

Abstract
Directed evolution methods were developed for Cu-containing nitrite reductase (NiR) from Alcaligenes faecalis S-6. The PCR cloning strategy allows for the efficient production of libraries of 100 000 clones by a modification of a megaprimer-based whole-plasmid synthesis reaction. The high-throughput screen includes colony lift onto a nylon membrane and subsequent lysis of NiR-expressing colonies in the presence of Cu2+ ions for copper incorporation into intracellularly expressed NiR. Addition of a chromogenic substrate, 3, 3′-diaminobenzidine (DAB), results in deposition of red, insoluble color at the site of oxidation by functional NiR. Twenty-thousand random variants of NiR were screened for improved function with DAB as a reductant, and five variants were identified. These variants were shuffled and screened, yielding two double variants. An analog of the DAB substrate, o-dianisidine, which is oxidized to a water-soluble product was used for functional characterization. The double variant M150L/F312C was most proficient at o-dianisidine oxidation with dioxygen as the electron acceptor (5.5X wt), and the M150L single variant was most proficient at o-dianisidine oxidation with nitrite as the electron acceptor (8.5X wt). The library generation and screening method can be employed for evolving new reductase functions in NiR and for screening of efficient folding of engineered NiRs.
Keywords: colony screen, crystallography, directed evolution, electrochemistry, nitrite reductase
Introduction
Under oxygen-limiting conditions, certain facultative anaerobic bacteria can respire with nitrate and nitrite as electron acceptors to produce the gases nitric oxide, nitrous oxide and dinitrogen (Averill, 1996). Copper-containing nitrite reductase (NiR) from Alcaligenes faecalis S-6 (AfNiR) catalyzes the single electron reduction of nitrite 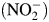 to nitric oxide (NO) in a process known as dissimilatory denitrification (Kakutani et al., 1981a). Energetically, respiratory denitrification is less efficient than oxygen respiration (Tran and Unden, 1998). Furthermore, the switch from anaerobic to aerobic growth for A. faecalis leads to a rapid inactivation of NiR (Kakutani et al., 1981c), a process that terminates the use of the less efficient terminal electron acceptor.
to nitric oxide (NO) in a process known as dissimilatory denitrification (Kakutani et al., 1981a). Energetically, respiratory denitrification is less efficient than oxygen respiration (Tran and Unden, 1998). Furthermore, the switch from anaerobic to aerobic growth for A. faecalis leads to a rapid inactivation of NiR (Kakutani et al., 1981c), a process that terminates the use of the less efficient terminal electron acceptor.
AfNiR is a 110 kDa homotrimer, the monomers of which consist of two Greek key β-barrels, with one type-1 and another type-2 copper site per monomer (Kukimoto et al., 1994). The type-1 copper is coordinated by His95, Cys136, His145 and Met150. Residues 136, 145 and 150 are part of a single ‘ligand loop’, and His95 is located on an adjacent β-strand. The type-2 copper is located between monomers and is coordinated by residues His100, His135 and His306, the last of which comes from an adjacent monomer. The type-1 and type-2 copper sites are connected via the peptide backbone between His135 and Cys136. In vivo, the small type-1 copper protein pseudoazurin is proposed to donate electrons to the type-1 copper (Kakutani et al., 1981b), followed by intramolecular electron transfer to the type-2 copper, the site of nitrite binding and reduction (Kukimoto et al., 1994).
NiR can catalyze other reactions. With the artificial reductants NADH and phenazine methosulfate, nitric oxide has been shown to be a substrate for NiR by the proposed overall reaction (Jackson et al., 1991).
In addition, Kakutani et al. (1981b) reported that NiR is able to reduce dioxygen to hydrogen peroxide and that prolonged production of H2O2 led to NiR inactivation. The addition of catalase abolished this inactivation. Concurrent reduction of AfNir by pseudoazurin is essential to the inactivation by H2O2, an observation that led Kakutani et al. to propose that the inactivation of the enzyme in this manner results from hydroxyl radical that is generated by further reduction of H2O2 at the type-2 copper (Kakutani et al., 1981b).
NiR is homologous to the multicopper oxidases (MCOs) and often is included in the same protein family, but rather than a mononuclear type-2 site, MCOs possess a trinuclear site that consists of type-2 and type-3 coppers. This trinuclear site is the catalytic center at which dioxygen is reduced to water (for a review comparing NiR to MCO see MacPherson and Murphy, 2007). MCOs such as laccase and ascorbate oxidase have reduction potentials among the highest of known type-1 copper proteins (330–780 mV versus SHE) (Shleev et al., 2005). These proteins catalyze the oxidation of various substrates at the type-1 copper site and the concomitant transfer of electrons to an active site trinuclear copper cluster (Solomon et al., 2001). Interestingly, studies of type-1 copper-depleted laccase have shown the production of a bridged peroxide intermediate (Shin et al., 1996) that is analogous to the peroxide product proposed for the reaction of NiR with dioxygen.
The NiR function of NiR has been the subject of numerous site-directed mutagenesis studies targeting residues surrounding the two copper sites (Kukimoto et al., 1994; Boulanger et al., 2000; Boulanger and Murphy, 2001; Boulanger and Murphy, 2003; Wijma et al., 2003; Wijma et al., 2006). Application of directed evolution to the study of NiR catalysis has been limited by the lack of an effective functional screen. Here, a high-throughput screen is developed that employs a chemical reductant of NiR to enable the study of dioxygen reduction. Characterization of variants obtained from high-throughput screening by spectroscopic enzyme assays and direct electrochemistry validates the screen method. As well, the variants provide insights into the mechanisms of dioxygen and nitrite reduction by NiR. The potential applicability of the screening method to other oxidoreductase systems is discussed.
