

Abstract
A new visualizing method was developed for monitoring protein-protein (P-P) interactions in live mammalian cells. P-P interactions are visualized by directing localization of a bait protein to endosomes. This method is sufficiently robust to analyze signal-dependent P-P interactions such as calcium-dependent protein interactions. We visualized interactions between activated calmodulin and calmodulin-binding proteins, and observed oscillatory interactions via time-lapse imaging. In addition, this new method can simultaneously monitor multiple P-P interactions in a single live cell, which allows comparison of interactions between several prey proteins and a single bait protein. We observed that CaMKK1 and CaMKIIα bind calmodulin with distinct binding affinities in live cell, which indicates that calcium signaling is fine-tuned by distinct activation patterns of CaM kinases. This method will enable investigation of cellular processes based on dynamic P-P interactions.
Keywords: calmodulin-dependent kinase, live cell imaging, Protein-Protein interaction, calcium, small GTPase
Analyzing protein-protein (P-P) interactions is a fundamental step toward understanding cellular processes such as proliferation, differentiation, migration, and apoptosis (1). Recently, several imaging methods have been developed for monitoring P-P interactions in live mammalian cells, including Bimolecular Fluorescence Complementation (BiFC) and FRET (2–4). Although these techniques are very useful, they require optimization of the proximity between binding partners and the orientation of fusion proteins. Therefore, such studies require a variety of fusion proteins that take into consideration the relative locations of the fluorescence and binding proteins (bait or prey), as well as the linker sequence (3, 5, 6). Moreover, BiFC is not appropriate for detecting dynamic P-P interactions that occur within minutes, since a long maturation time (approximately 1 h) is required to form the fluorophore product as readout.
Calcium signaling is involved in many important cellular functions including the cell cycle, apoptosis, and gene expression (7). In resting cells, cytosolic Ca2+ levels are approximately 10,000-fold lower than extracellular Ca2+. Thus, Ca2+ functions as a second messenger in response to various extracellular stimuli, which promote the influx of extracellular Ca2+ or its release from intracellular compartments such as the endoplasmic reticulum (8). Ca2+ is released in elemental aliquots called sparks, which have a specific magnitude and frequency depending upon the extent of the Ca2+ signal, and decoding proteins such as calmodulin translate this code of Ca2+ sparks into a cellular signal (9). To decode the signal and initiate the calcium signaling cascade, Ca2+-bound calmodulin undergoes a conformational change that induces interaction with various calmodulin-binding proteins, which in turn relay the signal (10). Calmodulin-dependent kinase IIα (CaMKIIα), one of the most studied calmodulin-binding proteins, is a multifunctional protein kinase that exists as a dodecameric complex; it decodes Ca2+ spikes to regulate various cellular processes (11, 12). Recently, CaMKIIα has been shown to function as a key regulator in synaptic plasticity, learning, and memory (13, 14). Despite their importance, dynamic interactions between calmodulin and CaMKIIα have not been visualized, primarily because the calmodulin response is both rapid and reversible. Elevated Ca2+ levels can also enable calmodulin binding to other CaM kinases (15, 16), which may exert contradictory effects. For example, although calmodulin-dependent kinase IV (CaMKIV) and CaMKIIα bind to the same site in calmodulin, they exert opposing effects, which can be explained by differences in phosphorylation (17, 18). However, the mechanisms by which two kinases are regulated by a single stimulus remain to be studied. Therefore, visualizing such interactions in live cells will be crucial to elucidating the underlying complexities of calcium signaling.
In this study, we set out to design a method for visualizing reversible calcium-dependent P-P interactions. In addition, we devised an approach that used simple cloning strategies for plasmid construction. In order to achieve this goal, it must be possible to investigate multiple prey proteins interacting with a single bait protein, either competitively or simultaneously. Following this conceptual design, we developed a localization-based method for detecting P-P interactions using endosomal localization as the readout. Endosomes can be categorized into early, recycling, and late endosomes, and they function as cellular storage compartments and transporters of organic molecules (19). Since cytosolic-located endosomes recruit limited signaling proteins compared to the plasma membrane, where most signal transduction pathways initiate, this subcellular compartment can function as an appropriate platform for localization-based imaging analysis of P-P interactions.
Results
Proof of Concept.
