed in CDH1+/+ cells (Fig. 6A). This result is unambiguous, demonstrating that Cdh1 is not essential, at least for the degradation of GFP-tagged C630.
One possible explanation for the above result could be that these iMEFs had adapted; because this cell lineage never contained Cdh1, perhaps they bypassed the requirement for Cdh1 by upregulation of alternative pathways. To address this, we immortalised MEFs harbouring a conditional CDH1 gene whereby exons 2 and 3 are flanked by LoxP sites (Garcia-Higuera et al., 2008). These iMEFs were then infected for 48 hours with an adenoviral vector expressing Cre to recombine the LoxP sites. GFP-C630 was transiently transfected into the Cdh1-null cells and analysed by time-lapse. Importantly, this allowed us to analyse the degradation of GFP-C630 in the first cell cycles following inactivation of CDH1. However, despite efficient inactivation of CDH1, GFP-C630 was degraded normally (data not shown), further supporting the notion that Cdh1 is not required for the post-anaphase degradation of Cenp-F.
APC/CCdc20 contributes to post-mitotic degradation of Cenp-F
The observation that GFP-C630 is degraded normally in Cdh1-null cells is somewhat perplexing as all the prior evidence is consistent with Cenp-F being an APC/CCdh1 substrate. Specifically, as shown above, Cenp-F is degraded after anaphase onset in a KEN-box-dependent manner, a feature believed to be typical of Cdh1-dependent proteolysis (Pfleger and Kirschner, 2000). However, these lines of evidence are circumstantial and evidence is emerging that the degradation of some mitotic regulators previously believed to be APC/CCdh1 substrates, such as Plk1, are in fact not dependent on Cdh1 (Floyd et al., 2008; Garcia-Higuera et al., 2008). Consistent with this, a GFP-Plk1 fusion was degraded normally in Cdh1-null iMEFs (Fig. 6B). One question immediately arises: if Cdh1 is not essential for Cenp-F degradation, then is the APC/C involved, and if so, is it also Cdc20-dependent?
To address whether the APC/C is required for Cenp-F degradation, we transfected RPE cells with siRNA duplexes to repress two APC/C subunits, namely APC2 and APC8. Because inhibition of APC/C was anticipated to induce mitotic arrest prior to the point in the cell cycle where Cenp-F is normally degraded, we used the CDK1 inhibitor Roscovitin to drive the cells out of mitosis. At various time points after mitotic exit, cells were harvested and analysed by immunoblotting. Importantly, under these conditions, APC/C-proficient cells degraded securin, cyclin B1 and Cenp-F: within 1 hour of adding Roscovitin, these proteins were undetectable (Fig. 7A). By contrast, in cells deficient for either APC2 or APC8, securin, cyclin B1 and Cenp-F were still clearly detectable after 2 hours. Thus, these observations demonstrate that Cenp-F degradation is APC/C-dependent.
Fig. 7.
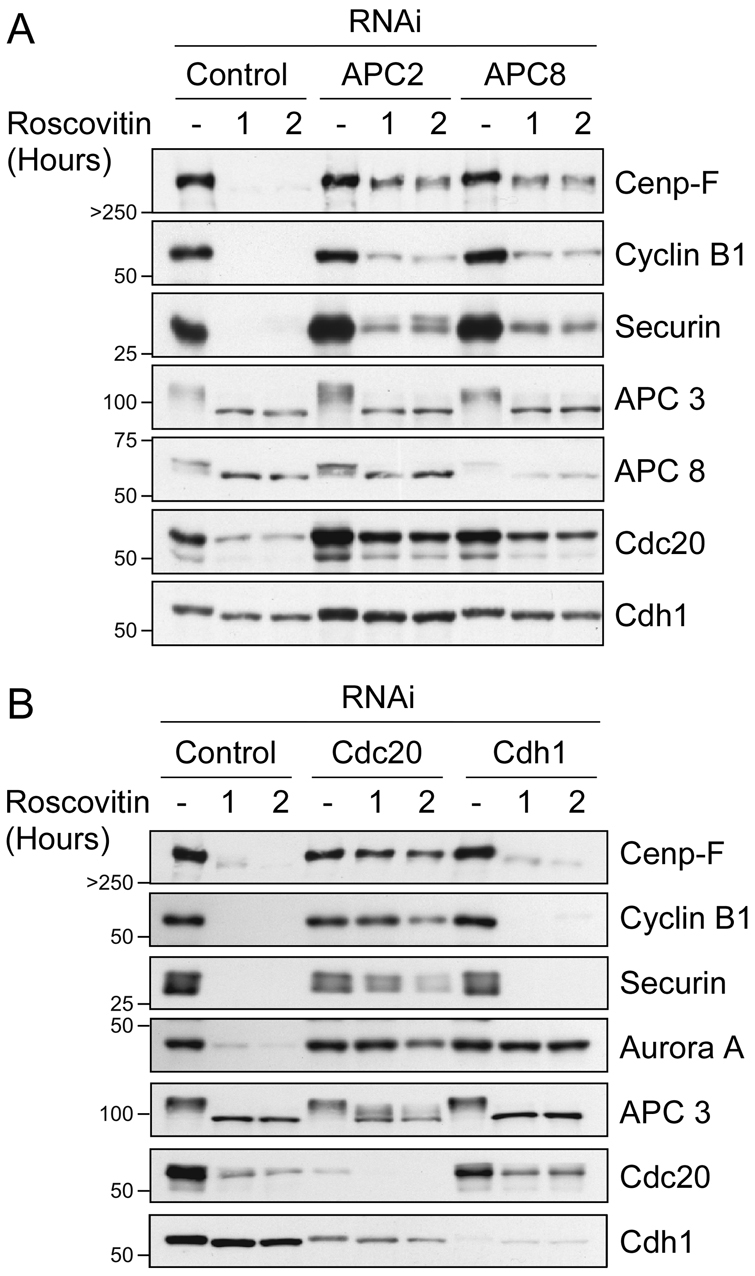
Inhibition of APCCdc20 blocks Cenp-F degradation. Immunoblot showing degradation of securin, cyclin B1, Aurora A and Cenp-F following RNAi repression of (A) APC 2 and APC 8, or (B) Cdc20 and Cdh1. The blots are derived from one experiment, representative of three independent experiments revealing highly comparable results.
