

Abstract
Microglial activation is common in several neurodegenerative disorders. In the present study, we used the murine BV-2 microglial cell line stimulated with γ-interferon and lipopolysaccharide to gain new insights into the effects of endogenously produced NO on mitochondrial respiratory capacity, iron regulatory protein activity, and redox-active iron level. Using polarographic measurement of respiration of both intact and digitonin-permeabilized cells, and spectrophotometric determination of individual respiratory chain complex activity, we showed that in addition to the reversible inhibition of cytochrome-c oxidase, long-term endogenous NO production reduced complex I and complex II activities in an irreversible manner. As a consequence, the cellular ATP level was decreased in NO-producing cells, whereas ATPase activity was unaffected. We show that NO up-regulates RNA-binding of iron regulatory protein 1 in microglial cells, and strongly reduces the labile iron pool. Together these results point to a contribution of NO derived from inflammatory microglia to the misregulation of energy-producing reactions and iron metabolism, often associated with the pathogenesis of neurodegenerative disorders.
Keywords: Aconitate Hydratase; antagonists & inhibitors; metabolism; Adenosine Triphosphate; Animals; Cell Line; Cell Respiration; drug effects; Cytochrome c Group; Electron Transport; physiology; Electron Transport Complex I; Electron Transport Complex II; Energy Metabolism; physiology; Enzyme Activation; Interferon-gamma; pharmacology; Intracellular Fluid; Iron; Iron-Regulatory Proteins; Iron-Sulfur Proteins; Lipopolysaccharides; Mice; Microglia; cytology; Multienzyme Complexes; NADH, NADPH Oxidoreductases; Nitric Oxide; Oxidoreductases; Protein Binding; RNA; RNA-Binding Proteins; Succinate Dehydrogenase
Keywords: iron regulatory proteins, labile iron pool, microglial cells, neurodegenerative diseases, nitric oxide, respiratory chain
INTRODUCTION
Microglial activation, oxidative stress, impairment of mitochondrial energy metabolism, and intracellular iron accumulation are features common to several neurodegenerative disorders (Beal, 1998; Hirsch, 2000; Murphy, 2000; Schapira, 1999). There is a consensus that these metabolic dysfunctions play an important part in the pathophysiology of these diseases, but their interplay is complex and difficult to define. Even though the major physiological roles of microglial cells are host defense and tissue repair by phagocytosis, when activated they can also generate high levels of inflammatory cytokines, nitric oxide (NO) and reactive oxygen species (ROS), which may exacerbate neuronal damage (Gonzalez-Scarano and Baltuch, 1999; Nakajima and Kohsaka, 1998; Stoll and Jander, 1999). Therefore, in Alzheimer’s disease (AD), microglia proliferate around the pathological lesions and are believed to be important in plaque formation and neuronal degeneration (Gonzalez-Scarano and Baltuch, 1999;Lee et al., 1999; Wa et al., 1996). Microglial activation and accumulation also occur in Parkinson’s disease (PD) (McGeer et al., 1988; Hirsch, 2000) and in animal models of neurodegeneration (Calingasan et al., 1998; Kurkowska-Jastrzebska et al., 1999; Liberatore et al., 1999).
NO, a signaling molecule responsible for a large number of physiological functions, may be harmful when produced in excessive concentrations and its implication in many neurodegenerative diseases has been suggested (Dawson and Dawson, 1998; Heales et al., 1999; Hirsch, 2000; Murphy, 2000; Toreilles et al., 1999). NO and/or peroxynitrite, a strong oxidant generated from the combination of NO and O2−, inhibit(s) several mitochondrial enzymes, including complexes of the respiratory chain and the Krebs cycle enzyme aconitase (Drapier, 1997; Clementi et al., 1998; Brown, 1999). Moreover, in parallel to impairment of mitochondrial functions, a marked dysregulation of iron metabolism is also a feature common to many neurodegenerative disorders including PD, AD and Friedreich’s ataxia (FA) (Bradley et al., 2000; Delatycki et al., 1999; Hirsch and Faucheux, 1998; Rötig et al., 1997; Schapira, 1999; Smith et al., 1997; Smith et al., 1998). Strikingly, NO production is responsible for alteration of iron metabolism in many cell types, and recent results including ours pointed to a drastic change in iron regulatory protein (IRP) activity induced by NO and peroxynitrite, especially in macrophages (reviewed in Drapier et al., 2000). We consider here whether NO generated by microglial cells exposed to inflammatory stimuli alters these cells’ mitochondrial and iron metabolisms. We found that NO production is responsible for alteration of specific sites of the electron transport chain associated with reduced ATP. In addition, we show for the first time that RNA-binding of IRP-1 is up-regulated by NO in microglial cells and that their labile iron pool (LIP) is decreased in an NO-dependent manner.
MATERIALS AND METHODS
Materials
Murine recombinant γ-interferon (IFN-γ, specific activity 2 × 107 units/mg) was from R & D Systems, Abingdon, UK. The pSPT-fer plasmid containing the iron responsive element (IRE) of human ferritin H-chain was kindly provided by Dr. Lukas C. Kühn (Institut Suisse de Recherches Experimentales sur le Cancer, Epalinges, Switzerland). The highly cell permeant iron chelator salicylaldehyde-isocotinoyl-hydrazone (SIH) was a generous gift from Dr. P. Ponka (McGill University, Montreal). Low endotoxin fetal calf serum and DMEM were from Life Technologies (Les Ulis, France). Calcein-acetoxymethylester (calcein-AM) from Molecular Probes was purchased from Interchim, (Montluçon, France). The NOS-2 specific inhibitors 1400W and L-NIL were from Cayman Chemicals (Spi-Bio, Massy, France). Calcein, Escherichia coli lipopolysaccharide (LPS), and all other chemicals were from Sigma (L’Isle d’Abeau, France).
Cell culture
The BV-2 murine microglial cell line established by Blasi et al., 1990 was kindly supplied by Dr. C. Demerlé-Pallardy (Institut Henri Beaufour, Les Ulis, France). Cells were routinely cultured in DMEM containing 1 g/l glucose, sodium pyruvate, pyridoxine, glutamax, and gentamycin antibiotic, and complemented with 10% fetal calf serum (FCS). For NO synthase (NOS) induction, semi-confluent cells were treated with 20 U/ml IFN-γ and 50 ng/ml LPS for 16 h.