We designed an assay that would allow visualization of interacting proteins recruited to the endosome. The assay uses two protein constructs, an endosome-localized bait and a cytosolic prey that colocalizes at the endosome due to signal-dependent P-P interactions. Constitutively active Rab5, which localizes to early endosomes and enhances their size and number, was employed as an “anchor” to the endosomes (20) (SI Text Fig. 1). To verify the concept, we examined colocalization between FKBP12-Rapamycin Binding protein (FRB) and FK506 Binding protein (FKBP), which form a heterodimer following induction by rapamycin (21) (Fig. 1A). The fluorescent fusion-proteins FRB-mCherry-Rab5 (red) and FKBP-EGFP (green) were coexpressed in HeLa cells. In the absence of induction, FRB localized to the enlarged endosomes, while FKBP-EGFP remained dispersed throughout the cytoplasm. As expected, treatment with rapamycin induced immediate FKBP-EGFP recruitment to endosomes. Since FRB-mCherry-Rab5 also locates to the surface of endosomes, this colocalization phenomenon generated the appearance of a “circle” that resembles a total eclipse (Fig. 1B, Movie S1). Therefore, this method was named ECLIPSE, i.e., Emerging CircLe of Interactive Proteins at Specific Endosomes. Quantitative analysis of colocalization indicated that recruitment began as an immediate response to rapamycin induction and became saturated within about 100 sec. (Fig. 1C).
Fig. 1.

The ECLIPSE concept. (A) Schematic diagram showing two-hybrid ECLIPSE assay. (B) In response to rapamycin, FKBP was recruited to the endosome where FRB was already located. (C) Quantitative analysis of colocalization between FKBP and FRB on selected endosomes (n = 5). Rapamycin induction was performed at time = 0 min. (D) Schematic diagram of a three-hybrid ECLIPSE assay showing the Cdc42-WASP interaction as a case of constitutive P-P interaction. (E) ECLIPSE shows binding between constitutively active (GTP) Cdc42 and the CRIB domain of WASP, but no binding between the CRIB domain and the Cdc42 dominant-negative (GDP) form. (F) Quantitative analysis of Cdc42h-WASP interactions between WASP and a constitutively active form of Cdc42 or a dominant-negative form of Cdc42. Colocalization was measured for 5 min after induction. RFU were normalized against the fluorescence before induction (n = 8 for GTP-Cdc42; n = 4 for GDP-Cdc42; error bars indicated S.E.).(Scale bars,10 μm). Abbreviations: R, mCherry; IM, ionomycin.
As rapamycin induction of FKBP-FRB heterodimerization could be clearly visualized in live mammalian cells, we examined use of the ECLIPSE method as a monitoring tool for constitutive P-P interactions. We tested a three-hybrid format that comprised the well-known binding partners Wiskott-Aldrich Syndrome Protein (WASP) and Cdc42 (22), as well as the inducible FKBP-rapamycin-FRB complex, in which Rab5-fused FRB functioned as the endosomal anchor binding the FKBP-conjugated bait, which in turn recruited cytosolic prey. The Cdc42/Rac-Interactive Binding (CRIB) domain of WASP (the bait) was fused to a tandem FKBP repeat, generating bait that could be recruited to the endosome by induction with rapamycin. A fusion was generated between EYFP (yellow fluorescent protein) and Cdc42 from which the CAAX motif was deleted, so that the fusion protein localized in the cytoplasm as prey. For ease of visualization, all three interacting proteins, i.e., the Rab5-FRB anchor, FKBP-WASPCRIB and Cdc42, were fused to different colors of fluorescent protein (mCherry, ECFP, and EYFP, respectively; Fig. 1D). After rapamycin induction, constitutively active Cdc42 (GTP-bound form) moved to the endosome and formed “Circles” where the CRIB domain was located; the dominant-negative form of Cdc42 (GDP-bound form) very weakly appeared to colocate (Fig. 1E, F). These results show clearly that constitutively active Cdc42 binds the CRIB domain of WASP, whereas a dominant-negative form of Cdc42 has very low binding-affinity (22, 23). Thus, we confirmed that ECLIPSE represents an effective method for monitoring constitutive P-P interactions in live mammalian cells.
Detection of Signal-Dependent Interactions.
We used ECLIPSE to detect calcium-dependent interactions between calmodulin as bait (fused to the FKBP tandem repeat and mTFP, teal fluorescence protein) and CaMKIIα as prey (fused to YFP). In the absence of induction, FRB localized to the enlarged endosomes, while FKBP-conjugated calmodulin and CaMKIIα remained dispersed throughout the cytoplasm. The endosomal “circles” of CaMKIIα emerged upon addition of both rapamycin and ionomycin, an ionophore that elevates cytosolic Ca2+ levels, which induce interactions between calmodulin and CaMKIIα. Neither ionomycin nor rapamycin alone was able to induce localization of CaMKIIα to endosomes (Fig. 2A, B). Consequently, these results confirm that ECLIPSE is a robust imaging assay for analyzing signal-dependent P-P interactions.