Next, we used a similar approach to determine whether degradation of Cenp-F was also dependent on Cdc20. RPE cells were transfected with siRNA duplexes designed to repress Cdc20 and Cdh1 and, once again, mitotically arrested cells were driven out of mitosis with Roscovitin and then analysed by immunoblotting. As expected, repression of Cdc20 stabilised two bona fide APC/CCdc20 substrates, namely securin and cyclin B1 (Fig. 7B). Both securin and cyclin B1 were, however, degraded following Cdh1 RNAi. Consistent with it being a Cdh1 substrate, Aurora A was stabilised following Cdh1 RNAi, but it was still degraded after inhibition of Cdc20. Note, however, that Aurora A degradation is delayed following Cdc20-RNAi; for reasons that are not entirely clear, Cdc20-RNAi cells are more resistant to Roscovitin-stimulated mitotic exit. Significantly, repression of Cdc20 stabilised Cenp-F, suggesting that Cdc20 does indeed play a role in Cenp-F degradation. If this is the case, then the notion that APC/C activation switches from being dependent on Cdc20 to being dependent on Cdh1 following anaphase would appear to be too simplistic. Our data suggest that APC/CCdc20 contributes to the post-anaphase-dependent degradation of Cenp-F. Consistent with the time-lapse data (Fig. 5C), degradation of Cenp-F did not go to completion following Cdh1 RNAi (Fig. 7B). Although the analysis of Cdh1-null MEFs demonstrates that Cdh1 is not essential for Cenp-F degradation (Fig. 6), taking both the RNAi data and the MEF-based data into account, we suggest that in a normal cell, degradation of Cenp-F is initiated by APC/CCdc20 and then driven to completion by APC/CCdh1.
Discussion
Cenp-F was previously shown to be degraded sometime during or after mitosis (Liao et al., 1995). However, exactly when, how and why Cenp-F is degraded remained unclear. Here, we show that degradation of GFP-tagged Cenp-F fusions initiates about 6 minutes after anaphase onset and is dependent on a C-terminal KEN-box. Although this suggests that Cenp-F is an APC/CCdh1 substrate, RNAi-mediated repression of Cdh1 in human cells only partially inhibits Cenp-F degradation. Moreover, mouse fibroblasts completely lacking Cdh1 degrade Cenp-F normally. By contrast, RNAi-mediated repression of Cdc20 stabilises Cenp-F, arguing that Cenp-F is in fact a Cdc20 substrate. Below, we rationalise these observations and, as previously suggested by Floyd, Pines and Lindon (Floyd et al., 2008), we argue that in contrast to the Cdc20/Cdh1 ‘anaphase-switch’ model, APC/CCdc20 does in fact contribute to post-anaphase degradation of mitotic regulators.
Both APC/CCdc20 and APC/CCdh1 contribute to Cenp-F degradation
Here, we used time-lapse microscopy to study the degradation of GFP-tagged Cenp-F fusion protein. This approach enabled the study of individual cells, thus providing a level of temporal resolution that can be correlated with chromosome alignment and segregation. To allow a direct comparison of various mutants we used a system that allows the generation of isogenic tetracycline-inducible stable cell lines. Nevertheless, a key issue is whether the behaviour of the exogenous GFP-tagged protein reflects that of endogenous Cenp-F. We believe that it does for two reasons. First, GFP—Cenp-F localises similarly to endogenous Cenp-F. During G2, it accumulates in the nucleus but is excluded from nucleoli, then goes to the nuclear envelope at the onset of prophase and finally binds kinetochores in mitosis (supplementary material Fig. S1). Second, analysis of endogenous Cenp-F expression by immunofluorescence microscopy and immunoblotting is entirely consistent with the data derived from the time-lapse studies (data not shown). Indeed, this approach has been extremely informative when studying the degradation of a number of mitotic regulators (Clute and Pines, 1999; Hagting et al., 2002; Lindon and Pines, 2004; Wolthuis et al., 2008).