Preparation of mitochondrial and cytosolic fractions
Cells were detached, washed once in phosphate buffered saline (PBS) and centrifuged at 400 × g for 10 min. Cells were resuspended in 0.25 M sucrose, 40 mM KCl, 2 mM EGTA, 20 mM TRIS, pH 7.2, and lysed with 0.007% digitonin for 5 min on ice. After centrifugation at 1,800 × g for 10 min, cytosol-free pellets (3 mg protein/ml) were dried and frozen at −80°C and were referred to as the mitochondrial fraction. Collected supernatants were ultracentrifuged at 150,000 × g for 60 min and the resultant cytosolic extracts (0.5 mg protein/ml) were kept at −80°C. The lactate dehydrogenase (LDH) and cytochrome-c oxidase (COX) activities were measured spectrophotometrically as markers of the cytoplasmic and mitochondrial fractions, respectively. The LDH activity was measured by the decline of NADH oxidation at 340 nm by using a commercially available kit (LD-L10, Sigma Diagnostics). LDH activity in the mitochondrial fraction was below 5% of total LDH activity (cytoplasmic + mitochondrial fraction) indicating that very little cytoplasmic material remained in the mitochondrial fraction. Conversely, the 95% of COX activity, which was measured as described below, was found in the mitochondrial fraction.
Polarographic measurement of cellular respiration and electron transport chain (ETC) activities
Oxygen consumption was measured with a Clarke-type electrode in a Peltier-controlled chamber (Hansatech oxytherm, EUROSEP, Cergy-St-Christophe, France). Polarography was used to determine activity of specific segments of the ETC using digitonin-permeabilized cells (1×107 cells/ml) in 0.25 M sucrose, 10 mM MgCl2, 2 mM KH2PO4, 1 mM EGTA, 7% BSA, 0.005% digitonin, 20 mM HEPES pH 7.2, as previously described (Drapier and Hibbs, 1988). Appropriate substrates and inhibitors were added as indicated. Acceptor control ratio was ~3.
Spectrophotometric determination of ETC activities
The activity of each complex of the ETC was assayed spectrophotometrically in the mitochondrial fraction (30 μg protein) as previously described (Rustin et al., 1994; Trounce et al., 1996). All enzymatic activities were measured at 37°C with a SAFAS UVmc2 spectrophotometer equipped with multikinetic analysis software (SAFAS, Monaco). Complex I, NADH-ubiquinone reductase, activity was followed through the decline of NADH oxidation at 340 nm in the presence of decylubiquinone as an electron acceptor. Complex II, succinate-ubiquinone reductase, activity was determined by the reduction of the electron donor 2,6-dichloroindophenol at 600 nm in the presence of decylubiquinone and rotenone. Complex II + III, succinate-cytochrome-c reductase, activity was measured by the reduction of oxidized cytochrome-c at 550 nm in the presence of succinate as substrate. Complex IV, COX, activity was measured by the oxidation of reduced cytochrome-c at 550 nm. Complex V, ATPase, activity was determined by coupling the reaction to a pyruvate kinase and LDH system, and followed by monitoring the decline of NADH oxidation at 340 nm.
Electrophoretic mobility shift assay (EMSA)
The IRP-IRE interaction was analyzed as described (Drapier and Hibbs, 1996), by incubating 2 μg protein of cell lysates with a molar excess of 32P-CTP-labeled ferritin IRE probe in 20 μl of 10 mM HEPES (pH 7.6), 40 mM KCl, 3 mM MgCl2, and 5% glycerol. In parallel experiments, samples were treated with 2% 2-mercaptoethanol before addition of the RNA probe to allow full expression of the IRE binding activity. After a 20-min incubation at room temperature, 1 μl of RNase T1 (1 unit/μl) and 2 μl of heparin (50 mg/ml) were sequentially added for 10 min each. The IRP-IRE complexes were resolved on 6% non-denaturing polyacrylamide gels and were quantified with the IMAGEQUANT software from Molecular Dynamics (Amersham-Pharmacia Biotech, Orsay, France).
Determination of the labile iron pool (LIP)
LIP was determined by spectrofluorimetry as previously described (Epsztejn et al., 1997) with slight modifications. Briefly, 2.106 cells were resuspended in 2 ml of prewarmed 20 mM HEPES, pH 7.3, 150 mM NaCl, 1% BSA buffer, and were incubated with 50 nM calcein-AM for 15 min at 37°C. After centrifugation and washing, cells were resuspended in the same buffer, and the calcein fluorescence intensity was recorded as a function of time. A stable baseline signal was recorded for 60 sec before the addition of 100 μM SIH, a highly permeant iron chelator, which caused a rise in fluorescence corresponding to the LIP. LIP concentration was calibrated and calculated as described (Epsztejn et al., 1997). The excitation and emission wavelengths were 483 nm and 505 nm, respectively.
Cytochrome-c immunostaining
Cells were grown in four-well Lab-Tek chamber slides (Nalgen Nunc International, Napperville, IL). After drug treatment, cells were fixed and permeabilized by 50 % v/v Permeafix (Ortho, Paris, France) in water for 1 h. Then, cells were washed with PBS buffer containing 1% BSA and 5% FCS, and incubated for 1 h with anti-cytochrome-c antibody diluted 1:300 in the same buffer. Then, cells were washed again three times and incubated with FITC-conjugated mouse Ig (Dako, Copenhagen, Denmark) diluted 1:50 in the above buffer. Immunostaining was performed at room temperature.
NOS activity assay
NOS activity of BV-2 cytosolic extracts was determined by the conversion of 3H-L-arginine in 3H-L-citrulline. Briefly, protein extract (50 μg) was incubated for 30 min at 37°C with 0.4 μCi of L-(2,3-3H)-arginine (specific activity 44.2 Ci/mmol, NEN, Les Ulis, France), 20 μM L-arginine, 0.5 mM NADPH, 4 μM tetrahydrobiopterin, 2 mM magnesium diacetate, 100 mM Tris-HCl, pH 7.5. The reaction mixture was then mixed with AG50W-X8 resin (Bio-Rad, Ivry-sur-Seine, France) and the radioactivity of the synthesized L-citrulline was counted in the supernatant.
Lactate measurement
BV2 cells were cultured in DMEM + 10% FCS for 16 hours. At this time, the culture medium was removed and the incubation continued with fresh medium for 5 hours. The medium was then removed, centrifuged, and aliquots of culture medium were assayed for lactate by monitoring the oxidation of L-lactic acid to pyruvate by NAD in the presence of lactate dehydrogenase (Sigma). NADH formation was monitored at 340 nm.