Fig. 2.

Monitoring signal-dependent interactions using a three-hybrid format. (A) A three-hybrid ECLIPSE showing that interaction between calmodulin and CaMKIIα is dependent upon cytosolic Ca2+ levels. Images were captured before and after induction with ionomycin, rapamycin, or both agents. (B) Quantitative analysis showing that both rapamycin and ionomycin are required for endosomal localization of CaMKIIα (n = 1). RFU were normalized against cell fluorescence before induction. (Scale bars,10 μm). Abbreviations: R, mCherry; IM, ionomycin.
While monitoring signal-dependent P-P interactions, we observed that the free cytosolic levels of CaMKIIα decreased as endosomal levels increased (Fig. 2A). However, it also appeared that cytoplasmic levels recovered slightly after reaching a minimum (Fig. S2), from which ECLIPSE might detect the reversible interaction of the calmodulin-CaMKIIα.
To investigate the reversibility of calcium-dependent interactions further, we simplified the monitoring system to a two-hybrid format, employing the calmodulin-Rab5 fusion protein (CaM-mCherry-Rab5) to reduce the assay complexity. This simpler design enabled diffusion of CaMKIIα after it dissociated from calmodulin. The Rab5-calmodulin fusion protein clearly localized to endosomes (Fig. 3A). In cells that expressed the Rab5-calmodulin fusion, the endosomes were smaller and concentrated in the perinuclear region, which provided a high intensity of fluorescence and, thus, a more reliable readout for assays. Additionally, the transformation of endosomes was revealed not to affect the cell viabilities (Fig. S3).
Fig. 3.

Monitoring signal-dependent interactions with ECLIPSE. (A) Two-hybrid ECLIPSE showing the dynamic and reversible interactions between calmodulin and CaMKIIα. (B) The reversible interaction between calmodulin and CaMKIIα was confirmed by line profiling at three time points. (C) Quantitative analysis of endosomal localization between CaM kinase IIα (YFP-CaMKIIα) and a calmodulin-Rab5 fusion (CaM-mCherry-Rab5) in HeLa cells. RFU were normalized against the fluorescence before induction (n = 1). Abbreviations: R, mCherry; IM, ionomycin.
The reversible interaction between calmodulin and CaMKIIα could be observed clearly by enhancing cytosolic Ca2+ levels (Fig. 3A) and quantitative analysis of colocalization confirmed these findings (Fig. 3B, C, and Movie S2). Endosomal-localized CaMKIIα levels were enhanced by elevated cytosolic Ca2+ levels, but this localization began to dissipate after 5 min. This spontaneous dissociation indicates that the interaction between calmodulin and CaMKIIα is reversible. These results were validated by using impaired binding partners, which did not show colocalization (Fig. S4). Moreover the calcium levels in the cytosol and the endosomal region, where the calmodulin and CaMKIIα interact, increased in the same manner by ionomycin (Fig. S5B). Thus, it appears that the simpler two-hybrid format is more appropriate for monitoring reversible interactions than the three-hybrid assay.
Detection of Oscillatory Interactions Between Calmodulin and CaMKIIα.
For further characterization of CaMKIIα recruitment, the interactions between calmodulin and CaMKIIα were monitored over a longer period of time, i.e., more than 30 min. We observed an interesting pattern in four of 15 independent experiments. After addition of ionomycin, the magnitude of CaMKIIα recruitment increased in four separate oscillations (Fig. 4A, Movie S3). Immediately after Ca2+induction, the first weak peak (1.5-fold increase) was observed, and then the second and third peaks showed 2- and 2.5-fold increases, respectively (Fig. 4B). This oscillatory interaction has been suggested previously in the decoding model, which predicts that calmodulin mediated activation of CaMKIIα will be dependent upon the frequency of Ca2+ oscillation (22). This interaction pattern could occur if there were higher binding than dissociation rates between calmodulin and CaMKIIα, and the increasing interactions caused by the high frequency and magnitude of Ca2+ spikes may have resulted in this situation.
Fig. 4.