Materials and methods
Library construction
Variant libraries were generated in a pET28a construct (pAfNiR28a) containing the coding region for the soluble fragment of NiR (Boulanger et al., 2000). Random PCR mutagenesis was performed by the method of Cadwell and Joyce (1992) using primers NIRFOR (5′GCAACTGCGGCAGAAATAGCA) and NIRREV (5′CGTGCCAGATGGTGCGA), which correspond to the first 21 and last 17 nucleotides of the coding sequence for soluble NiR. Reaction conditions were 10 mM Tris–Cl pH 8.3, 50 mM KCl, 7 mM MgCl2, 0.15 mM MnCl2, 200 µM dNTPs, 0.4 µM of each primer, 30 ng template and 5 U Taq polymerase in 100 µl total volume. The PCR product was cloned into pET28a using a variation of the method described by Miyazaki, 2003. Briefly, pAfNiR28a was used as template, and the mutagenic PCR product used as a megaprimer for whole plasmid synthesis. The major variation was cutting of the template pAfNiR28a plasmid at a unique EcoR1 site within the NiR gene. Reactions contained 1X Platinum Pfx buffer (Invitrogen), 1 mM MgSO4, 300 µM dNTPs, 75 ng freshly cut template, 700 ng mutagenic PCR product, 4% DMSO and 2.5 U Platinum Pfx polymerase (Invitrogen). Reactions were cycled between 95°C (30 s) and 68°C (7 min) for 12–24 cycles. The reaction product was dialyzed, electroporated into E. coli HMS174(de3) and plated onto 2YT agar plates containing kanamycin (25 µg ml−1) and IPTG (66 µM).
Shuffling of mutant genes was performed by the Stemmer method (Stemmer, 1994), and the DNA encoding the enzyme variants identified by screening as the most efficient in color development was subjected to controlled digestion by DNAase I. Approximately 50 base pair fragments were reassembled by 30X thermal cycling of 94°C (30 s), 40°C (30 s) and 72°C (4 min) extensions. A final PCR with the NIRFOR and NIRREV primers amplified the shuffled library. Shuffled mutants were introduced into pET28a using the above-described modification of the Miyazaki method.
Screening method
The colony lysis protocol was adapted from a method described by Kadono-Okuda and Andres (1997). Plates were incubated for 15 h at 33–35°C. The resulting colonies were lifted onto Biodyne-A 0.45 µm nylon membranes (Pall) and placed, colony side up, onto Whatman filter paper saturated with a lysis solution (10 mM Tris pH 7, 2% SDS, 0.3% Tween-20 and 50 µM CuSO4) and incubated at 50°C for 30 min. The membrane was then washed gently in 10 mM Tris pH 7.5, 100 mM NaCl for 5–10 min. The membranes were blotted dry and submerged in screening reagent (0.76 mM 3,3′diaminobenzidine tetrahydrochloride (Sigma) and 0.5 µM horseradish peroxidase (Sigma) in 100 mM sodium phosphate pH 7.4). In the screen, DAB acts as both the chromogenic electron donor for NiR as well as a chromogenic substrate for horseradish peroxidise, which consumes NiR-generated hydrogen peroxide for further DAB oxidation. Red spots, representing colonies expressing active NiR, were identified visually on the membrane. The spots appearing most rapidly were selected and mapped to the original plate. Variants were rescreened to compare variants from different plates, as each plate contained only ∼2000 clones. The original kanamycin/IPTG plates were incubated at 30°C to allow colonies to re-grow, and selected colonies were picked, grown overnight for plasmid isolation and sequenced to define the mutations.
Protein expression and purification
NiR variants containing a carboxyl-terminal his-tag were expressed in E. coli BL21(de3) from pET28a vectors as described previously (Wijma et al., 2003). One litre cultures were inoculated with 3 ml overnight culture and grown at 30°C to an OD600 of 1.0. Following growth for 30 additional minutes at 25°C, the cultures were induced with 0.5 mM IPTG and grown for 16–18 h at 25°C. The cells were pelleted and resuspended in nickel column-binding buffer (20 mM sodium phosphate pH 7.8, 500 mM NaCl) supplemented with 1 mM CuSO4 and lysed with an Emulsiflex C-5 homogenizer (Avestin). The soluble fraction was applied to a nickel metal chelate Sepharose column (GE Healthcare) for purification, and the desired protein was eluted in 20 mM sodium phosphate pH 6.0, 500 mM NaCl and 500 mM imidazole, resulting in greater than 95% purity. All variants expressed at a minimum of 200 mg l−1 culture and were sufficiently stable to allow concentration to >30 mg ml−1 with minimal precipitation (<1%).
Pseudoazurin was expressed and isolated as described previously (Wijma et al., 2004). Briefly, E. coli BL21(de3) containing the expression construct ppAz24c (Boulanger, 2001) was grown at 30°C to an OD600 of 2.0. Following growth for 30 additional minutes at 25°C, the culture was induced with 0.5 mM IPTG, and grown for 12–14 h at 25°C. The cells were pelleted and resuspended in 20 mM sodium phosphate pH 6.3 supplemented with 10 mM CuSO4, lysed with an Emulsiflex C-5 homogenizer (Avestin). The soluble fraction was applied to CM-sepharose (GE Healthcare) and eluted with increasing NaCl in 20 mM sodium phosphate pH 6.3. Typical yields were 100–150 mg of pseudoazurin per litre culture.
Activity assays
Pseudoazurin-based assays for nitrite reduction were performed as described by Wijma et al. (2004). Pseudoazurin was reduced with excess ascorbate and then buffer-exchanged against N2-saturated 100 mM MES-HEPES, pH 7.0. Reactions containing 315 µM reduced pseudoazurin and 2.5 mM NaNO2 in MES-HEPES pH 7.0 were started by the addition of NiR to the final concentration of 450 pM. The absorbance at 593 nm, corresponding to the amount of oxidized pseudoazurin (ε = 2900 M−1 cm−1), was monitored with a Cary 50 Bio UV-Vis spectrophotometer in a cell maintained at 25°C with circulating water.
Pseudoazurin-based assays for oxygen reduction were performed under similar conditions as for nitrite reduction, but 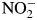 was omitted. NiR was added to a final concentration of 9–90 nM to initiate the assays. Bovine liver catalase (Sigma) was added to a final concentration of 1 µM.
was omitted. NiR was added to a final concentration of 9–90 nM to initiate the assays. Bovine liver catalase (Sigma) was added to a final concentration of 1 µM.
o-Dianisidine (3,3′-dimethoxybenzidine dihydrochloride), a structural analog of 3,3′-diaminobenzidine (DAB) with a water-soluble oxidation product, was also used as an electron donor to NiR. Note, both o-dianisidine and 3,3′diaminobenzidine tetrahydrochloride are suspected carcinogens. Oxygen reduction assays were performed in 100 mM MES-HEPES pH 7.0 containing 0.95 mM o-dianisidine and 1 µM catalase. Nitrite reduction assays contained 0.95 mM o-dianisidine and 2.5 mM sodium nitrite. Oxidation of o-dianisidine was monitored by the increase in absorbance at 460 nm (ϵ = 11 300 M−1 cm−1).