Other assays
The accumulation of the NO-end product nitrite in the culture media was determined spectrophotometrically at 543 nm by using the Griess reagent with final concentrations of 0.5% sulfanilamide and 0.05% N-(1-naphthyl)ethylenediamine hydrochloride in 45% acetic acid. The intracellular ATP was measured in cell lysates by chemiluminescence with the ATP-bioluminescence-assay-kit HS-II (Roche Diagnostics, Meylan, France) and following the supplier’s instructions. Aconitase activity was measured in both cytoplasmic and mitochondrial fractions by following the disappearance of cis-aconitate at 240 nm at 37°C as described previously (Drapier and Hibbs, 1996). Units are nmoles of substrate consumed/min. The protein content of both the mitochondrial fraction and cytoplasmic extracts was determined at 595 nm by using the Bio-Rad protein assay with BSA as a standard.
Statistics
Data represent the mean the mean ± S.D. of at least three independent experiments. Statistical significance was tested with one-way ANOVA with post-hoc Student-Newman-Keuls comparison (p<0.01 or p<0.05).
RESULTS
Stimulation of BV-2 microglial cells induces NO-synthesis and cytochrome-c release
The combination of 20 U/ml IFN-γ plus 50 ng/ml LPS was chosen because it induced a time-dependent NO production in the BV-2 cell line without triggering toxicity. Nitrite accumulation in media was detectable as early as 4 h after stimulation, and the increase was subsequently linear for the next 20 h. Increasing IFN-γ and/or LPS concentration did not increase nitrite production, whereas lower concentrations were not fully efficient (data not shown). The NOS inhibitors L-NMMA (2 mM) and S-ethylisothiourea (SEIT, 100 μM) reduced nitrite accumulation by an average of 82% and 90%, respectively (Fig. 1A). In addition, nitrite production was fully inhibited by the NOS-2 specific inhibitors 1400W (100 μM) and L-NIL (500 μM). NOS activity was further assayed by the production of (3H)-citrulline from (3H)-L-arginine in cytosol. NOS activity was induced by IFN-γ plus LPS treatment and was fully inhibited by the presence of L-NMMA during the stimulation period (Fig. 1B)
FIG. 1.
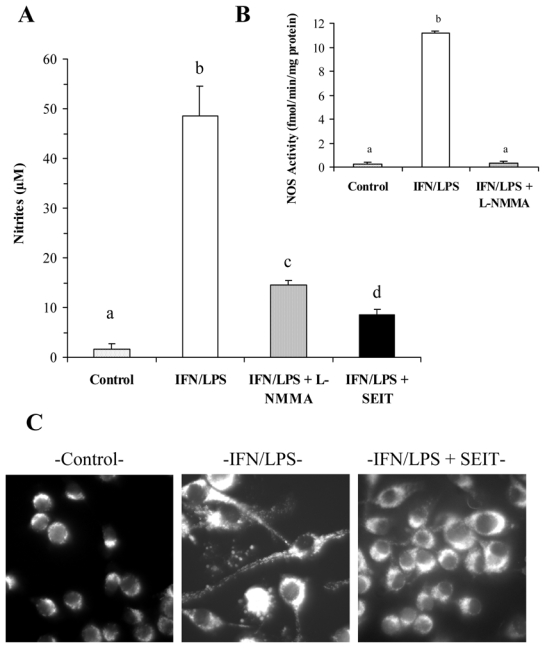
NO synthesis and cytochrome-c release induced by IFN-γ plus LPS stimulation of BV-2 microglial cells. A, nitrite accumulation in the culture medium was determined according to the Griess reaction after 16 h of stimulation in the presence, or absence (Control), of 20 U/ml IFN-γ and 50 ng/ml LPS (IFN/LPS), together with the NOS inhibitors L-NMMA (2 mM) or SEIT (100 μM). B, NOS activity was determined in BV-2 cytosolic extracts from control or stimulated cells by the conversion of 3H-L-arginine in 3H-L-citrulline. A, B, Data are the mean ± S.D. of at least three independent experiments. Statistical significance was tested with one-way ANOVA with post-hoc Student-Newman-Keuls comparison; columns with different letters within each substrate group differ from each other (p<0.01). C, Cells were cultured as described in A and cytochrome-c release was analysed by fluorescence microscopy after immunostaining with anti-cytochorome-c antibody and FITC-conjugated secondary antibody.
It is worthy of note that apoptosis signalling occurred during the course of the 18-h stimulation of BV-2 cells by IFN-γ and LPS as testified by cytochrome-c release. Indeed, whereas immunostaining of cytochrome-c was mainly punctual and perinuclear in control cells, the fluorescence was more diffuse in the whole cytoplasm of stimulated cells and, in some of them, fluorescence was overspread (Fig. 1C). This phenomenon was NO-dependent since the presence of SEIT efficiently inhibited the cytochrome-c release (Fig. 1C).
NO-mediated inhibition of cellular respiration
The effect of NO production on the different segments of the ETC was studied in digitonin-permeabilized cells to which were added exogenous substrates and selective inhibitors of the different complexes which compose the chain. The oxygen consumption rate in state 3, which is dependent on either complex I (malate/glutamate as substrates) or complex II (succinate as substrate in the presence of rotenone), was substantially decreased in permeabilized cells which had been previously exposed to IFN-γ plus LPS (Fig. 2). BV2 cells stimulated in the presence of L-NMMA, an inhibitor of NOS, did not exhibit such a decrease.
FIG. 2.
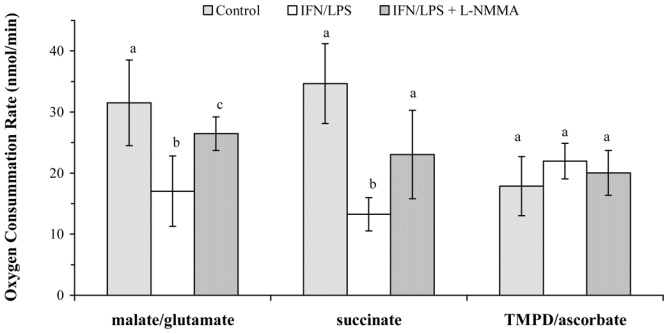
Activity of ETC segments in permeabilized BV-2 cells. Oxygen consumption of digitonin-permeabilized cells was recorded by polarography as described in Materials and Methods in the presence of either complex I substrates (malate 5 mM/glutamate 5 mM), complex II substrate (succinate 5 mM) and rotenone (3 nM), or complex IV substrates (TMPD 0.2 mM/ascorbate 10 mM). Cells were previously stimulated, or not, for 16 h with IFN-γ (20 U/ml) plus LPS (50 ng/ml) in the presence or absence of L-NMMA (2 mM). Data are the mean ± S.D. of five independent experiments. Statistical significance was tested with one-way ANOVA with post-hoc Student-Newman-Keuls comparison; columns with different letters within each substrate group differ from each other (p<0.05).