ECLIPSE monitoring of an oscillatory interaction. (A) The oscillatory interaction between calmodulin and CaMKIIα was validated via ECLIPSE: * indicates the 1st peak. Images were captured at 20 sec. intervals for 30 min. (Scale bars,10 μm). (B) Quantitative analysis confirmed that the oscillatory interaction between calmodulin and CaMKIIα strengthened over time. RFU were normalized against the fluorescence before induction (n = 1). Abbreviations: R, mCherry; IM, ionomycin.
Detection of Simultaneous Interactions in a Single Live Cell.
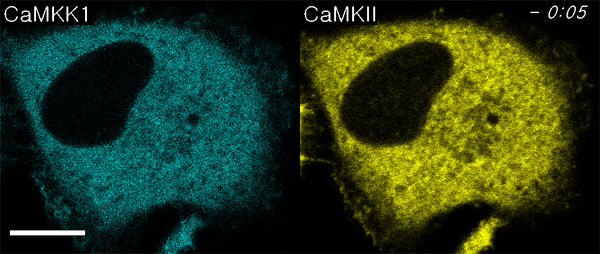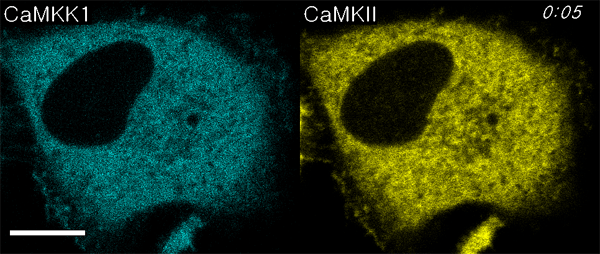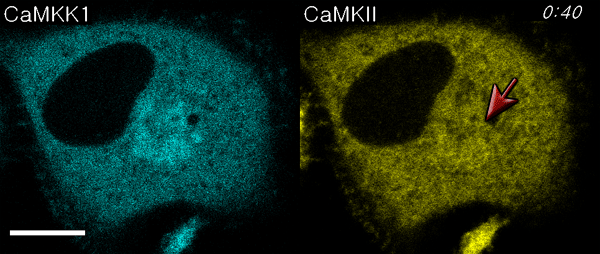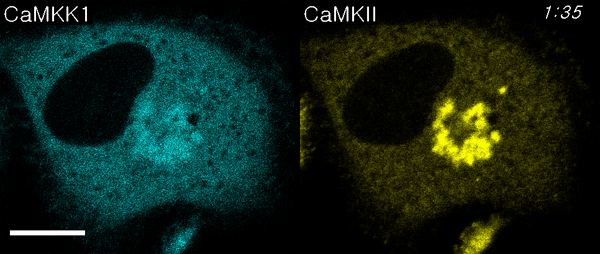
We next used ECLIPSE to visualize interactions between calmodulin (bait) and two prey species, CaM kinase kinase 1 (CaMKK1) and CaMKIIα, in a single cell. Like CaMKIIα, CaMKK1 is a well-known calmodulin-binding partner (24, 25). Surprisingly, this assay not only demonstrated binding to calmodulin at the endosomes, but also sequential recruitment of CaMKK1 and CaMKIIα to the endosomes (Fig. 5A, Movie S4). This sequential recruitment was validated by independent experiments. Quantitative analyses clearly indicated that the two kinases showed different initial rates of colocalization, and CaMKK1 bound calmodulin faster than CaMKIIα (Fig. 5B). It has been reported that CaMKK1 activates CaMKVI and not CaMKIIα (10). Therefore, this result might reflect the distinct binding affinities of two kinases that bind calmodulin independently. In contrast, when ECLIPSE was performed using CaMKVI in place of CaMKIIα, CaMKVI and CAMKK1 were recruited simultaneously to the calmodulin at endosomes (Fig. S6). These results could be caused from the difference of protein bulkiness; CaMKK1 is monomeric while CaMKIIα is dodecamic. Because the bulkiness of proteins can affect the mobility of themselves, an alternative test with the FKBP-conjugated CaM kinases was performed (Fig. S7). In these experiments, the endosomal localization of CaM kinase was controlled by the FRB-Rapamycin-FKBP interaction, not by the calmodulin-CaM kinase interactions. Therefore the mobility of monomeric CaMKK1 and dodecamic CaMKIIα was compared under the same force heading to endosomes. Both monomeric CaMKK1 and dodecamic CaMKIIα showed the same initial timing of colocalization to the endosome, implying that the difference of protein bulkiness may not be a critical factor for the translocation timing in the case of the two kinases. Consistently, it had been reported that the particles of from 10 to 100 Å in radius have indistinguishable diffusion rate in cytoplasm (26).