Because degradation of GFP—Cenp-F occurred after anaphase, and in a KEN-box-dependent manner, our initial hypothesis was that Cenp-F was an APC/CCdh1 substrate. Consistently, Cdh1 RNAi inhibited Cenp-F degradation. However, although Cdh1 RNAi almost completely blocked Aurora A degradation, the effect on GFP—Cenp-F was only partial. One explanation for this could be that residual Cdh1 allows APC/C-mediated ubiquitylation to continue, but only of its preferred or more processive substrates (Rape et al., 2006). If Cenp-F is a particularly processive substrate, then the residual APC/CCdh1 activity left following RNAi might be sufficient to partially degrade Cenp-F, although having no effect on Aurora A. However, analysis of iMEFs clearly shows that in the complete absence of Cdh1, GFP—Cenp-F is degraded normally, even though GFP—Aurora-B is stabilised (Fig. 6). This effect is not limited to Cenp-F because GFP-Plk1 was also degraded normally in Cdh1-null iMEFs, consistent with recent reports (Floyd et al., 2008; Garcia-Higuera et al., 2008). Although this raises the possibility that another E3 ligase might be involved in Cenp-F ubiquitylation, we suspect that this is not the case because inhibition of either APC2 or APC8 stabilised Cenp-F. Taken together with the observation that Cdc20 RNAi also stabilised Cenp-F, our data argues that APC/CCdc20 plays a role in Cenp-F degradation. Although this notion is inconsistent with the idea that APC/C switches from Cdc20- to Cdh1-mediated degradation at anaphase onset, it is consistent with a recent study showing that even though Cdh1 RNAi inhibited degradation of Aurora A and B, there was little effect on other presumed Cdh1 substrates such as Plk1 (Floyd et al., 2008).
On the basis of the above, we therefore favour the idea that APC/CCdc20 plays a key role in the post-anaphase degradation of Cenp-F. However, if this is the case, how can we explain the fact that Cdh1 RNAi partially inhibits degradation of GFP—Cenp-F? We suggest that in unperturbed cells, both APC/CCdc20 and APC/CCdh1 are involved in ensuring the complete degradation of Cenp-F. Thus, shortly after anaphase, Cdc20 mediates the initial rapid degradation of Cenp-F, and then as Cdh1 becomes active it continues the process through to completion. Following Cdh1 RNAi, we suggest that APC/CCdc20 functions normally, explaining the initial phase of Cenp-F degradation. However, we suspect that the residual levels of Cdh1 following RNAi are sufficient to result in Cdc20 destruction, thus bringing the degradation of Cenp-F to a halt at about 60 minutes after anaphase. Indeed, it was previously shown that Cdc20 is degraded normally in Cdh1 RNAi cells (Floyd et al., 2008). However, in Cdh1-null cells, where there is no APC/CCdh1 activity to inactivate Cdc20, APC/CCdc20-mediated degradation of Cenp-F continues unabated. In a normal cell, however, Cenp-F degradation continues for at least 120 minutes after anaphase, presumably long after Cdc20 has been degraded. This suggests that when Cdh1 is present, it is responsible for Cenp-F degradation once Cdc20 has been destroyed: otherwise, in a non-RNAi cell, degradation of Cenp-F would stop within about 60 minutes post-anaphase. This explanation suggests that either Cdc20 or Cdh1 can recruit Cenp-F to the APC/C after anaphase onset. Alternatively, perhaps Cenp-F binds the APC/C directly, independently of Cdc20 or Cdh1, so that it is then ubiquitylated once the APC/C is activated by either co-factor. As we discuss below, degradation of Cenp-F is dependent on it being farnesylated. Although there is no evidence to support this notion at present, one possibility is that Cenp-F interacts with the APC via a novel farnesylation-dependent mechanism and it is then ubiquitylated irrespective of which activating co-factor is bound.
Cenp-F needs to be farnesylated to be degraded
Several years ago it was reported that FTIs induce chromosome alignment and segregation defects during mitosis (Crespo et al., 2001; Schafer-Hales et al., 2007). Because Cenp-E and Cenp-F have been shown to be farnesylated, these FTI-induced effects might be due to dysfunction of Cenp-E and/or Cenp-F (Ashar et al., 2000; Schafer-Hales et al., 2007). Previously we showed that FTIs inhibit Cenp-F turnover: however, whether the FTI affected turnover of Cenp-F during interphase or mitosis was unclear. Here, we show that farnesyl transferase activity is required for the post-anaphase degradation of Cenp-F. Importantly, this effect of the FTI does not appear to be indirect, for example via inhibition of the ubiquitin proteasome pathway Time-lapse and immunoblot analysis showed that in the presence of the FTI, degradation of securin, securinΔD and Aurora A occur normally (data not shown). Furthermore, degradation of Cenp-F is absolutely dependent on its CAAX farnesylation motif. Importantly, this phenomenon is not a peculiarity of Cenp-F; we show here for the first time that the Cenp-E tail also needs to be farnesylated in order to be degraded.