Electrochemistry
Reduction potentials of the variants were measured by a method described by Kohzuma et al. in which apo-pseudoazurin is used as an electrode modifier (Kohzuma et al., 1994). Apo-pseudoazurin was prepared by dialysis against 6 mM KCN overnight. This treatment was accompanied by a loss of blue color from the sample as expected with removal of type-1 Cu. The apo-pseudoazurin (30 µm) and variant NiR (10–100 µM) in 100 mM sodium phosphate, pH 7 were added to an electrochemical cell containing an edge-oriented pyrolytic graphite working electrode that had been polished with alumina (0.3 µM). The reference electrode was saturated calomel, and the counter electrode was platinum wire. An Eco-Chemie Autolab potentiostat (PGStat30) was used to control the potential of the working electrode (−300 to 300 mV) with a scan rate of 20 mV s−1. Typical experiments resulted in a peak separation of 59–60 mV.
Electronic absorption spectroscopy
A Cary 50 spectrophotometer was used for wavelength scans of protein sample in 20 mM MOPS pH 7.0. Protein concentration was determined by absorbance at 280 nm (ε = 39 400 M−1 cm−1, Boulanger, 2001). NiR proteins were centrifuged for 10 min to reduce light scattering. Scans were obtained for wavelength range 350–850 nm.
Crystal structures
Crystals of variant NiRs were grown in hanging-drop format. Mother liquor consisted of 100 mM sodium acetate pH 4.5, 8–12% PEG 6000. Crystals were transferred to mother liquor supplemented with 30% glycerol as a cryoprotectant and immersed in liquid nitrogen. Home source X-rays were used for data collection of M62L variant, whereas data sets of the M94T/F312C and M150L/F312C variants were collected on beamline 9–2 at the Stanford Synchrotron Radiation Lightsource (SSRL). For refinement, the starting point was the 1.4 Å resolution structure of nitrite-bound native NiR (PDB code 1SJM) after removal of nitrite. The M62L and M94T/F312C variants are isomorphous with the native structure (space group P212121), and the starting model was used directly in refinement. These structures have the complete NiR trimer in the asymmetric unit. M150L/F312C crystallized in space group R3, and chain A of the starting model was selected and used for molecular replacement using MOLREP (Vagin and Teplyakov, 1997). All variant structures were refined with Refmac (Murshudov et al., 1997) from the CCP4 package (Collaborative Computational Project, N, 1995), and manual model building was performed with Xfit (Collaborative Computational Project, N, 1995). For the structure of M150L/F312C, water molecules were added by using the coot:findwaters feature of the CCP4 package. A summary of data collection and refinement statistics can be found in Table I.
Table I.
Crystallographic data collection and refinement statistics
| Crystal | M62L | M94T/F312C | M150L/F312C |
|---|---|---|---|
| Space group | P212121 | P212121 | R3 |
| Cell dimensions (Å) | a = 61.13 | a = 61.30 | a = 126.72 |
| b = 102.22 | b = 102.08 | b = 126.72 | |
| c = 146.15 | c = 146.34 | c = 65.06 | |
| Resolution (Å) | 84.5–2.1 | 84.5–1.6 | 63.4–1.5 |
| R-merge | 0.100 (0.356)a | 0.068 (0.232) | 0.084 (0.234) |
| {I}/{σ(I)}b | 9.8 (2.6) | 21.0 (3.9) | 21.3 (4.7) |
| Completeness (%) | 97.3 (97.6) | 89.4 (71.8) | 99.6 (99.1) |
| Unique reflections | 50 095 | 103 196 | 54 872 |
| Working R-factor | 0.161 | 0.181 | 0.179 |
| Free R-factor | 0.217 | 0.225 | 0.204 |
| RMSD bond length (Å) | 0.011 | 0.012 | 0.012 |
| Overall B-factor (Å2) | 26.2 | 23.2 | 24.3 |
| Water molecules | 1122 | 1347 | 414 |
| PDB entry code | 3H4F | 3H4H | 3H56 |
aValues in parenthesis are for the highest resolution shell. b{I}/{σ(I)} is the average intensity divided by the average estimated error in intensity.
Results
Directed evolution
Cloning efficiency is often a limiting factor in high-throughput colony screening. This limitation is especially prevalent with notoriously difficult cloning vectors such as pET. For this reason, the megaprimer method of Miyazaki et al. was used in this study with some modifications. Particularly, cleaving of the template plasmid DNA within the NiR gene significantly improved the polymerase-based cloning reaction (Fig. 1). The rationale for this modification was the assumption that megaprimer annealing is rate limiting when the target template is coiled-circular DNA. Denaturation at 95°C undoubtedly melts the strands of plasmid DNA; however, strand separation fails due to their closed circular nature. Denaturation of singly cut template plasmid results in two separate ssDNA strands without restricted access to their termini (Fig. 1), which makes annealing of megaprimers more efficient. As the megaprimers are consumed in the polymerization reaction, fewer will be available to bind both template and product, resulting in the circularization of the major product and formation of a viable plasmid (Fig. 2).
Fig. 1.

Schematic representation of the megaprimer-based cloning method. Green bands correspond to the NiR gene in the original pET28a plasmid. Orange bands correspond to the mutagenic PCR product (megaprimers). The plasmid is represented by black lines and arrows (not drawn to scale).
Fig. 2.

Typical megaprimer cloning reactions from various cycle numbers (12–24). Products were electrophoresed on a 1% agarose gel. The band proposed to be nicked circular product corresponds to successfully cloned variant. Note that the megaprimer is consumed as the cycle number increases. The appearance of the proposed nicked circular product corresponds to colony-forming units upon transformation.