Fig. 5.

Comparison of CaM kinase calmodulin-binding affinities. (A) Interactions between bait (calmodulin) and two prey molecules (CaMKK1 and CaMKIIα) were monitored in a single cell using ECLIPSE: ** and *** indicate the first endosomal localizations of CaMKK1 and CaMKIIα, respectively. (B) CaMKK1 and CaMKIIα exhibit distinct responses to elevated Ca2+ levels. (C) Interaction rates between calmodulin and CaM kinases (CaMKIIα and CaMKK1) at various concentrations of ionomycin in HeLa cells. Interactions were measured by ECLIPSE assay. The addition of ionomycin was considered to be time = 0 min. RFU were normalized against the fluorescence before induction (n = 5). Abbreviations: R, mCherry; IM, ionomycin.
To validate the distinct calmodulin-binding affinities of CaMKIIα and CaMKK1, we next compared the binding patterns of calmodulin and CaM kinases at various concentrations of ionomycin. Increasing ionomycin concentrations corresponded to increasing cytosolic Ca2+ levels, which were measured with the Ca2+-chelating dye Fluo-4 (Fig. S5A). It is likely that the increased interactions between calmodulin and CaMKIIα were indeed due to increased cellular Ca2+ levels. We monitored the effects of varying intracellular Ca2+ levels on P-P interactions in the cell using the ECLIPSE method. As expected, the magnitude and duration of interactions increased with increasing ionomycin concentration (Fig. 5C). Additionally, the recruitment of CaMKK1 was detected at lower cytosolic Ca2+ levels than required for CaMKIIα, which demonstrates that the calmodulin-CaMKK1 interaction is more sensitive to cytosolic Ca2+ levels than calmodulin-CaMKIIα. Taken together, these results indicate that CaMKK1 binds calmodulin more rapidly than CaMKIIα, owing to its higher sensitivity to conformational change of calmodulin by elevated cytosolic Ca2+ level.
Discussion
In this study, we introduced ECLIPSE as a simple method for monitoring dynamic P-P interactions in live mammalian cells, especially calcium-dependent interactions. As the design requires less consideration of component structures and proximities, it may be performed more easily than other visualizing methods. Moreover, we confirmed the value of this simple method for detecting simple signal-dependent interactions, as well as visualizing oscillatory interactions between calmodulin and CaMKIIα (Fig. 3, 4). In addition, we were able to compare the binding affinities of different calmodulin-dependent kinases for calmodulin (Fig. 5).
It has been proposed that calmodulin and CaMKIIα may exhibit an oscillatory interaction, since kinase activity is dependent upon the frequency of Ca2+ spikes (12, 16). According to the decoding hypothesis, the rate of association between CaMKIIα and calmodulin increases following an elevated frequency of Ca2+ spikes. However, the dissociation rate remains constant and new associations can begin before dissociation ends completely (12, 16). But the oscillatory interaction has not been shown under low frequency Ca2+ spikes. The oscillatory interactions of CaMKII proposed by the decoding hypothesis, has not been confirmed. However, as suggested by the decoding hypothesis, our results have shown the existence of an oscillating interaction pattern between CaMKIIα and calmodulin under native cellular conditions.
Understanding the different binding affinities of proteins is important for elucidating complex cellular processes. For example, in a situation where one protein is activated by an upstream signal and then regulates at least two downstream proteins, it can be difficult to understand the signaling steps accurately without understanding the binding affinity or sensitivity of regulators that control the interactions. Notably, by employing one bait protein and two prey proteins, our method can distinguish two different P-P interactions in a single cell. In this study, we compared the binding kinetics of CaMKIIα and CaMKK1 to calmodulin. Since our localization-based method does not require consideration of proximity or orientation of fusion proteins, we could use calmodulin (one bait) and two different calmodulin-dependent kinases (two prey) to compare prey binding affinities in a single live cell.