Protein farnesylation is typically involved in targeting proteins to membranes and, as far as we are aware, farnesylation has not previously been implicated as a signal for ubiquitin-mediated degradation. One possibility is that farnesylation targets Cenp-E and Cenp-F to membranes and that this in turn allows them to be degraded. Although Cenp-F binds the nuclear membrane just prior to mitosis (Hussein and Taylor, 2002), there is no evidence to suggest that Cenp-E associates with membranes, so we suspect that this explanation is unlikely. Also, the C-terminal 103 amino acids of Cenp-F, which contains both the KEN-box and the CAAX motif, is degraded after anaphase without any apparent localisation to the nuclear membrane or any other specific location within the cell (data not shown). Note that farnesylation of Cenp-E and Cenp-F is not just required for their degradation because their correct localisation during mitosis also requires farnesylation (Hussein and Taylor, 2002; Schafer-Hales et al., 2007). Furthermore, although expression of C630 delays progression through G2 and/or M phase, expression of the CAAX mutant does not (Hussein and Taylor, 2002). We suspect therefore that farnesylation of Cenp-E and Cenp-F plays a more significant role in their overall functions. Interestingly, farnesylation of Ras2 in budding yeast increases its affinity for adenylyl cyclase by 100-fold, independent of any membrane localisation (Kuroda et al., 1993). In addition, farnesylation of Ykt6 increases its α-helical content, inducing a more compact and stable conformation (Pylypenko et al., 2008). Perhaps farnesylation of Cenp-E and Cenp-F similarly promotes conformational changes and, in doing so, increases their affinity for various binding partners. This, in turn, regulates their localisation, function and degradation.
The role of Cenp-F degradation
Elevated Cenp-F expression has been associated with poor prognosis in patients with breast cancer, leading to the suggestion that Cenp-F overexpression might contribute to tumorigenesis (O'Brien et al., 2007). Surprisingly, we observed no obvious mitotic or cell-cycle defects following overexpression of various non-degradable Cenp-F mutants (data not shown) and as such the role of Cenp-F degradation remains unclear. One possibility is that Cenp-F degradation plays roles in contexts outside of mitosis. For example, Cenp-F inhibits the transcription factor ATF4 (Zhou et al., 2005), which has been implicated in numerous post-mitotic phenomenon including long-term memory and synaptic plasticity (Ameri and Harris, 2008). Cenp-F can also bind to Rb (Rattner et al., 1993), which apart from its well-characterised role in the cell cycle, also activates transcription of muscle-specific genes (Novitch et al., 1996; Novitch et al., 1999). Cenp-F orthologues in mouse and chicken have been implicated in muscle differentiation (Papadimou et al., 2005; Robertson et al., 2008), raising the possibility that Cenp-F might regulate Rb during this process. Finally, Cenp-F also interacts with the dynein adapters Ndel1, Nde1 and Lis1 (Liang et al., 2007; Stehman et al., 2007; Vergnolle and Taylor, 2007). These proteins are essential for regulating nuclear translocation during neuronal migration and for neuron positioning during cortical development (Shu et al., 2004). Because APC/CCdh1 also has important roles in post-mitotic neurons (Stegmuller and Bonni, 2005), this raises the possibility that Cenp-F degradation might be important during neuron migration and positioning.
Materials and Methods
Expression constructs encoding APC/C substrates
The securin and Plk1 cDNAs were kind gifts from Jonathon Pines (Gurdon Institute, Cambridge, UK) and René Medema (University Medical Center, Utrecht, Netherlands), respectively. Aurora A and B plasmids were as previously described (Girdler et al., 2006). A Cenp-F cDNA encoding amino acids 632-3113 was a kind gift from Wen-Hwa Lee (University of Texas Health Science Center, San Antonio, TX). cDNA encoding the N-terminal 750 amino acids was generated by RT-PCR amplification of mRNA from HeLa cells and was then subcloned into a vector encoding the C-terminal 2363 amino acids. All cDNAs were then cloned into a pcDNA5/FRT/TO-based vector (Invitrogen) modified to include a N- or C-terminal GFP tag. Some constructs also contained a Myc-tag. ΔKEN and C:S mutants were created by site-directed mutagenesis (QuickChange, Stratagene).