NiR variants were obtained by high-throughput screening on the basis of their ability to catalyze the dioxygen-dependent oxidation of DAB. A central component of the high-throughput screening was the colony lift and lysis method, which exposed the cytoplasmically expressed NiRs to Cu (free Cu ion availability is significantly limited in the cytoplasm (Rensing and Grass, 2003)) and allowed for NiR interaction with DAB. DAB oxidation resulted in the deposition of red color at lysed colonies, indicating turnover by the variant NiRs (Fig. 3).
Fig. 3.

Picture of a screened membrane. About 2000 clones are represented, half are not apparent due to loss of activity.
In this study, the library construction method typically yielded 1000–2000 colonies per μl reaction (Fig. 1). Thus, as many as 100 000 clones could be obtained from a 50 µl reaction. Since only ∼20 000 variants were screened in this study, cloning efficiency was not a limiting factor. Based on the sequencing of five randomly selected variants, a mutation rate of three to four substitutions per clone was obtained using a MnCl2 concentration of 0.15 mM in the mutagenic PCR reactions. The mutation rate defined by limited sequencing is consistent with the approximately 50% rate of inactivation observed in the screen.
Plasmid DNA from the 12 initial variants selected from the screen was isolated and re-transformed, and a second round of colony screening yielded five single-site variants. These variants were shuffled and screened yielding two double variants. A list of variants obtained in this study is found in Table II.
Table II.
Reduction potentials and relative apparent reaction rates of variant NiRs
| Variant | Oxygen reduction (o-dianisidine) | Nitrite reduction (o-dianisidine) | Oxygen reduction (pseudoazurin) | Nitrite reduction (pseudoazurin) | E1/2 (mV)f |
|---|---|---|---|---|---|
| Wt | 1a | 1b | 1c | 1d | 250 |
| F312C | 2.70 ± 0.04 | 0.54 ± 0.05 | 0.23 ± 0.02 | 0.22 ± 0.02 | 250 |
| M94T | 0.73 ± 0.03 | 0.92 ± 0.14 | 1.1 ± 0.1 | 0.59 ± 0.04 | 236 |
| M62L | 1.30 ± 0.02 | 3.1 ± 0.1 | 0.81 ± 0.04 | 2.1 ± 0.0 | 272 |
| M150L | 3.4 ± 0.7 | 8.5 ± 0.6 | 0.38 ± 0.03 | 0.30 ± 0.01 | 275 |
| N96S | 1.3 ± 0.1 | 5.9 ± 0.1 | 0.15 ± 0.01 | 0.41 ± 0.01 | 261 |
| F312C/M94T | 2.7 ± 0.5 | 0.64 ± 0.06 | 0.20 ± 0.03 | 0.62 ± 0.07 | 215 |
| F312C/M150L | 5.5 ± 0.3 | 6.8 ± 0.3 | 0.12 ± 0.01 | 0.22 ± 0.01 | 265 |
| D98N | 0.90 ± 0.25 | 1.3 ± 0.1 | 0.05 ± 0.03 | 0.01 ± 0.00 | e |
aWt apparent rate of oxygen reduction using o-dianisidine as the electron donor: 0.0042 ± 0.0003 s−1.
bWt apparent rate of nitrite reduction using o-dianisidine as electron donor: 0.15 ± 0.01 s−1.
cWt apparent rate of oxygen reduction using pseudoazurin as the electron donor: 14.0 ± 0.3 s−1.
dWt apparent rate of nitrite reduction using pseudoazurin as the electron donor: 401 ± 23 s−1.
eReduction potential not measured in this study but presumed to be similar to wt.
fReducton potentials are measured with an uncertainty of ±1 mV.
ο-Dianisidine and pseudoazurin oxidation
ο-Dianisidine is structurally similar to DAB; however, the oxidized form is also water soluble, enabling spectrophotometric determination of oxidation rates. ο-Dianisidine can serve as the reductant for reduction of both dioxygen and nitrite by NiR (Table II). With the exception of M94T, all variants identified by screening oxidized o-dianisidine more rapidly than did wt NiR (0.0042 s−1) when dioxygen was the electron acceptor. Expressed as dioxygen reduction rates relative to the wt protein (nX wt), the functional consequences of the individual substitutions F312C (2.7X wt) and M150L (3.4X wt) were additive in the shuffled variant F312C/M150L NiR (5.5X wt). Asp98 is an active site residue that forms an H-bond to nitrite bound to the type-2 Cu site (Boulanger and Murphy, 2001). By comparison, the site-directed variant D98N NiR oxidized o-dianisidine at a rate similar to that of wt NiR (0.9X wt).
With sodium nitrite (2.5 mM) as the electron acceptor, different activity patterns were observed among these proteins (Table II). Firstly, wt NiR reduced nitrite at a significantly greater rate (0.15 s−1) using o-dianisidine as reductant, roughly 35 times faster than with the rate of dioxygen reduction. All but one of the variants identified by screening were more efficient in reducing nitrite than in reducing dioxygen with o-dianisidine as the electron source. The F312C NiR variant, on the other hand, exhibited lower relative activity (0.54X wt) in nitrite reduction than in reduction of dioxygen (2.7X wt). The variant M150L NiR showed the greatest activity in reducing nitrite with o-dianisidine as a donor (8.5X). However, the rate exhibited by the M1150L/F312C NiR double variant (6.8X wt) was consistent with the negative influence of the F312C substitution on nitrite reduction observed for the F312C single variant.
When dioxygen reduction was measured with pseudoazurin as the reductant, wt NiR exhibited a rate constant of 14 s−1, a value ∼3000 times faster than observed with o-dianisidine as the reductant (Table II). The relative rate for D98N NiR is 20X lower than that for wt NiR. Interestingly, with the exception of M94T, all of the variants obtained from high-throughput screening displayed lower relative activity values at reducing oxygen with pseudoazurin. Most notably, M150L reduced oxygen with pseudoazurin at 38% of the wt rate.
With one exception, nitrite reduction with reduced pseudoazurin as the electron donor was most rapid with wt NiR. Only the M62L NiR variant exhibited greater activity 2.1X than the wt enzyme (Table II). Again, the F312C NiR variant showed diminished pseudoazurin/nitrite activity (0.22X wt), the M150L NiR variant exhibited similarly diminished activity (0.30X wt), and the double variant M150L/F312C NiR had 0.22X activity compared with wt NiR.