CaM kinases are known as regulators of gene expression, such as cAMP Response Element Binding protein (CREB)-mediated transcription. In CREB-mediated transcription, the c-fos promoter, one of first identified promoters regulated by CREB, is regulated by CaMKI, IV and II (15). Interestingly, CaMKI and IV can stimulate the expression of the c-fos gene, while CaMKIIα inhibits transcription by phosphorylation of CBP and decreasing binding affinity for CREB (17, 18, 27). How does one signal, such as elevated level of cytosolic Ca2+ stimulate and inhibit expression of the same gene? Comparison of binding affinity can help to answer this question. We confirmed that CaMKK1 has a higher affinity for calmodulin than CaMKIIα by visualizing sequential recruitment to calmodulin located on endosomes. Moreover, we observed that CaMKIV and CaMKK1 were recruited onto endosomes at the same time (Fig. S6). Experiments showed that the calmodulin-binding affinities of CaMKI and CaMKIV are higher than CaMKII (28–30). CaMKK1 is known as an activator of CaMKI and IV, but not CaMKII (25). Therefore, it has been suggested that CaMKIV becomes activated more rapidly than CaMKII in live cells (15). This would result in stimulation of CREB-mediated transcription prior to inhibition by the CaMKIIα-regulated mechanism, providing a fine control of gene expression. Thus, the ability to compare binding affinity in live cells will contribute to our understanding of intriguing signaling mechanisms, such as calcium signaling.
In this study, we showed that interactions between two prey proteins and a bait protein can be performed in the same cell. Additional prey proteins may be employed by removing the fluorescent protein from a bait protein and by means of stable cell lines. Moreover, comparing different combinations of prey proteins may enable determination of an order of relative priority for each prey protein interacting with the same bait protein. This type of application could also provide valuable insights into the assembly of protein complexes, such as signaling complexes based on scaffold proteins and hetero-oligomeric complexes.
Since this method is based on native endosomes, proteins that localize normally to this subcellular compartment are not appropriate as prey proteins. However, if endosomal localization is controlled by a specific localization motif, removal of the motif can allow controlled endosomal localization of proteins to be studied. Indeed, we demonstrated that ECLIPSE could be used to examine the interaction between Cdc42 and WASP by deleting the Cdc42 CAAX peptide, which is a localization motif for the plasma membrane and endosomes (Fig. 1E, F). Moreover, utilization of other subcellular compartments, such as the Golgi apparatus or other types of endosomes, will enlarge the spectrum of P-P interactions that can be investigated using ECLIPSE.
In this paper, two kinds of ECLIPSE assay, three-hybrid format using rapamycin as inducible system and two-hybrid format for reversible interaction, were demonstrated. In the three-hybrid format, free bait and prey proteins interacted in the cytoplasm, and then moved to endosomes by the addition of rapamycin. The three-hybrid format of ECLISPE assay is valuable to detect various constitutive P-P interactions because false-positive readouts are excluded by the inducible translocation to the endosome through rapamycin. However, the kinetics of P-P interaction may not be analyzed by the three-hybrid format. On the other hand, in two-hybrid format to observe the dynamic interactions including reversible interaction, the kinetics of P-P interaction can be analyzed because endosomal localization is induced by signal-dependent P-P interaction. Moreover comparison of kinetics of two different P-P interactions is able to be performed by two-hybrid format. Therefore, the simpler two-hybrid format is more appropriate for monitoring reversible P-P interactions and comparing two different P-P interactions than the three-hybrid assay.
In conclusion, ECLIPSE is an effective tool for monitoring dynamic P-P interactions, especially signal-dependent interactions in live cells. For example, calcium-dependent oscillatory interactions between calmodulin and CaMKIIα were visualized via ECLIPSE and this method can concomitantly monitor multiple interactions in a single live cell. This feature makes it possible to compare the binding affinities of different prey for the same bait protein and to monitor the real-time assembly of signaling complexes. Although this assay is unlikely to be effective for the investigation of endosomal proteins, ECLIPSE represents an innovative method for monitoring transient and reversible P-P interactions. The removal of localization motifs like the CAAX peptide of Cdc42, and the utilization of other subcellular compartments, will increase the types of interactions that can be investigated in future studies.
Materials and Methods
Vector Construction.