After cell permeabilization and in the absence of exogenously added L-arginine and NOS cofactors, the NO flux was stopped during the measurement of oxygen consumption rate. Therefore, in agreement with the fact that inhibition of complex IV activity by NO is reversible, the oxygen consumption using TMPD plus ascorbate as substrates was not markedly inhibited under these conditions (Fig. 2).
Inhibition of complexes I and II isolated activities by endogenous NO
The measurement of the activity of complexes I–III by O2 consumption relies on the assumption that the downstream segment (complex IV) is not limiting. As the latter is sensitive to NO, it is worth measuring the activities of complexes I–III individually. Accordingly, the precise site of the NO-dependent blockade in the respiratory chain was assessed by measuring the activity of each complex of the chain, by means of spectrophotometry using mitochondrial fractions prepared from control and stimulated BV-2 cells. The results depicted in Fig. 3 show that NO production by BV2 cells was associated with an average 45% inhibition of complex I and 59% inhibition of complex II. In addition, the activity of complexes II + III was inhibited to the same extent (57%) than that of complex II, implying that complex III was left intact. Furthermore, in the mitochondrial fraction from cells previously stimulated in the presence of L-NMMA, the average activity of complexes I and II was 89% and 103% of the control, respectively (Fig. 3), indicating that the inhibitions observed in stimulated cells were NO-dependent. The activity of complex IV was not inhibited in the mitochondrial fraction from stimulated cells, which can be explained, as mentioned above, by the absence of NO flux during the assay.
FIG. 3.
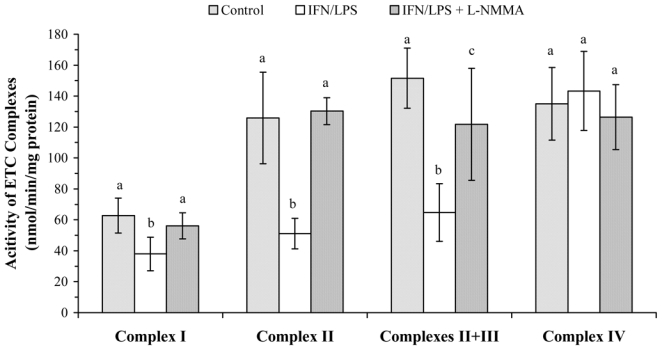
Activity of individualized complexes of the respiratory chain in BV-2 mitochondrial fractions. Cells were stimulated, or not, for 16 h with IFN-γ (20 U/ml) plus LPS (50 ng/ml) in the presence or absence of L-NMMA (2 mM). Mitochondrial fractions were prepared as described in the Materials and Methods and used for the spectrophotometric determination of each complex activity. Data are the mean ± S.D. of ten independent experiments. Statistical significance was tested with one-way ANOVA with post-hoc Student-Newman-Keuls comparison; columns with different letters within each complex group differ from each other (p<0.01).
NO-mediated decrease of intracellular ATP and mitochondrial aconitase activity
Intracellular ATP was measured in either control or NO-producing BV-2 cells. ATP was decreased by an average of 67% in IFN-γ plus LPS treated cells and maintained at control level when L-NMMA was present during stimulation (Fig. 4A). Nevertheless, ATPase activity was unchanged in the mitochondrial fraction from control and stimulated cells (Fig. 4B), suggesting that energetic impairment occurred upstream of complex V.
FIG. 4.
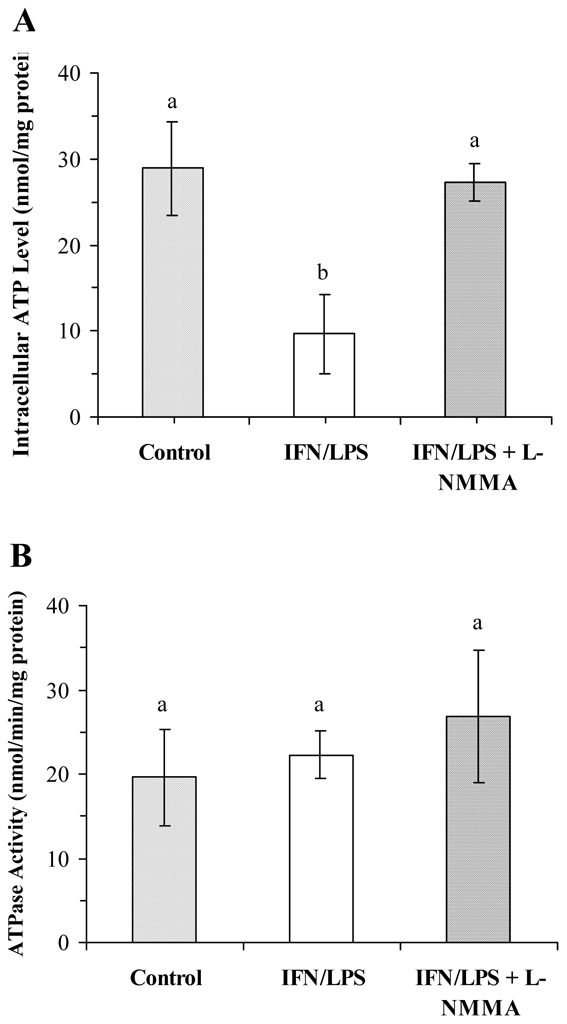
NO-mediated decrease of intracellular ATP and ATPase activity in BV-2 cells. Cells were stimulated, or not, for 16 h with IFN-γ (20 U/ml) plus LPS (50 ng/ml) in the presence or absence of L-NMMA (2 mM). A, intracellular ATP was determined by chemiluminescence after cell lysis. B, ATPase activity was measured in mitochondrial fraction by using a spectrophotometric LDH-pyruvate kinase coupled assay. Data are the mean ± S.D. of three independent experiments. Statistical significance was tested with one-way ANOVA with post-hoc Student-Newman-Keuls comparison; columns with different letters differ from each other (p<0.01).
In addition, the activity of the Krebs cycle enzyme aconitase was strongly inhibited in the mitochondrial fraction from NO-producing cells (1.11 ± 0.52 U/mg protein versus 42.36 ± 7.33 U/mg in control mitochondrial fraction, n=3, p<0.01). By contrast, the glycolysis pathway was not affected since lactate levels were not significantly changed in IFN-γ plus LPS treated cells (1.37 ± 0.08 mM versus 1.24 ± 0.18 mM in control cells medium, n=3, p>0.3).