Type-1 Cu site reduction potentials and electronic spectra
Reduction potentials of the type-1 copper sites of wt and variant NiRs were measured with cyclic voltammetry. The potential of wt NiR, 250 mV (versus SHE), agreed well with previously published values of 240 (Kohzuma et al., 1994) and 260 mV (Wijma et al., 2003). Reduction potentials for the variants ranged from 215 mV for F312C/M94T to 275 mV for M150L.
Visible spectra of the variants differed significantly from that of wt NiR. The spectra of the M150L NiR variant and the double variant, M150L/F312C differ the most from that of wt NiR in that they exhibit a significantly increased intensity in the absorption maximum at ∼600 nm and minimal absorption at 460 nm (Fig. 4). The spectrum of M62L NiR also had a distinctly different absorption spectrum, with the 458 nm maximum of wt NiR shifted to 464 nm and an A464/A589 ratio (1.7) greater than that (1.3) of wt NiR. This change in spectrum changes the appearance of the M62L variant from the olive green of wt NiR to grass green.
Fig. 4.

Electronic spectra of wt and variant NiRs. Wt, solid black; M62L, orange; F312C, pink; M94T, light blue; N96S, purple; M150L, red; M150L/F312C, green; M94T/F312C, dark blue.
Crystal structures
The residue positions of all five single-variants are mapped to the wt NiR structure to show their relative positions in Fig. 5. Crystal structures were solved for three variants: M62L, F312C/M94T and F312C/M150L to define the structural changes in these variants with improved o-dianisidine oxidation activity (Fig. 5). In wt NiR, Met62 is located in close proximity to two type-1 copper site ligands, His145 and Met150. Two structural rearrangements occur in the M62L variant relative to the wt NiR structure. Specifically, the imidazole moiety of His145 is rotated 20° about χ2 such that the copper coordinating Nδ1 atom is displaced in the direction of Leu62 by ∼0.2–0.3 Å (over the three monomers). A second significant change in the M62L NiR structure is at Trp144. The tryptophan side chain rotates about χ2 by ∼7° such that the indole ring moves closer to the type-1 copper.
Fig. 5.

(A) Residue positions of positively screened variants mapped to the wt NiR structure (PDB code 1SJM). Labels at magenta-colored residues refer to the substitution obtained at that residue position. In addition to Met150, type-1 Cu site His and Cys ligands are shown as green sticks. Type-2 Cu His ligands are also shown as green sticks. Copper ions are spheres. (B) Stereo representation of the M62L NiR crystal structure at the site of the substitution. Wt NiR (PDB code 1SJM, green) and M62L (slate) are superposed. Cu ions are spheres. (C) Superposition of the wt NiR (green) with the double variant M94T/F312C (salmon) crystal structure. (D) Superposition of the wt NiR (green) with double variant M150L/F312C (light blue) crystal structures. Oxygen atoms are colored red and nitrogen atoms are colored dark blue. ‘Resi’ refers to a residue position that is mutated.
The crystal structures of F312C/M94T and F312C/M150L variants of NiR have the substitution F312C in common (Fig. 5). Both structures show an oxidized cysteine (sulfinoalanine) at position 312. No other significant structural changes are attributed to this substitution. In wt NiR Met94 is located at the molecular surface close to the type-1 copper site. Replacement of this residue with threonine results in a new hydrogen bond (2.7 Å) formed between the threonine hydroxyl and Asn115 side-chain carbonyl. To accomplish this interaction, the Asn115 amide rotates ∼180° about χ2. Additionally, the conformation of Glu113 changes to occupy space taken by Met94 in wt NiR, forming a new hydrogen bond (2.9 Å) between the Glu113 carboxylate and the side chain amide nitrogen of Asn96.
Met150 is the axial ligand to the type-1 copper site in wt NiR. Replacement with leucine shifts the type-1 copper ∼0.6 Å into the plane of the other three ligands, His95, Cys136 and His145 giving rise to a trigonal planar geometry (Fig. 5).
Discussion
Library generation and colony screening
Mutagenic library generation and high-throughput screening enabled isolation of variants with altered specificity for both the electron donor and acceptor. The use of DAB as the electron donor and as a screening agent fulfills the requirements for a colorimetric colony screen. In particular, DAB oxidation results in the deposition of red, water-insoluble end product at lifted colonies with NiR-catalyzed oxygen reducing activity. Increased sensitivity is achieved since DAB is both a chromogenic electron donor for NiR and a chromogenic substrate for peroxidase that consumes NiR-generated H2O2. Addition of horseradish peroxidase to the screening reagent ensured removal of H2O2, which inactivates NiR. Other reductants could serve as electron donors to NiR, while retaining DAB/peroxidase as H2O2 indicators. However, the use of stronger chemical reductants such as ascorbate and dithionite diminished the peroxidase-catalyzed oxidation of DAB as well as other chromogenic peroxidase substrates (data not shown); therefore, the screen is limited in terms of electron donors. Nonetheless, reduced pseudoazurin has been shown to greatly increase the rate of red color formation in this colony lift screen, using DAB and horseradish peroxidase as H2O2 indicators (data not shown). Alternative protein electron donors such as reduced cytochrome c have been used successfully in NiR assays and may be expected to substitute for pseudoazurin (Hulse et al., 1988).
The colony lift and lysis allows cytoplasmically expressed protein to bind copper and be functional. The screen is broadly applicable to non-native MCOs expressed in the cytoplasm of E. coli. Color formation by oxidation of DAB is expected to be a robust process for many MCOs due to their efficient oxidation of o-dianisidine (up to 400 s−1) (Schosinsky et al., 1974; Hassett et al., 1998; Larrondo et al., 2003; Quaratino et al., 2007). We have verified the broad applicability of the colony screen (without horseradish peroxidase in the screening reagent) with an MCO from Arthrobacter. Indeed, color formation occurs in less than 10 s for this enzyme (unpublished results). Thus, the mutagenesis and screening methods described here could allow for evolution of soluble, heterologously expressed variant MCOs with initial poor yields in E. coli. The detection limit of the screen for NiR, which expresses at ∼300 mg/l culture, is estimated to less than 0.004 s−1 based on oxidation of o-dianisidine (Table II). By extrapolation, the high sensitivity of DAB as a substrate enables the accurate detection of MCO activity with even minimal expression (<50 µg l−1 culture for an enzyme with an apparent rate of 200 s−1). Thus, the library generation and screening method can be used as a folding screen, provided that mutations do not influence the rate of DAB oxidation. Current research is aimed at randomizing specific regions of NiR and using the high-throughput method to screen for variants that allow proper folding and expression.