Sequences encoding FRB and FKBP were amplified by PCR and then cloned into pmCherry-C1 and pECFP-N1 (Clonetech Inc.) to generate pFRB-mCherry and pFKBPx2-CFP, respectively. Sequence encoding FRB-mCherry was then digested with NheI and BsrGI, and inserted into DNA encoding CFP-RAB5BQ79L, which was obtained from a small GTPase library (20, 31); the resulting construct was pFRB-mCherry-Rab5. DNA encoding the CRIB domain of WASP was PCR-amplified from a human cDNA library and digested with XhoI and BamHI. The fragment was then ligated into the corresponding sites in DNA encoding the C-terminal region of FKBPx2-ECFP, which generated the expression construct pFKBPx2-ECFP-WASP (CRIB). Plasmids expressing YFP fused to constitutively active Cdc42Q61L or the dominant-negative Cdc42T17N were obtained from a small GTPase library (20). The construct pFKBPx2-mTFP was generated by replacing DNA encoding ECFP with mTFP. pNCS-mTFP1 (Allele Biotechnology) was used as a template for PCR amplification of DNA encoding mTFP. The calmodulin expression vectors FKBP-TFP-CaM and CaM-mCherry-Rab5 were generated by PCR amplification from a human cDNA library. All plasmids were prepared using DNA-Spin™ plasmid purification kit (iNtRON Biotechnology). The expression vectors encoding CaM kinases, including CaM kinase IIα, CaM kinase IV, and CaM kinase kinase 1, originated from the Alliance For Cellular Signaling (AFCS) ID: A000456, A000461, and A000462, respectively. PCR primers used for generating these constructs are listed in Table S1.
Cell Culture and Electroporation.
HeLa cells were maintained in DMEM (#11955-065) containing 10% FBS (#10437-028) at 37 °C and 5% CO2. Both reagents were purchased from Invitrogen Life Science Inc. Electroporation was performed using a Microporator™ (MP-100; Digital Bio Technology) following the manufacturer’s instructions. The optimized conditions for HeLa cells were 2 pulses of 1080 V for 35 milliseconds. For cell imaging, shocked cells were aliquotted into 96-well black plates with glass-bottoms (Metrical Inc.) and, after 24 h, cells were used for assays.
ECLIPSE.
Twenty-four hours after electroporation, media from transfected cells were replaced by prewarmed Opti-MEM® Reduced-Serum Medium (#31985-062; Invitrogen Life Technologies). For induction, stock concentrations of 500 μM rapamycin (B0395) and 1 mM ionomycin (I9657) (Sigma Life Science) were diluted 500-fold in Opti-MEM® Reduced-Serum Medium for x2 induction media. Both chemical agents were dissolved in DMSO as x1000 stocks and stored at -80 °C. Unless stated otherwise, media to induce bait movement contained 500 nM of rapamycin or 1 μM of ionomycin.
Measurement of Cytosolic Calcium Levels.
HeLa cells were incubated with serum free DMEM containing 1 μM of Fluo-4 AM ester or Fluo-3 AM ester at 37 °C. Fluo-4 AM ester (F14201) and Fluo-3 AM ester (F1201) were purchased from Invitrogen Life Science Inc. Cells were incubated for 1 h, washed twice with PBS, and then resuspended in OPTI-MEM before imaging. Cells were treated with various concentrations of ionomycin (0.125–2 μM) in order to enhance cytosolic Ca2+ levels.
Cell Imaging.
Cell imaging was performed using a confocal microscope (A1R, Nikon). Each image was captured with a CFI Plan Apochromat VC objective lens (60×/1.40 Oil) at a resolution of 512 × 512 using digital zooming. During two-hybrid assays, two color images were captured at the same time when single image and time-lapse images were obtained. In three-hybrid assays, red fluorescence, i.e., mCherry, was captured first to focus correctly on the endosomes within cells, and then cyan and yellow fluorescence images were captured at the same focal distance. For time-lapse imaging, cyan and yellow fluorescence images from bait and prey were captured at the same time, but red fluorescence images were only obtained before and after induction. All images were stored as JPG2000 or ND files, which are standard file formats for Nikon A1R confocal microscope.
Image Analysis.
Cell images were used for quantitative analysis, ratio view and video-making. These processes were performed with Nikon imaging software (NIS-element AR 64-bit version 3.00, Laboratory Imaging) and MetaMorph offline version 7.6.0.0, MDS Analytical Technologies. Image file formats were transferred from ND and JPG2000 files to ICS and TIFF formats, respectively, using NIS-element software. Quantitative analysis of endosomal colocalization was performed using the “Time-measurement” tool for “Region Of Intensity” (ROI) in the NIS-element software, or the “Total measurement” tool for the ROI in the MetaMorph software. After ROIs were defined according to localization of Rab5b-chimera proteins, localization of other components was measured using the defined ROIs for both analytic programs. Relative Fluorescence Units (RFU) were normalized against the initial intensity of each ROI prior to treatment, and then plotted using Microsoft® Office Excel 2007. Ratio view and videos were obtained using MetaMorph software.
Supplementary Material
Acknowledgments.
We thank professors Daesoo Kim, Daeyoup Lee and Walton Jones (KAIST, Korea) for helpful discussions and reading this manuscript. This research was supported by Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program and the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science, and Technology (M10871040001-08N7104-00110).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911262107/DCSupplemental.