Increased IRE-binding activity of IRP-1 in NO-producing BV-2 cells
The effect of NO on IRP-1 and IRP-2 was investigated in the cytosol from control and IFN-γ plus LPS -γ-treated BV-2 cells by an EMSA. IRE-binding activity of IRP-1 was strongly increased in NO-producing BV-2 cells (Fig. 5), and, as a counterpart, cytosolic aconitase activity of IRP-1 which was 100.85 ± 37.85 U/mg protein in control cytosol, was totally inhibited. The results of Fig. 5 also show that IRP-2 RNA binding was not significantly modified.
FIG. 5.
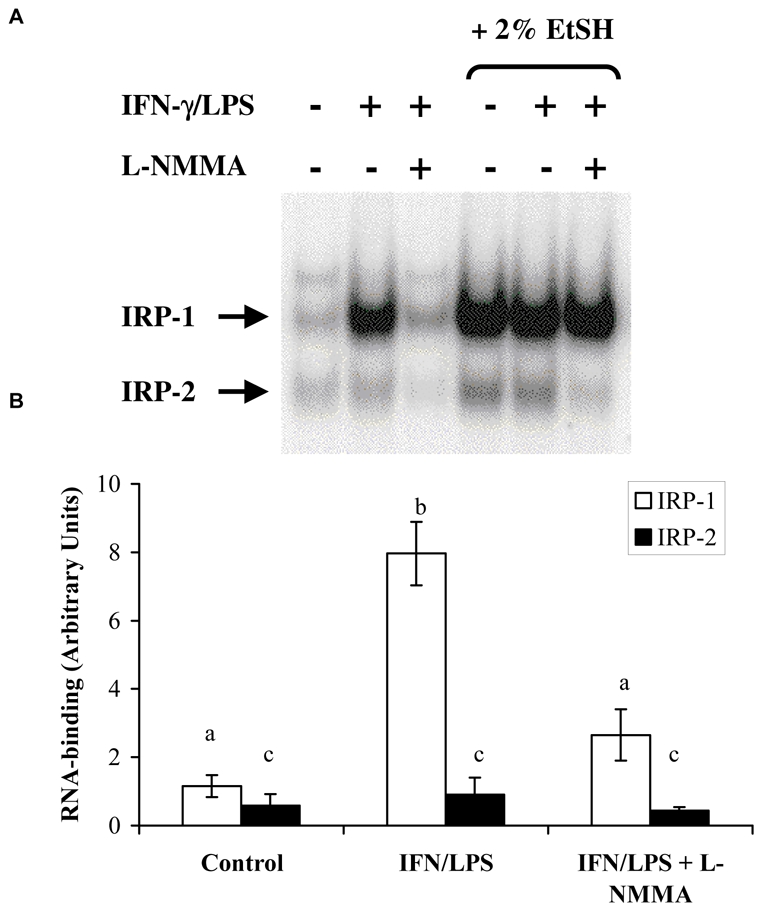
IRE-binding activity of IRPs in BV-2 cells. Cells were stimulated, or not, for 16 h with IFN-γ (20 U/ml) plus LPS (50 ng/ml) in the presence or absence of L-NMMA (2 mM). A, equal amount of protein from cytosolic extracts were analysed for IRE binding activity by EMSA. Data from a typical experiment representative of three. B, radioactivity associated with IRPs/IRE complexes was quantified by PhosphorImaging. Data are the mean ± S.D. of three independent experiments. Statistical significance was tested with one-way ANOVA with post-hoc Student-Newman-Keuls comparison; columns with different letters differ from each other (p<0.05).
Decreased LIP in NO-producing BV-2 cells
Whether NO production and IRP-1 modulation affect intracellular iron and particularly the LIP was then assessed. LIP was determined by the method described by Epsztejn et al., 1997. This procedure takes advantage of the quenching of the calcein fluorescence by iron and its enhancing in the presence of the highly permeant iron chelator SIH. Results from LIP determination in BV-2 cells indicate that stimulation of BV-2 cells by IFN-γ plus LPS clearly decreased the LIP as early as 8 h (1.74 ± 0.37 μM versus 2.35 ± 0.53 μM in control, n=3, p<0.05), and low LIP was maintained after 16 h, showing a significant (42%) reduction in LIP level (Fig. 6). The presence of the NOS inhibitor SEIT during the stimulation period overcame this diminution of LIP to a large extent, indicating that it was NO-dependent. In parallel experiments, incubation of microglial cells with deferoxamine, the prototypic iron chelator, led to a 31% reduction in LIP level after 16 h of culture (1.88 ± 0.34 μM versus 2.71 ± 0.40 μM in control, p<0.01).
FIG. 6.
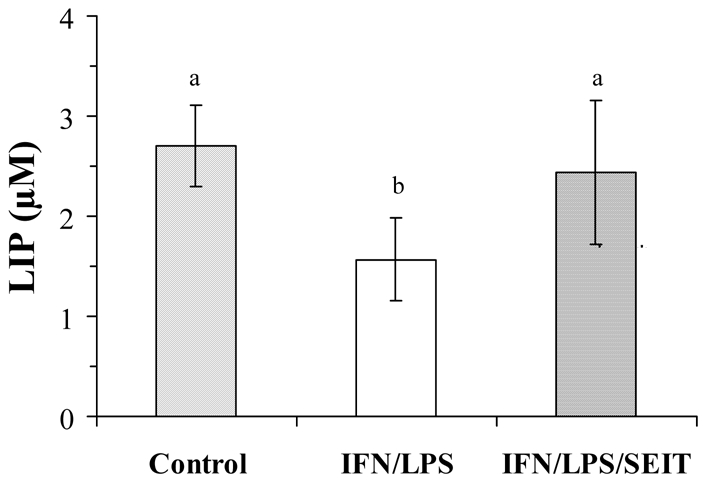
LIP decrease in NO-producing BV-2 cells. Cells were stimulated, or not, for 16 h with IFN-γ (20 U/ml) plus LPS (50 ng/ml) in the presence or absence of the NOS inhibitor SEIT (100 μM). After centrifugation and washing, cells were incubated with calcein-AM for 15 min and the fluorescence intensity was recorded before and after iron chelator (SIH) addition, as described in Materials and Methods. Data are the mean ± S.D. of five independent experiments. Statistical significance was tested with one-way ANOVA with post-hoc Student-Newman-Keuls comparison; columns with different letters differ from each other (p<0.05).