Cell culture and generation of stable cell lines
DLD-1 cells and MEFs were cultured as described previously (Taylor et al., 2001). Thymidine (Sigma) was used at 2 mM, SCH 66336 (Hussein and Taylor, 2002) at 1 μM, AGOH (Troutman et al., 2005) at 30 μM, and tetracycline (Sigma) at 1 μg/ml. An open reading frame (ORF) encoding histone H2B (Morrow et al., 2005) was cloned into pDsRed-Monomer-N1 to create a histone-H2B-dsRed ORF (BD Biosciences). This plasmid was then linearised and transfected into DLD-1 LacZeo/TO cells (Girdler et al., 2006) using Lipofectamine Plus (Invitrogen). Transfected cells were selected in 8 ug/ml Blasticidin (Melford), 60 ug/ml Zeocin (Invitrogen) and 0.75 mg/ml G418 (Sigma). DsRed-positive cells were then isolated by flow cytometry using a FACSAria (BD Biosciences) and seeded into 96-well plates to generate clonal cell lines. The pcDNA5/FRT/TO vectors encoding GFP-tagged APC/C substrates were then co-transfected with pOG44 into DLD1 LacZeo/TO Histone-H2B-dsRed cells as previously described (Tighe et al., 2004).
Antibody techniques
Immunofluorescence analysis was performed essentially as described (Taylor et al., 2001) using the following antibodies: 4A6 mouse anti-Myc, (Upstate, 1:1000); sheep anti-Bub1 (Taylor et al., 2001). Fluorescence microscopy was performed using a Zeiss Axiovert 200 equipped with epifluorescence and a Photometrics Cool Snap HQ CCD camera driven by Metamorph software (Universal Imaging). Immunoblot analysis was done as previously described (Taylor et al., 2001), with proteins transferred onto either nitrocellulose or PVDF membranes. Cenp-F immunoblots were transferred to nitrocellulose membranes as previously described (Tighe et al., 2004). The following antibodies were used: 4A6 mouse anti-Myc (1:1000; Upstate); sheep anti-Bub3 (1:1000; SB3.1), sheep anti-BubR1 (Taylor et al., 2001), rabbit anti-GFP (1:5000; Abcam), DH01 mouse anti-Cdh1 (1:200; Abcam) mouse anti-cyclin B1 (1:100; GNS1, Abcam).
Farnesylation
To analyse protein farnesylation, DLD1 cells were treated with the AGOH analogue for 38-48 hours as previously described (Troutman et al., 2005). The cells were then lysed and soluble extracts prepared as described (Morrow et al., 2005). Anti-Myc (4A6) or anti-IgG antibodies coupled to protein G Sepharose (Amersham) were added to cell lysates for 1-2 hours and collected by centrifugation. The beads were washed with lysis buffer, then the proteins eluted in SDS sample buffer. Samples were resolved by SDS-PAGE, transferred to nitrocellulose and blotted with mouse anti-Myc and mouse anti-AGOH antibodies (Troutman et al., 2005).
Time-lapse microscopy
For time-lapse imaging, cells were cultured in multi-well chamber slides (Lab-Tek, Nunc). Images were taken using a manual Axiovert 200 (Zeiss) microscope equipped with a PZ-2000 automated stage (Applied Scientific Instrumentation) and an environmental control chamber (Solent Scientific), which maintained cells at 37°C in a humidified stream of 5% CO2. Shutters, filter wheels and point visiting were driven by Metamorph software (MDS analytical technologies) and images taken using a CoolSNAP HQ camera (Photmetrics). Images were taken every two minutes for 24 hours, with a 40× objective. The time-lapse images were analysed using Metamorph sofware in which boxes were drawn around individual cells, pixel intensities taken for each time frame, and background intensities subtracted. The GFP fluorescence was then normalised to the maximum intensity before or around anaphase for each individual cell and plotted as a percentage against time, with 0 minutes being set as anaphase onset.
Cdh1 RNAi
To repress Cdh1, approximately 2×104 DLD1 cells were treated with 50 nM ON-Target smartpool siRNA duplexes (5′-CCACAGGAUUAACGAGAAU-3′, 5′-GGAACACGCUGACAGGACA-3′, 5′-GCAACGAUGUGUCUCCCUA-3′, 5′-GAAGAAGGGUCUGUUCACG-3′, Dharmacon) as per the manufacturer's instructions. Briefly, siRNA duplexes were mixed with 2 μl Interferin (Autogen Bioclear) in media without serum and antibiotics, incubated for 15 minutess, then added to the cells with media containing serum and antibiotics.
Mouse embryo fibroblasts
Immortalised CDH1+/+ and CDH1−/− MEFs were cultured as previously described (Garcia-Higuera et al., 2008). iMEFs were transiently transfected with the GFP-tagged constructs using ProFection calcium phosphate transfection kit (Promega) as per the manufacturers instructions. Briefly, 1.35×104 cells were transfected with approximately 400 ng of DNA. DNA precipitates were then washed away after 24 hours.