Oxygen reduction in NiR
Facultative denitrifying bacteria transition between using dioxygen and nitrate/nitrite as electron acceptors in oxidative phosphorylation (Ingledew and Poole, 1984). The heme cd1 containing NiRs are able to reduce dioxygen to water and are insensitive to an aerobic environment (Lam and Nicholas, 1969). In contrast, copper-containing NiRs are characterized as being sensitive to dioxygen under reducing conditions due to the production of hydrogen peroxide and, possibly, more reactive hydroxyl radicals (Kakutani et al., 1981b). Notably, catalase protects NiR during catalytic turnover in the presence of dioxygen and a significant rate of dioxygen reduction, 14 s−1, was determined for wt NiR as monitored by the reoxidation of pseudoazurin (Table II). The nitrite reduction activity of wt NiR under the same conditions is 30X greater. If the coupling of electrons to H2O2 production is high, then the expected rate of H2O2 production is 7 s−1, as this reduction is a two-electron process. The presence of excess catalase results in the conversion of H2O2 to water and dioxygen, so dioxygen is partially replenished in the assay.
Asp98 is an absolutely conserved residue that forms a hydrogen bond to nitrite and nitric oxide bound to the type-2 copper site (Tocheva et al., 2004). According to the proposed mechanism of nitrite reduction (Tocheva et al., 2004), deprotonated Asp98 forms a hydrogen bond with HNO2, poising it for reduction and addition of a second proton to yield a copper nitrosyl intermediate and the release of a water molecule. The D98N substitution decreased the specific NiR activity of NiR to 1% of wt (Boulanger et al., 2000). Additionally, crystal structures of D98N NiR show a loss of interaction of this residue with nitrite or water (in the nitrite-free structure) (Boulanger and Murphy, 2001), suggesting that this interaction is a key requirement for catalysis. To gain insight into the mechanism of dioxygen reduction, D98N NiR was also assayed and found to have 5% of wt NiR activity with pseudoazurin as the electron donor (Table II). Strongly diminished oxygen reduction by D98N NiR suggests a role for the carboxylate group of Asp98 in this reaction as well. As suggested in the proposed nitrite reduction mechanism, Asp98 could serve to stabilize the singly protonated, two-electron reduced, Cu(II)-hydroperoxo intermediate. A second proton acquisition would result in hydrogen peroxide formation. Secondly, Asp98 could relay protons to reduced dioxygen to form hydrogen peroxide.
Dioxygen and nitrite reduction with DAB and o-dianisidine
Wt NiR oxidizes o-dianisidine in the presence of dioxygen with a rate constant of 0.0042 s−1, a value <0.1% of the rate constant observed with reduced pseudoazurin as the electron donor. With the exception of M94T, variants obtained from high throughput screening improved the rate of o-dianisidine oxidation with dioxygen as the electron acceptor up to 5.5-fold. The greatest increase in activity was for the double variant M150L/F312C. Met150 is the axial ligand for the type-1 copper site in NiR and replacement with leucine alters the site geometry from tetragonal to planar trigonal (Fig. 5) and thus is expected to have a significant impact on the reduction potential. Indeed, the reduction potential of M150L NiR was determined to be 275 mV, 25 mV greater than wt (Table II). Substitution of the axial methionine with leucine in the homologous NiR from Alcaligenes xylosoxidans increased the reduction potential of this protein by 96 mV (to 336 mV) as determined by potentiometric titration (Hough et al., 2005). The discrepancy in reduction potentials of this variant of AfNiR and the corresponding variant of A. xylosoxidans NiR may be explained in part by the different methods used for determination of potentials. A similar increase of 97 mV is measured by potentiometric titration for the methionine-to-leucine variant of CotA, an MCO (Durao et al., 2006). For A. xylosoxidans NiR, the rate of nitrite reduction was found to be 1.7X that of the wt enzyme with azurin as the reductant. This increase is in sharp contrast to the ∼3-fold loss of activity observed for AfNiR using pseudoazurin (Table II). Note that absolute rates for A. xylosoxidans NiR were not provided and may be significantly lower than those measured for AfNiR.
Of particular interest are the reduction potentials of the different NiR copper sites and substrates. By detailed electrochemical methods, the reduction potential of the type-1 copper of AfNiR has been determined to be 260 mV (Wijma et al., 2003), a relatively low value compared with other type-1 copper proteins, but greater than the reduction potential of the half reaction, 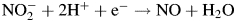 (calculated at neutral pH with NO in the aqueous phase (189 mV) (Wijma et al., 2004)), and in the same range as the cupredoxin pseudoazurin (270 mV) from which NiR receives electrons (Kukimoto et al., 1995; Wijma et al., 2004). The reduction potential of the type-2 copper site has proven more difficult to measure. Studies on a homologous NiR from Rhodobacter sphaeroides have suggested that the reduction potential of the active site without nitrite bound is significantly lower than that of the type-1 site, by at least 40 mV (Olesen et al., 1998). It was suggested that the binding of nitrite to the type-2 copper could raise the reduction potential to favor electron transfer from the type-1 copper (Olesen et al., 1998). The standard potential for the reduction of oxygen to hydrogen peroxide at neutral pH is 280 mV (Sawyer, 1991), close to that measured for the NiR type-1 Cu.