DISCUSSION
There is accumulating evidence that activated glial cells, by producing inflammatory regulators like tumor necrosis factor, certain eicosanoids or nitrogen oxides, contribute to neuronal injury (Gonzalez-Scarano and Baltuch, 1999; Hirsch, 2000; Knott et al., 2000; Liberatore et al., 1999; Nakajima and Kohsaka, 1998). For example, it has been shown that activation of microglia by β-amyloid peptide or inflammatory cytokines such as IFN-γ and interleukin-1β leads to high NO synthesis in rodents (Araujo and Cotman, 1992; Boje and Arora, 1992; Chao et al., 1992; Goodwin et al., 1995; Zielasek et al., 1992) as well as in humans (Colasanti et al., 1995; Ding et al., 1997; Meda et al., 1995). In this report, we focused on the relationship between NO synthesis in microglial cells and two parameters crucial for cell survival: mitochondrial metabolism and the level of redox-active iron. Stimulation of BV2 microglial cells by a combination of IFN-γ and LPS induced NOS activity, and a marked flux of NO was released from cells for several hours. Several metabolic dysregulations dealing with cellular iron-related functions and specific sites of mitochondrial energy production were observed.
Over the last twenty years, several approaches have been followed to assess the NO-dependent dysfunction of the mitochondrial respiration. Originally, cells were harvested and permeabilized before measuring the oxygen uptake by polarography. These experimental conditions allowed demonstration that complex I and complex II activities were significantly reduced soon after the beginning of physiological production of NO (Granger and Lehninger, 1982; Drapier and Hibbs, 1986; Drapier and Hibbs, 1988). It was also noticed that electron flow was reversibly inhibited at the complex IV level (Brown, 1999; Heales et al., 1999). Alternatively, each segment of the electron transport chain can be individually assessed by spectrophotometry (Rustin et al., 1994) to avoid possible caveats linked to measurement of oxygen consumption. Our present study combined these different approaches in the same experimental cell model. Our data underlined that persistent exposure of microglial cells to endogenous NO or derivatives led to reduction of complex I and complex II activities. Few reports have addressed the question of whether NO-dependent reduction of mitochondrial respiration is accompanied by inhibition of ATP synthesis. It has been recently reported that short exposure of brain cells to NO gas is responsible for reversible inhibition of ATP synthesis by mitochondria (Brookes et al., 1999). Here, we provide evidence that long-term blockade of ETC dependent on enzymatically-produced NO, combined with a decrease in the Krebs cycle aconitase activity, was reflected by a loss of chemical energy. The pool of cellular energy (ATP) was strongly decreased, showing that the stimulated microglial cells did not compensate for the energy loss by glycolysis.
The pattern of metabolic dysfunctions we have described, i.e. inhibition of respiration, loss of mitochondrial aconitase activity or ATP depletion, is often associated with iron overload within neuronal cells. This proved true in the Parkinsonian substantia nigra (Gu et al., 1998; Hirsch and Faucheux, 1998; Schapira, 1999), mitochondria of heart cells of FA patients (Bradley et al., 2000; Delatycki et al., 1999; Rötig et al., 1997) and hippocampus of AD patients (Smith et al., 1997, 1998), and it has been proposed that oxidative stress triggered by iron accumulation could be responsible for the deficiency of the respiratory chain Fe-S enzymes (Rötig et al., 1997). The pool of potentially toxic iron is finely tuned by two cytoplasmic regulatory proteins, namely iron regulatory proteins (IRP-1 and IRP-2). IRPs control, via binding to IRE or IRE-like motifs, the translation of proteins playing a relevant part in energy metabolism, namely the Krebs cycle aconitase, succinate dehydrogenase subunit b (at least in insect) and the 75 kDa subunit of complex I (Lin et al., 2001), and of proteins involved in iron uptake or storage, like ferritin, transferrin receptor, divalent metal transporter (DMT) -1 (see Cairo and Pietrangelo, 2000 for a review). In BV-2 microglial cells, IRP1 contributes a higher proportion of total IRP activity. We showed that IRP-1 (but not IRP-2) activity was markedly increased in NO-producing BV2 cells, thus implying that IRE-containing mRNAs were subject to reduced translation or higher stability. NO-dependent modulation of IRPs that has previously been described in macrophages, neurons and fibroblasts (Drapier et al., 2000; Jaffrey et al., 1994; Pantopoulos and Hentze, 1995; Wardrop and Richardson, 2000) is reported here in microglial cells for the first time. A potential consequence of NO-dependent IRP-1 activation in glial cells would be repression of ferritin which has an IRE in the 5′ UTR of its mRNA, and increased stability of transferrin receptor mRNA which has 5 IREs in its 3′ UTR. As a consequence, increased iron uptake coupled to lessened storage may contribute to the cell damage often seen in mitochondria-related neurological disorders (Hirsch and Faucheux, 1998; Rötig et al., 1997; Smith et al., 1998).
It is worth stressing that IRP-1 is itself an aconitase which converts citrate into isocitrate in the cytosol. When activating the IRE-binding activity of IRP-1, NO inhibits its aconitase activity. The aconitase function of IRP-1 is still elusive but a recent genetic study in yeast pointed to a possible role of its aconitase activity in glutamate synthesis and in NADPH replenishment of the anti-oxidant systems glutathione and thioredoxin/thioredoxin reductase (Narahari et al., 2000). It is likely that the aconitase form of IRP-1 contributes to NADPH production in partnership with the cytosolic NADP-dependent isocitrate dehydrogenase in resting (i.e. not producing NO) cells. We checked that NO synthesis in BV2 microglial cells also led to loss of mitochondrial aconitase. In mitochondria, isocitrate dehydrogenase allows regeneration of NADPH required for reduction of glutathione disulfide (Vogel et al., 1999) and thioredoxin reductase (Watabe et al., 1999). Altogether, NO-dependent inhibition of cytosolic and mitochondrial aconitases, by limiting isocitrate supply, may contribute to modulation of the NADPH-dependent redox balance in both cell compartments.
A parameter relevant to oxidative stress and cellular iron homeostasis which often accompanies various neurological disorders is the fraction of intracellular iron that is not present in protein prosthetic groups or stored in ferritin. This pool of iron is still ill-defined but recent progress has been made in evaluating its level (Epsztejn et al., 1997) and it is now referred to as the labile iron pool or LIP. In this study, we showed that LIP reduction was dependent on NO synthesis and was even more pronounced than that exhibited by deferroxamine-exposed cells. Such a 40% change of LIP level was as high as what has been previously reported in the literature in the case of regulation by ferritin (Kakhlon et al., 2001; Picard et al., 1998). Interestingly, this NO-dependent LIP decrease was significantly different from control as early as 8 h after the beginning of the activation, i.e. shortly after the beginning of significant NO production. This result indicates that LIP decrease is not the consequence of ferritin and transferrin receptor modulation through the IRP/IRE system. Rather, reduction of LIP may contribute to IRP-1 enhancement in cooperation with a direct effect of NO. Elucidation of the exact mechanism by which NO affects LIP requires further investigation but, as NO has strong affinity for FeII and yields EPR-detectable complexes with non heme iron in NO-producing or NO-exposed cells (Drapier, 1997; Kennedy et al., 1997; Lancaster et al., 1994), it can be proposed that formation of such nitrosyl-iron complexes may represent a “sink” for LIP or favor its intracellular transit and/or metabolism. Alternatively, the significance of decreased LIP could be the reduction of the level of the ROS-production catalyst iron as a resistance mechanism in the NO/ROS-producing cells.