CENP-E analysis
A CENP-E tail cDNA encoding amino acids 1572-2606 was subcloned into a pcDN5/FRT/TO-based vector modified to contain an N-terminal Myc-LAP tag. The LAP tag is comprised of GFP—Tev-S-peptide as described previously (Cheeseman et al., 2004). For DNA content measurements, the CENP-E tail was induced for 24 hours, the cells then fixed in 70% ethanol over night, followed by staining with propidium iodide. The cells were then analysed using a Becton Dickinson LSR II. For immunofluorescence, cells were grown on acid-washed, glass coverslips coated with poly-L-lysine (Sigma-Aldrich), pre-extracted for 90 seconds in MTSB (100 mM PIPES pH 6.8, 0.1% Triton X-100, 0.1 mM CaCl2, 1 mM MgCl2) and fixed in 4% formaldehyde in MTSB. Cells were blocked in 2.5% FBS, 2 M glycine, 0.1% Triton X-100 in PBS for 1 hour. Antibody incubations were conducted in the blocking solution for 1 hour. The DNA was detected using DAPI and cells were mounted in Prolong Antifade (Invitrogen). Images were collected using a Deltavision Core system (Applied Precision) controlling an interline charge-coupled device camera (Cooldsnap, Raper). Images were deconvolved and maximum intensity 2D projections were assembled for each image using softWoRx (Applied Precision). Where indicated cells were treated with FTI-277 (Calbiochem).
APC and Cdc20 RNAi
hTERT-RPE1 cells (a kind gift from Kip Sluder, University of Massachusetts, Worcester, MA) were cultured in D-MEM plus 8% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin. ON-TARGET-plus SMARTpools of siRNA (Dharmacon) targeting APC2 (L-003200-00), APC8 (Cdc23, L-009523-00), Cdc20 (L-003225-00) and Cdh1 (FZR1, L-015377-00) were transfected using Lipofectamine 2000 according to manufacturer's instructions. Cells were then arrested in mitosis using nocodazole at 250 ng/ml for 15 hours. Mitotic cells were harvested by selective detachment in PBS at 37°C. 50 μM Roscovitin was then added for 1 hour. Cell lysates were then separated by SDS-PAGE, proteins blotted on to nitrocellulose membranes then probed with antibodies against CENP-F (Abcam), cyclin B1 (GNS1, Santa Cruz Biotechnology), Securin (Abcam), APC3/Cdc27 (Transduction Labs), APC8 (BioLegend), Cdc20 (E7, Santa Cruz Biotechnology) and Cdh1 (NeoMarkers).
Supplementary Material
Acknowledgments
The authors are grateful to Jonathon Pines (Cambridge), René Medema (Utrecht) and Wen-Hwa Lee (Texas) for reagents. We thank David Perera for comments on the manuscript. M.D.J.G. is supported by a studentship from the Wellcome Trust, S.S.T. is supported by a Senior Fellowship from Cancer Research UK. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/3/321/DC1
References
- Ameri K., Harris A. L. (2008). Activating transcription factor 4. Int. J. Biochem. Cell Biol. 40, 14-21 [DOI] [PubMed] [Google Scholar]
- Ashar H. R., James L., Gray K., Carr D., Black S., Armstrong L., Bishop W. R., Kirschmeier P. (2000). Farnesyl transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of CENP-E with the microtubules. J. Biol. Chem. 275, 30451-30457 [DOI] [PubMed] [Google Scholar]
- Bomont P., Maddox P., Shah J. V., Desai A. B., Cleveland D. W. (2005). Unstable microtubule capture at kinetochores depleted of the centromere-associated protein CENP-F. EMBO J. 24, 3927-3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K. D., Coulson R. M., Yen T. J., Cleveland D. W. (1994). Cyclin-like accumulation and loss of the putative kinetochore motor CENP-E results from coupling continuous synthesis with specific degradation at the end of mitosis. J. Cell Biol. 125, 1303-1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman I. M., Niessen S., Anderson S., Hyndman F., Yates J. R., 3rd, Oegema K., Desai A. (2004). A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev. 18, 2255-2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P., Pines J. (1999). Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol. 1, 82-87 [DOI] [PubMed] [Google Scholar]
- Crespo N. C., Ohkanda J., Yen T. J., Hamilton A. D., Sebti S. M. (2001). The farnesyltransferase inhibitor, FTI-2153, blocks bipolar spindle formation and chromosome alignment and causes prometaphase accumulation during mitosis of human lung cancer cells. J. Biol. Chem. 276, 16161-16167 [DOI] [PubMed] [Google Scholar]
- den Elzen N., Pines J. (2001). Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153, 121-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore B., Pines J. (2007). Emi1 is needed to couple DNA replication with mitosis but does not regulate activation of the mitotic APC/C. J. Cell Biol. 177, 425-437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd S., Pines J., Lindon C. (2008). APC/C Cdh1 targets aurora kinase to control reorganization of the mitotic spindle at anaphase. Curr. Biol. 18, 1649-1658 [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I., Manchado E., Dubus P., Canamero M., Mendez J., Moreno S., Malumbres M. (2008). Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 10, 802-811 [DOI] [PubMed] [Google Scholar]