(calculated at neutral pH with NO in the aqueous phase (189 mV) (Wijma et al., 2004)), and in the same range as the cupredoxin pseudoazurin (270 mV) from which NiR receives electrons (Kukimoto et al., 1995; Wijma et al., 2004). The reduction potential of the type-2 copper site has proven more difficult to measure. Studies on a homologous NiR from Rhodobacter sphaeroides have suggested that the reduction potential of the active site without nitrite bound is significantly lower than that of the type-1 site, by at least 40 mV (Olesen et al., 1998). It was suggested that the binding of nitrite to the type-2 copper could raise the reduction potential to favor electron transfer from the type-1 copper (Olesen et al., 1998). The standard potential for the reduction of oxygen to hydrogen peroxide at neutral pH is 280 mV (Sawyer, 1991), close to that measured for the NiR type-1 Cu.
In NiR, the M150L substitution gives an improved catalytic rate with o-dianisidine but has detrimental effects with pseudoazurin as the electron donor. The Met150 residue is buried and the M150L substitution results in minimal change to the surface characteristics of the type-1 Cu site, suggesting that the substitution is not likely to affect the affinity for the chemical reductants. Leucine is preferentially found in place of the axial methionine in the type-1 sites of many laccases, MCOs that catalyze the oxidation of phenolic substrates as well as benzidine-based molecules such as DAB and o-dianisidine. Notably, DAB and o-dianisidine have higher reduction potentials (480 mV and 340 mV, respectively) than the type-1 Cu of wt NiR (Chung et al., 2000). In addition to the increase of 25 mV in type-1 Cu reduction potential measured for the M150L variant, several other studies of type-1 Cu sites have shown that replacement of the axial methionine with leucine raises the reduction potential (Hall et al., 1999; Hough et al., 2005; Durao et al., 2006). Therefore, the large increase in overall rate for o-dianisidine oxidation observed with the M150L variant is best attributed to the increase in reduction potential, and suggests that reduction of the type-1 site by DAB and o-dianisidine is rate limiting. That type-1 Cu reduction by o-dianisidine is rate limiting is also consistent with the similar rate of o-dianisidine oxidation by D98N NiR using either oxygen or nitrite as the electron acceptor.
Conversely, the same increase in reduction potential could be expected to slow electron transfer to the type-2 Cu, due to a decrease in thermodynamic driving force for electron transfer between the Cu sites. This model explains the lower rates observed for dioxygen and nitrite reduction with pseudoazurin (approximately 4 and 120 s−1, respectively) for the M150L variant. However, these values are still high compared with o-dianisidine (0.014 and 1.24 s−1, respectively), which suggests that intramolecular electron transfer between type-1 and type-2 Cu sites of the M150L variant is not limiting for catalysis with o-dianisidine. Higher reduction potentials at the type-1 Cu site in MCOs are favorable because of the relatively higher reduction potential of the trinuclear active site (390–780 mV) (Reinhammar and Vanngard, 1971; Reinhammar, 1972).
Phe312 is located at the surface of the deep pocket which houses the active site of NiR. This hydrophobic residue is also surrounded by several other hydrophobic residues, Val142, Val304 and Leu308. This hydrophobic patch has been proposed to be the route of nitric oxide egress during nitrite reduction (Adman et al., 1995). Substitution with cysteine greatly increases the size as well as the hydrophilicity of this pocket. The positive impact on o-dianisidine oxidation (2.7× wt) with dioxygen reduction and negative impact (0.54 X wt) with nitrite reduction suggests a specific role for the F312C substitution in dioxygen reduction using o-dianisidine. This role could involve redox cycling of the cysteine, in which hydrogen peroxide reacts with the reduced form to generate the oxidized form, and then o-dianisidine re-reduces it. Another possible explanation is an opening of the hydrophobic pocket for release of the more hydrophilic H2O2, although this does not explain why F312C and double variants are less effective than wt at dioxygen reduction with reduced pseudoazurin as the source of reducing equivalents. Opening of the hydrophobic pocket could aid in o-dianisidine interaction directly with an oxygen-bound intermediate at the active site. More specifically, faster sequential reduction of dioxygen with o-dianisidine could be facilitated from two directions, one electron via the traditional type-1 site/type-2 site relay and the other via the F312C pocket.
Funding
This research was funded by a Natural Sciences and Engineering Research Council (NSERC) Discovery Grant (M.E.P.M.) and a Canada Research Chair (A.G.M.). I.S.M was a recipient of a NSERC Postgraduate Scholarship. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and the National Institute of General Medical Sciences.
Footnotes
Edited by Albert Berghuis
References
- Adman E.T., Godden J.W., Turley S. J. Biol. Chem. 1995;270:27458–27474. doi: 10.1074/jbc.270.46.27458. [DOI] [PubMed] [Google Scholar]
- Averill B. Chem. Rev. 1996;96:2951–2964. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- Boulanger M.J. The Molecular Mechanism of Copper-Containing Nitrite Reductase. Vancouver: University of British Columbia; 2001. p. 158. [Google Scholar]
- Boulanger M.J., Murphy M.E. Biochemistry. 2001;40:9132–9141. doi: 10.1021/bi0107400. [DOI] [PubMed] [Google Scholar]