In conclusion, our work shows that NOS-2 expression upon inflammatory stimulation of microglial cells leads to severe reduction in ATP supply and to misregulation of iron metabolism. Irreversible i.e. deleterious metabolic impairments resulting from NOS-2-dependent (i.e., long-term) production of NO are essentially inhibition of mitochondrial aconitase, inhibition of complex I and II, and increase in both IRP-1 activity and LIP. It is striking that these dysfunctions are closely related to the energetic and iron-linked neurological disorders such as PD, AD or HD. The present results indicate that inappropriate NO production in brain during the course of an inflammatory process could exaggerate the initial defects associated with neurological disorders.
Acknowledgments
This work was supported in part by a joint grant from ARERS and the French Ligue Nationale contre le Cancer, Comité de l’Aisne, and by the Association pour la Recherche contre le Cancer (Grant n°5856). We are grateful to Dr. Caroline. Demerlé-Pallardy for providing us with BV-2 cells and to Dr. Prem Ponka for the generous gift of SIH. We thank Dr. David Chauvier for expert advice on the cytochrome-c release experiments, Marie-Jeanne Chauveau for skilful technical assistance and Dr. Cécile Bouton for critical reading of the manuscript.
Abbreviations used
- AD
Alzheimer’s disease
- Calcein-AM
calcein-acetoxy-methylester
- COX
cytochrome-c oxidase
- EMSA
electrophoretic mobility shift assay
- ETC
electron transport chain
- FA
Friedreich’s ataxia
- FITC
fluoresceine-isothiocyanate
- IFN-γ
γ-interferon
- IRE
iron responsive element
- IRP
iron regulatory protein
- LDH
lactate dehydrogenase
- LIP
labile iron pool
- L-NIL
N6-(1-imminoethyl)-L-lysine
- L-NMMA
N-monomethyl- L-arginine
- LPS
lipopolysaccharide
- NO
nitric oxide
- NOS
nitric oxide synthase
- PBS
phosphate-buffered saline
- PD
Parkinson’s disease
- ROS
reactive oxygen species
- SEIT
S-ethylisothiourea
- SIH
salicylaldehyde-isocotinoyl-hydrazone
- UTR
untranslated region
References
- Araujo DM, Cotman CW. Beta-amyloid stimulates glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer’s disease. Brain Res. 1992;569:141–145. doi: 10.1016/0006-8993(92)90380-r. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AHV. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum Mol Genet. 2000;9:275–282. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Bolaños JP, Heales SJR. The assumption that nitric oxide inhibits mitochondrial ATP synthesis is correct. FEBS Lett. 1999;446:261–263. doi: 10.1016/s0014-5793(99)00217-3. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- Cairo G, Pietrangelo A. Iron regulatory proteins in pathobiology. Biochem J. 2000;352:241–250. [PMC free article] [PubMed] [Google Scholar]
- Calingasan NY, Park LCH, Calo LL, Trifiletti RR, Gandy SE, Gibson GE. Induction of nitric oxide synthase and microglial response precede selective cell death induced by chronic impairment of oxidative metabolism. Am J Pathol. 1998;153:599–610. doi: 10.1016/S0002-9440(10)65602-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colasanti M, Persichini T, Di Pucchio T, Gremo F, Lauro GM. Human ramified microglial cells produce nitric oxide upon Escherichia coli lipopolysaccharide and tumor necrosis factor α stimulation. Neurosci Lett. 1995;200:144–146. doi: 10.1016/0304-3940(95)12101-9. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog Brain Res. 1998;118:215–229. doi: 10.1016/s0079-6123(08)63210-0. [DOI] [PubMed] [Google Scholar]
- Delatycki MB, Camakaris J, Brooks H, Evans-Whipp T, Thorburn DR, Williamson R, Forrest SM. Direct evidence that mitochondrial iron accumulation occurs in Friedreich Ataxia. Ann Neurol. 1999;45:673–675. [PubMed] [Google Scholar]
- Ding M, St Pierre BA, Parkinson JF, Medberry P, Wong JL, Rogers NE, Ignarro LJ, Merrill JE. Inducible nitric oxide synthase and nitric oxide production in human fetal astrocytes and microglia. J Biol Chem. 1997;272:11327–11335. doi: 10.1074/jbc.272.17.11327. [DOI] [PubMed] [Google Scholar]
- Drapier JC. Interplay between NO and (Fe-S) clusters: relevance to biological systems. Methods. 1997;3:319–329. doi: 10.1006/meth.1996.0426. [DOI] [PubMed] [Google Scholar]
- Drapier JC, Hibbs JB., Jr Murine cytotoxic activated macrophages inhibit aconitase in tumor cells. Inhibition involves the iron-sulfur prosthetic group and is reversible. J Clin Invest. 1986;78:790–797. doi: 10.1172/JCI112642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapier JC, Hibbs JB., Jr Differentiation of murine macrophages to express nonspecific cytotoxicity for tumor cells results in L-arginine-dependent inhibition of mitochondrial iron-sulfur enzymes in the macrophage effector cells. J Immunol. 1988;140:2829–2838. [PubMed] [Google Scholar]
- Drapier JC, Hibbs JB., Jr Aconitases: a class of metalloproteins highly sensitive to nitric oxide synthesis. Methods Enzymol. 1996;269:26–36. doi: 10.1016/s0076-6879(96)69006-5. [DOI] [PubMed] [Google Scholar]
- Drapier JC, Bouton C, Oliveira L. Redox modulation of iron regulatory proteins by nitric oxide and peroxynitrite. In: Ignarro LJ, editor. Nitric oxide, Biology and Pathology. Academic Press; San Diego, CA: 2000. pp. 315–328. [Google Scholar]
- Epsztejn S, Kakhlon O, Glickstein H, Breuer W, Cabantchik ZI. Fluorescence analysis of the labile iron pool of mammalian cells. Anal Biochem. 1997;248:31–40. doi: 10.1006/abio.1997.2126. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Granger DL, Lehninger AL. Sites of inhibition of mitochondrial electron transport in macrophages-injured neoplastic cells. J Cell Biol. 1982;95:527–535. doi: 10.1083/jcb.95.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin JL, Uemura E, Cunnick JE. Microglial release of nitric oxide by the synergistic action of beta-amyloid and IFN-gamma. Brain Res. 1995;692:207–214. doi: 10.1016/0006-8993(95)00646-8. [DOI] [PubMed] [Google Scholar]