- Gil-Bernabé A. M., Romero F., Limón-Mortés M. C., Tortolero M. (2006). Protein phosphatase 2A stabilizes human securin, whose phosphorylated forms are degraded via the SCF ubiquitin ligase. Mol. Cell Biol. 26, 4017-4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdler F., Gascoigne K. E., Eyers P. A., Hartmuth S., Crafter C., Foote K. M., Keen N. J., Taylor S. S. (2006). Validating Aurora B as an anti-cancer drug target. J. Cell Sci. 119, 3664-3675 [DOI] [PubMed] [Google Scholar]
- Hagting A., Den Elzen N., Vodermaier H. C., Waizenegger I. C., Peters J. M., Pines J. (2002). Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J. Cell Biol. 157, 1125-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hames R. S., Wattam S. L., Yamano H., Bacchieri R., Fry A. M. (2001). APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 20, 7117-7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland A. J., Taylor S. S. (2006). Cyclin-B1-mediated inhibition of excess separase is required for timely chromosome disjunction. J. Cell Sci. 119, 3325-3336 [DOI] [PubMed] [Google Scholar]
- Holt S. V., Vergnolle M. A., Hussein D., Wozniak M. J., Allan V. J., Taylor S. S. (2005). Silencing Cenp-F weakens centromeric cohesion, prevents chromosome alignment and activates the spindle checkpoint. J. Cell Sci. 118, 4889-4900 [DOI] [PubMed] [Google Scholar]
- Huang J. N., Park I., Ellingson E., Littlepage L. E., Pellman D. (2001). Activity of the APC(Cdh1) form of the anaphase-promoting complex persists until S phase and prevents the premature expression of Cdc20p. J. Cell Biol. 154, 85-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein D., Taylor S. S. (2002). Farnesylation of Cenp-F is required for G2/M progression and degradation after mitosis. J. Cell Sci. 115, 3403-3414 [DOI] [PubMed] [Google Scholar]
- Irniger S., Piatti S., Michaelis C., Nasmyth K. (1995). Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell 81, 269-278 [DOI] [PubMed] [Google Scholar]
- King R. W., Peters J. M., Tugendreich S., Rolfe M., Hieter P., Kirschner M. W. (1995). A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81, 279-288 [DOI] [PubMed] [Google Scholar]
- Kramer E. R., Scheuringer N., Podtelejnikov A. V., Mann M., Peters J. M. (2000). Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell 11, 1555-1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda Y., Suzuki N., Kataoka T. (1993). The effect of posttranslational modifications on the interaction of Ras2 with adenylyl cyclase. Science 259, 683-686 [DOI] [PubMed] [Google Scholar]
- Laoukili J., Kooistra M. R., Bras A., Kauw J., Kerkhoven R. M., Morrison A., Clevers H., Medema R. H. (2005). FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 7, 126-136 [DOI] [PubMed] [Google Scholar]
- Li M., Shin Y. H., Hou L., Huang X., Wei Z., Klann E., Zhang P. (2008). The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat. Cell Biol. 10, 1083-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Yu W., Li Y., Yu L., Zhang Q., Wang F., Yang Z., Du J., Huang Q., Yao X., et al. (2007). Nudel modulates kinetochore association and function of cytoplasmic dynein in M phase. Mol. Biol. Cell 18, 2656-2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H., Winkfein R. J., Mack G., Rattner J. B., Yen T. J. (1995). CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J. Cell Biol. 130, 507-518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindon C., Pines J. (2004). Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J. Cell Biol. 164, 233-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow C. J., Tighe A., Johnson V. L., Scott M. I., Ditchfield C., Taylor S. S. (2005). Bub1 and aurora B cooperate to maintain BubR1-mediated inhibition of APC/CCdc20. J. Cell Sci. 118, 3639-3652 [DOI] [PubMed] [Google Scholar]
- Nilsson J., Yekezare M., Minshull J., Pines J. (2008). The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat. Cell Biol. 10, 1411-1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch B. G., Mulligan G. J., Jacks T., Lassar A. B. (1996). Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J. Cell Biol. 135, 441-456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch B. G., Spicer D. B., Kim P. S., Cheung W. L., Lassar A. B. (1999). pRb is required for MEF2-dependent gene expression as well as cell-cycle arrest during skeletal muscle differentiation. Curr. Biol. 9, 449-459 [DOI] [PubMed] [Google Scholar]
- O'Brien S. L., Fagan A., Fox E. J., Millikan R. C., Culhane A. C., Brennan D. J., McCann A. H., Hegarty S., Moyna S., Duffy M. J., et al. (2007). CENP-F expression is associated with poor prognosis and chromosomal instability in patients with primary breast cancer. Int. J. Cancer 120, 1434-1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman S., Fox D. T., Wahl G. M. (1991). Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 251, 1351-1355 [DOI] [PubMed] [Google Scholar]