- Boulanger M.J., Murphy M.E. Protein Sci. 2003;12:248–256. doi: 10.1110/ps.0224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger M.J., Kukimoto M., Nishiyama M., Horinouchi S., Murphy M.E. J. Biol. Chem. 2000;275:23957–23964. doi: 10.1074/jbc.M001859200. [DOI] [PubMed] [Google Scholar]
- Cadwell R., Joyce G. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- Chung K.T., Chen S.C., Wong T.Y., Li Y.S., Wei C.I., Chou M.W. Toxicol. Sci. 2000;56:351–356. doi: 10.1093/toxsci/56.2.351. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. Acta Crystallogr. D. 1995;50:760–763. [Google Scholar]
- Durao P., Bento I., Fernandes A.T., Melo E.P., Lindley P.F., Martins L.O. J. Biol. Inorg. Chem. 2006;11:514–526. doi: 10.1007/s00775-006-0102-0. [DOI] [PubMed] [Google Scholar]
- Hall J.F., Kanbi L.D., Strange R.W., Hasnain S.S. Biochemistry. 1999;38:12675–12680. doi: 10.1021/bi990983g. [DOI] [PubMed] [Google Scholar]
- Hassett R.F., Yuan D.S., Kosman D.J. J. Biol. Chem. 1998;273:23274–23282. doi: 10.1074/jbc.273.36.23274. [DOI] [PubMed] [Google Scholar]
- Hough M.A., Ellis M.J., Antonyuk S., Strange R.W., Sawers G., Eady R.R., Samar Hasnain S. J. Mol. Biol. 2005;350:300–309. doi: 10.1016/j.jmb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Hulse C.L., Tiedje J.M., Averill B.A. Anal. Biochem. 1988;172:420–426. doi: 10.1016/0003-2697(88)90464-2. [DOI] [PubMed] [Google Scholar]
- Ingledew W.J., Poole R.K. Microbio. Rev. 1984;48:222–271. doi: 10.1128/mr.48.3.222-271.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M., Tiedje J., Averill B. FEBS Lett. 1991;291:41–44. doi: 10.1016/0014-5793(91)81099-t. [DOI] [PubMed] [Google Scholar]
- Kadono-Okuda K., Andres D. Anal. Biochem. 1997;254:187–191. doi: 10.1006/abio.1997.2422. [DOI] [PubMed] [Google Scholar]
- Kakutani T., Beppu T., Arima K. Agric. Biol. Chem. 1981a;45:23–28. [Google Scholar]
- Kakutani T., Watanabe H., Arima K., Beppu T. J. Biochem. (Tokyo) 1981b;89:463–472. doi: 10.1093/oxfordjournals.jbchem.a133221. [DOI] [PubMed] [Google Scholar]
- Kakutani T., Watanabe H., Arima K., Beppu T. J. Biochem. (Tokyo) 1981c;89:453–461. doi: 10.1093/oxfordjournals.jbchem.a133220. [DOI] [PubMed] [Google Scholar]
- Kohzuma T., Shidara S., Suzuki S. Bull. Chem. Soc. Jap. 1994;67:138–143. [Google Scholar]
- Kukimoto M., Nishiyama M., Murphy M.E., Turley S., Adman E., Horinouchi S., Beppu T. Biochemistry. 1994;33:5246–5252. doi: 10.1021/bi00183a030. [DOI] [PubMed] [Google Scholar]
- Kukimoto M., Nishiyama M., Ohnuki T., Turley S., Adman E.T., Horinouchi S., Beppu T. Protein Eng. 1995;8:153–158. doi: 10.1093/protein/8.2.153. [DOI] [PubMed] [Google Scholar]
- Lam Y., Nicholas D.J. Biochim. Biophys. Acta. 1969;180:459–472. doi: 10.1016/0005-2728(69)90025-5. [DOI] [PubMed] [Google Scholar]
- Larrondo L.F., Salas L., Melo F., Vicuna R., Cullen D. Appl. Environ. Microbiol. 2003;69:6257–6263. doi: 10.1128/AEM.69.10.6257-6263.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson I.S., Murphy M.E. Cell Mol. Life Sci. 2007;64:2887–2899. doi: 10.1007/s00018-007-7310-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K. Meth. Mol. Biol. (Clifton, NJ) 2003;231:23–28. doi: 10.1385/1-59259-395-X:23. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. Acta Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Olesen K., Veselov A., Zhao Y., Wang Y., Danner B., Scholes C.P., Shapleigh J.P. Biochemistry. 1998;37:6086–6094. doi: 10.1021/bi971603z. [DOI] [PubMed] [Google Scholar]
- Quaratino D., Federici F., Petruccioli M., Fenice M., D'Annibale A. Antonie van Leeuwenhoek. 2007;91:57–69. doi: 10.1007/s10482-006-9096-4. [DOI] [PubMed] [Google Scholar]
- Reinhammar B.R. Biochim. Biophys. Acta. 1972;275:245–259. doi: 10.1016/0005-2728(72)90045-x. [DOI] [PubMed] [Google Scholar]
- Reinhammar B.R., Vanngard T.I. Eur. J. Biochem./FEBS. 1971;18:463–468. doi: 10.1111/j.1432-1033.1971.tb01264.x. [DOI] [PubMed] [Google Scholar]
- Rensing C., Grass G. FEMS Microbiol. Rev. 2003;27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- Sawyer D.T. Oxygen Chemistry. New York: Oxford University Press; 1991. [Google Scholar]
- Schosinsky K.H., Lehmann H.P., Beeler M.F. Clin. Chem. 1974;20:1556–1563. [PubMed] [Google Scholar]
- Shin W., Sundaram U.M., Cole J.L., Zhang H.H., Hedman B., Hodgson K.O., Solomon E.I. J. Am. Chem. Soc. 1996;118:3202–3215. [Google Scholar]
- Shleev S., Tkac J., Christenson A., Ruzgas T., Yaropolov A.I., Whittaker J.W., Gorton L. Biosens. Bioelectron. 2005;20:2517–2554. doi: 10.1016/j.bios.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Solomon E.I., Chen P., Metz M., Lee S.K., Palmer A.E. Angew. Chem. Int. Ed. Engl. 2001;40:4570–4590. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Stemmer W.P. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- Tocheva E.I., Rosell F.I., Mauk A.G., Murphy M.E. Science. 2004;304:867–870. doi: 10.1126/science.1095109. [DOI] [PubMed] [Google Scholar]
- Tran Q.H., Unden G. Eur. J. Biochem./FEBS. 1998;251:538–543. doi: 10.1046/j.1432-1327.1998.2510538.x. [DOI] [PubMed] [Google Scholar]
- Vagin A., Teplyakov A. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- Wijma H.J., Boulanger M.J., Molon A., Fittipaldi M., Huber M., Murphy M.E., Verbeet M.P., Canters G.W. Biochemistry. 2003;42:4075–4083. doi: 10.1021/bi027270+. [DOI] [PubMed] [Google Scholar]
- Wijma H.J., Canters G.W., de Vries S., Verbeet M.P. Biochemistry. 2004;43:10467–10474. doi: 10.1021/bi0496687. [DOI] [PubMed] [Google Scholar]
- Wijma H.J., Macpherson I., Alexandre M., Diederix R.E., Canters G.W., Murphy M.E., Verbeet M.P. J. Mol. Biol. 2006;358:1081–1093. doi: 10.1016/j.jmb.2006.02.042. [DOI] [PubMed] [Google Scholar]