- Gu M, Owen AD, Toffa SEK, Cooper JM, Dexter DT, Jenner P, Marsden CD, Schapira AHV. Mitochondrial function, GSH and iron in neurodegeneration and Lewy body diseases. J Neurol Sci. 1998;158:24–29. doi: 10.1016/s0022-510x(98)00095-1. [DOI] [PubMed] [Google Scholar]
- Heales SJR, Bolaños JP, Stewart VC, Brookes PS, Land JM, Clark JB. Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta. 1999;1410:215–228. doi: 10.1016/s0005-2728(98)00168-6. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Faucheux BA. Iron metabolism and Parkinson’s disease. Mov Disord. 1998;13(suppl 1):39–45. [PubMed] [Google Scholar]
- Hirsch EC. Glial cells and Parkinson’s disease. J Neurol. 2000;247(suppl 2):58–62. doi: 10.1007/pl00007762. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Cohen NA, Rouault TA, Klausner RD, Snyder SH. The ironresponsive element binding protein: a target for synaptic actions of nitric oxide. Proc Natl Acad Sci USA. 1994;91:1294–1298. doi: 10.1073/pnas.91.26.12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhlon O, Gruenbaum Y, Cabantchik ZI. Repression of the heavy ferritin chain increases the labile iron pool of human K562 cells. Biochem J. 2001;356:311–316. doi: 10.1042/0264-6021:3560311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MC, Antholine WE, Beinert H. An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J Biol Chem. 1997;272:20340–20347. doi: 10.1074/jbc.272.33.20340. [DOI] [PubMed] [Google Scholar]
- Knott C, Stern G, Wilkin GP. Inflammatory regulators in Parkinson’s disease: iNOS, Lipocortin-1, and cyclooxygenases-1 and –2. Mol Cell Neurosci. 2000;16:724–739. doi: 10.1006/mcne.2000.0914. [DOI] [PubMed] [Google Scholar]
- Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61. doi: 10.1006/exnr.1998.6993. [DOI] [PubMed] [Google Scholar]
- Lancaster JR, Jr, Werner-Felmayer GH, Wachter H. Coinduction of nitric oxide synthesis and intracellular nonheme iron-nitrosyl complexes in murine cytokine-treated fibroblasts. Free Radic Biol Med. 1994;16:869–870. doi: 10.1016/0891-5849(94)90206-2. [DOI] [PubMed] [Google Scholar]
- Lin E, Graziano JH, Freyer GA. Regulation of the 75 kDa subunit of mitochondrial complex I by iron. J Biol Chem. 2001;276:27685–27692. doi: 10.1074/jbc.M100941200. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nature Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Murphy S. Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia. 2000;29:1–13. doi: 10.1002/(sici)1098-1136(20000101)29:1<1::aid-glia1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Functional roles of microglia in the central nervous system. Hum Cell. 1998;11:141–155. [PubMed] [Google Scholar]
- Narahari J, Ma R, Wang M, Walden WE. The aconitase function of iron regulatory protein 1. Genetic studies in yeast implicate its role in iron-mediated redox regulation. J Biol Chem. 2000;275:16227–16234. doi: 10.1074/jbc.M910450199. [DOI] [PubMed] [Google Scholar]
- Pantopoulos K, Hentze MW. Nitric oxide signaling to iron-regulatory protein: direct control of ferritin mRNA translation and transferrin receptor mRNA stability in transfected fibroblasts. Proc Natl Acad Sci U S A. 1995;92:1267–71. doi: 10.1073/pnas.92.5.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard V, Epsztejn S, Santambrogio P, Cabantchik ZI, Beaumont C. Role of ferritin in the control of the labile iron pool in murine erythroleukemia cells. J Biol Chem. 1998;273:15382–15386. doi: 10.1074/jbc.273.25.15382. [DOI] [PubMed] [Google Scholar]
- Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nature Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- Rustin P, Chrétien D, Bourgeron T, Gérard B, Rötig A, Saudubray JM, Munnich A. Biochemical and molecular investigation in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Schapira AHV. Mitochondrial involvement in Parkinson’s disease, Huntington’s disease, hereditary spastic paraplegia and Friedreich’s ataxia. Biochim Biophys Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- Smith MA, Harris PLR, Sayre LM, Perry G. Iron accumulation in Alzheimer’s disease is a source of redox-generated free radicals. Proc Natl Acad Sci USA. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Wehr K, Harris PLR, Siedlak SL, Connor JR, Perry G. Abnormal localization of iron regulatory protein in Alzheimer’s disease. Brain Res. 1998;788:232–236. doi: 10.1016/s0006-8993(98)00002-x. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58:233–247. doi: 10.1016/s0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- Torreilles F, Salman-Tabcheh S, Guérin MC, Torreilles J. Neurodegenerative disorders: the role of peroxynitrite. Brain Res Rev. 1999;30:153–163. doi: 10.1016/s0165-0173(99)00014-4. [DOI] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- Vogel R, Wiesinger H, Hamprecht B, Dringen R. The regeneration of reduced glutathione in rat forebrain mitochondria identifies metabolic pathways providing the NADPH required. Neurosci Lett. 1999;275:97–100. doi: 10.1016/s0304-3940(99)00748-x. [DOI] [PubMed] [Google Scholar]
- Wardrop SL, Richardson DR. Nitrogen monoxide activates iron regulatory protein 1 RNA-binding activity by two possible mechanisms: effect on the (4Fe-4S) cluster and iron mobilization from cells. Biochemistry. 2000;39:2748–2758. doi: 10.1021/bi991099t. [DOI] [PubMed] [Google Scholar]
- Watabe S, Makino Y, Ogawa K, Hiroi T, Yamamoto Y, Takahashi SY. Mitochondrial thioredoxin reductase in bovine adrenal cortex its purification, properties, nucleotide/amino acid sequences, and identification of selenocysteine. Eur J Biochem. 1999;264:74–84. doi: 10.1046/j.1432-1327.1999.00578.x. [DOI] [PubMed] [Google Scholar]
- Zielasek J, Tausch M, Toyka KV, Hartung HP. Production of nitrite by neonatal rat microglial cells/brain macrophages. Cell Immunol. 1992;141:111–120. doi: 10.1016/0008-8749(92)90131-8. [DOI] [PubMed] [Google Scholar]