References
- 1.Stelzl U, et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell. 2005;122(6):957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 2.Shyu YJ, Suarez CD, Hu CD. Visualization of AP-1 NF-kappaB ternary complexes in living cells by using a BiFC-based FRET. Proc Natl Acad Sci U S A. 2008;105(1):151–156. doi: 10.1073/pnas.0705181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1(3):1278–1286. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eyckerman S, et al. Reverse MAPPIT: Screening for protein-protein interaction modifiers in mammalian cells. Nat Methods. 2005;2(6):427–433. doi: 10.1038/nmeth760. [DOI] [PubMed] [Google Scholar]
- 5.Kolossov VL, et al. Engineering redox-sensitive linkers for genetically encoded FRET-based biosensors. Exp Biol Med (Maywood) 2008;233(2):238–248. doi: 10.3181/0707-RM-192. [DOI] [PubMed] [Google Scholar]
- 6.Ha JS, et al. Design and application of highly responsive fluorescence resonance energy transfer biosensors for detection of sugar in living Saccharomyces cerevisiae cells. Appl Environ Microbiol. 2007;73(22):7408–7414. doi: 10.1128/AEM.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1(1):11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 8.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: Dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 9.Chin D, Means AR. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000;10(8):322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 10.Hoeflich KP, Ikura M. Calmodulin in action: Diversity in target recognition and activation mechanisms. Cell. 2002;108(6):739–742. doi: 10.1016/s0092-8674(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 11.Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron. 1994;12(5):943–956. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- 12.Soderling TR, Chang B, Brickey D. Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2001;276(6):3719–3722. doi: 10.1074/jbc.R000013200. [DOI] [PubMed] [Google Scholar]
- 13.Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6(4):267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- 14.Soderling TR. CaM-kinases: Modulators of synaptic plasticity. Curr Opin Neurobiol. 2000;10(3):375–380. doi: 10.1016/s0959-4388(00)00090-8. [DOI] [PubMed] [Google Scholar]
- 15.Hook SS, Means AR. Ca(2 + )/CaM-dependent kinases: From activation to function. Annu Rev Pharmacol Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- 16.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364(Pt 3):593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matthews RP, et al. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14(9):6107–6116. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8(21):2527–2539. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- 19.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2(2):107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 20.Heo WD, et al. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314(5804):1458–1461. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Zheng XF, Brown EJ, Schreiber SL. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci U S A. 1995;92(11):4947–4951. doi: 10.1073/pnas.92.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung DW, Rosen MK. The nucleotide switch in Cdc42 modulates coupling between the GTPase-binding and allosteric equilibria of Wiskott-Aldrich syndrome protein. Proc Natl Acad Sci U S A. 2005;102(16):5685–5690. doi: 10.1073/pnas.0406472102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leung DW, Otomo C, Chory J, Rosen MK. Genetically encoded photoswitching of actin assembly through the Cdc42-WASP-Arp2/3 complex pathway. Proc Natl Acad Sci U S A. 2008;105(35):12797–12802. doi: 10.1073/pnas.0801232105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oldroyd GE, Downie JA. Calcium, kinases, and nodulation signalling in legumes. Nat Rev Mol Cell Biol. 2004;5(7):566–576. doi: 10.1038/nrm1424. [DOI] [PubMed] [Google Scholar]
- 25.Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24(6):232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson K, Wojcieszyn J. The Translational Mobility of Substances within the Cytoplasmic Matrix. P Natl Acad Sci-Biol. 1984;81(21):6747–6751. doi: 10.1073/pnas.81.21.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheng M, Greenberg ME. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4(4):477–485. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- 28.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256(5060):1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 29.Cruzalegui FH, Means AR. Biochemical-Characterization of the Multifunctional Ca2+/Calmodulin-Dependent Protein-Kinase Type-Iv Expressed in Insect Cells. J Biol Chem. 1993;268(35):26171–26178. [PubMed] [Google Scholar]
- 30.Miyano O, Kameshita I, Fujisawa H. Purification and characterization of a brain-specific multifunctional calmodulin-dependent protein kinase from rat cerebellum. J Biol Chem. 1992;267(2):1198–1203. [PubMed] [Google Scholar]
- 31.Heo WD, Meyer T. Switch-of-function mutants based on morphology classification of Ras superfamily small GTPases. Cell. 2003;113(3):315–328. doi: 10.1016/s0092-8674(03)00315-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}