- Papadimou E., Menard C., Grey C., Puceat M. (2005). Interplay between the retinoblastoma protein and LEK1 specifies stem cells toward the cardiac lineage. EMBO J. 24, 1750-1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. M. (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell. Biol. 7, 644-656 [DOI] [PubMed] [Google Scholar]
- Pfleger C. M., Kirschner M. W. (2000). The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14, 655-665 [PMC free article] [PubMed] [Google Scholar]
- Pines J., Hunter T. (1991). Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J. Cell Biol. 115, 1-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylypenko O., Schonichen A., Ludwig D., Ungermann C., Goody R. S., Rak A., Geyer M. (2008). Farnesylation of the SNARE protein Ykt6 increases its stability and helical folding. J. Mol. Biol. 377, 1334-1345 [DOI] [PubMed] [Google Scholar]
- Rape M., Reddy S. K., Kirschner M. W. (2006). The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell 124, 89-103 [DOI] [PubMed] [Google Scholar]
- Rattner J. B., Rao A., Fritzler M. J., Valencia D. W., Yen T. J. (1993). CENP-F is a.ca 400 kDa kinetochore protein that exhibits a cell-cycle dependent localization. Cell Motil. Cytoskeleton 26, 214-226 [DOI] [PubMed] [Google Scholar]
- Robertson J. B., Zhu T., Nasreen S., Kilkenny D., Bader D., Dees E. (2008). CMF1-Rb interaction promotes myogenesis in avian skeletal myoblasts. Dev. Dyn. 237, 1424-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer-Hales K., Iaconelli J., Snyder J. P., Prussia A., Nettles J. H., El-Naggar A., Khuri F. R., Giannakakou P., Marcus A. I. (2007). Farnesyl transferase inhibitors impair chromosomal maintenance in cell lines and human tumors by compromising CENP-E and CENP-F function. Mol. Cancer Ther. 6, 1317-1328 [DOI] [PubMed] [Google Scholar]
- Shu T., Ayala R., Nguyen M. D., Xie Z., Gleeson J. G., Tsai L. H. (2004). Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron 44, 263-277 [DOI] [PubMed] [Google Scholar]
- Sorensen C. S., Lukas C., Kramer E. R., Peters J. M., Bartek J., Lukas J. (2000). Nonperiodic activity of the human anaphase-promoting complex-Cdh1 ubiquitin ligase results in continuous DNA synthesis uncoupled from mitosis. Mol. Cell. Biol. 20, 7613-7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmuller J., Bonni A. (2005). Moving past proliferation: new roles for Cdh1-APC in postmitotic neurons. Trends Neurosci. 28, 596-601 [DOI] [PubMed] [Google Scholar]
- Stehman S. A., Chen Y., McKenney R. J., Vallee R. B. (2007). NudE and NudEL are required for mitotic progression and are involved in dynein recruitment to kinetochores. J. Cell Biol. 178, 583-594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V., Ganoth D., Dahan A., Heller H., Hershko J., Luca F. C., Ruderman J. V., Hershko A. (1995). The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 6, 185-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S. S., Hussein D., Wang Y., Elderkin S., Morrow C. J. (2001). Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J. Cell Sci. 114, 4385-4395 [DOI] [PubMed] [Google Scholar]
- Tighe A., Johnson V. L., Taylor S. S. (2004). Truncating APC mutations have dominant effects on proliferation, spindle checkpoint control, survival and chromosome stability. J. Cell Sci. 117, 6339-6353 [DOI] [PubMed] [Google Scholar]
- Troutman J. M., Roberts M. J., Andres D. A., Spielmann H. P. (2005). Tools to analyze protein farnesylation in cells. Bioconjug Chem. 16, 1209-1217 [DOI] [PubMed] [Google Scholar]
- Vergnolle M. A., Taylor S. S. (2007). Cenp-F links kinetochores to Ndel1/Nde1/Lis1/dynein microtubule motor complexes. Curr. Biol. 17, 1173-1179 [DOI] [PubMed] [Google Scholar]
- Visintin R., Craig K., Hwang E. S., Prinz S., Tyers M., Amon A. (1998). The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell 2, 709-718 [DOI] [PubMed] [Google Scholar]
- Wolthuis R., Clay-Farrace L., van Zon W., Yekezare M., Koop L., Ogink J., Medema R., Pines J. (2008). Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol. Cell 30, 290-302 [DOI] [PubMed] [Google Scholar]
- Yang Z., Guo J., Chen Q., Ding C., Du J., Zhu X. (2005). Silencing mitosin induces misaligned chromosomes, premature chromosome decondensation before anaphase onset, and mitotic cell death. Mol. Cell. Biol. 25, 4062-4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W., Schwab M., Nasmyth K., Seufert W. (1998). Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 282, 1721-1724 [DOI] [PubMed] [Google Scholar]
- Zhou X., Wang R., Fan L., Li Y., Ma L., Yang Z., Yu W., Jing N., Zhu X. (2005). Mitosin/CENP-F as a negative regulator of activating transcription factor-4. J. Biol. Chem. 280, 13973-13977 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}