

Abstract
Ca2+ binding proteins are essential for regulating the role of Ca2+ in cell signaling and maintaining Ca2+ homeostasis. Negatively charged residues such as Asp and Glu are often found in Ca2+ binding proteins and are known to influence Ca2+ binding affinity and protein stability. In this paper, we report a systematic investigation of the role of local charge number and type of coordination residues in Ca2+ binding and protein stability using de novo designed Ca2+ binding proteins. The approach of de novo design was chosen to avoid the complications of cooperative binding and Ca2+-induced conformational change associated with natural proteins. We show that when the number of negatively charged coordination residues increased from 2 to 5 in a relatively restricted Ca2+-binding site, Ca2+ binding affinities increased by more than 3 orders of magnitude and metal selectivity for trivalent Ln3+ over divalent Ca2+ increased by more than 100-fold. Additionally, the thermal transition temperatures of the apo forms of the designed proteins decreased due to charge repulsion at the Ca2+ binding pocket. The thermal stability of the proteins was regained upon Ca2+ and Ln3+ binding to the designed Ca2+ binding pocket. We therefore observe a striking tradeoff between Ca2+/Ln3+ affinity and protein stability when the net charge of the coordination residues is varied. Our study has strong implications for understanding and predicting Ca2+-conferred thermal stabilization of natural Ca2+ binding proteins as well as for designing novel metalloproteins with tunable Ca2+ and Ln3+ binding affinity and selectivity.
PACS codes: 05.10.-a
1. Background
Electrostatic interactions have been shown to be important for many biological processes [1]. Additionally, they are known to be important for the stability and folding of proteins, though their net effects can vary greatly from protein to protein, depending on the relative contributions of direct Coulomb interactions between charges and the desolvation cost [2]. A series of studies on surface charge variants of a cold shock protein has revealed that the context of mutations rather than the net protein charge is a critical determinant of protein stability [3-5]. If net charge were an important factor, one would expect proteins with small net charges to be more stable than proteins with large net charges. However, Henzl and Graham reported that β-parvalbumin, with a net charge of -16, displays a 7°C higher thermal transition temperature than a close homologue, α-parvalbumin, with a net charge of -5 [6,7].
In contrast to the variable effects of charged surface residues, Ca2+-binding to charged coordination residues almost always increases protein stability, simply due to selective ion binding to the folded state over the denatured state [8]. In the absence of Ca2+, calmodulin (CaM) is highly dynamic and labile even at room temperature [9,10]. Upon cooperatively binding four Ca2+ ions, the Tm of CaM increased to over 90°C. Additionally, proteins such as thermolysin, α-amylase, cadherin, subtilisin, and protein S bind Ca2+ to enhance thermal and proteolytic stability [11-14]. Many of the recent advances in the development of thermally stable enzymes for industrial use require the use of Ca2+ for stability [12,15].
In addition to their effect on thermal stability, electrostatic interactions contribute greatly to Ca2+-binding affinity [16-19]. A survey has shown that 106 of 165 EF-hand Ca2+ binding sites contain 4 negatively charged coordination residues [20]. Our recent comprehensive analysis of the available structures in the Protein Data Bank also reveals that three and four negative charges are frequently observed in the coordination shells of natural Ca2+-binding sites [21]. In general, removal of charged coordination residues is found to reduce Ca2+-binding affinity, though theories about the role of charge effects in Ca2+-binding affinity [8,16], such as the acid-pair hypothesis [18], are often inconsistent with experimental observations of Ca2+ binding affinity in mutant Ca2+-binding proteins, such as EF-hand proteins [22,23]. Replacements of Asp and Glu at the conserved loop positions 1 and 12 with non-charged residues significantly decreased Ca2+ binding affinity of several EF-hand proteins [24]. Additionally an Asp-to-Asn mutation at Site III of CaM led to a decrease in both Ca2+ binding affinity and cooperativity at the C-terminal domain [16,25]. On the other hand, the same mutation at Site II of CaM led to the opposite effect at the N-terminal domain. Henzl et al. have shown that increasing the net negative charge in the coordination sphere of rat α- and β-parvalbumins from -4 to -5 increased Ca2+ binding affinity [26,27], while Falke et al. have shown that in galactose-binding protein, increasing the net negative charge from -3 to -4 decreased affinity for Ca2+ but increased affinity for trivalent metal ions [28]. Factors that may explain the variations in observed effects of charge mutations on metal-binding affinity include cooperation between different metal-binding sites on the same protein, conformational entropy, and contribution of the protein environment. To separate out the contribution of charged coordination residues to Ca2+ binding affinity, extensive work has also been conducted utilizing peptide fragments encompassing the Ca2+ binding sites of several proteins. Unfortunately, this approach suffers from the limitations of ill-defined structures in solution and peptide aggregation [8,16,29-31]. To date, a quantitative description of local charge effects on Ca2+ binding and stability has not been established.
We have developed a design strategy to overcome the confounding factors noted above, allowing for a comprehensive study of local determinants for Ca2+ binding affinity and thermal stability [8,19,32]. Our first designed Ca2+-binding protein did not possess a stable structure at room temperature [8]. However, since then, several proteins with stable structures have been designed and engineered. Among them, CD2.trigger undergoes Ca2+-dependent conformational change (unpublished data) while 7E15 [32], 6D15 (also termed Ca.CD2) [19], and 6D79 [33] do not exhibit global conformational changes upon Ca2+ binding. These designed proteins are excellent model systems for gaining insight into the tradeoff between Ca2+ binding affinity and protein stability without the limitations associated with natural Ca2+-binding proteins [32]. In this paper, we report our systematic investigation of the roles of local charge and coordination residue type on Ca2+ binding affinity and Ca2+-conferred thermal stabilization. To investigate the effects of local charges without the interference of protein global conformational change, CD2.7E15 (Fig. 1) was chosen as the template, primarily because it tolerates up to five negative charges at the binding site instead of four negative charges as in 6D15 and 6D79 [32,33]. Additionally, CD2.7E15 retains a CD2-like fold and no significant conformational changes are observed due to binding of Ca2+ (Kd ~ 0.1 mM) in the designed pocket. Using various biophysical measurements as well as theoretical calculations, we show that, when the number of charged coordination residues increases from 2 to 5, Ca2+ binding affinities and metal selectivity for trivalent Ln3+ over divalent Ca2+ increases, while the thermal transition temperatures of the proteins in the absence of cations decrease. Ca2+ or Ln3+ binding to the designed Ca2+-binding pocket, however, allows the proteins to regain thermal stability. The results suggest that in a relatively restricted Ca2+-binding site, more negative charges facilitate binding of Ca2+ and Ln3+ accompanied by a tradeoff in protein stability due to significant repulsion among the negatively charged coordination residues.
Figure 1.
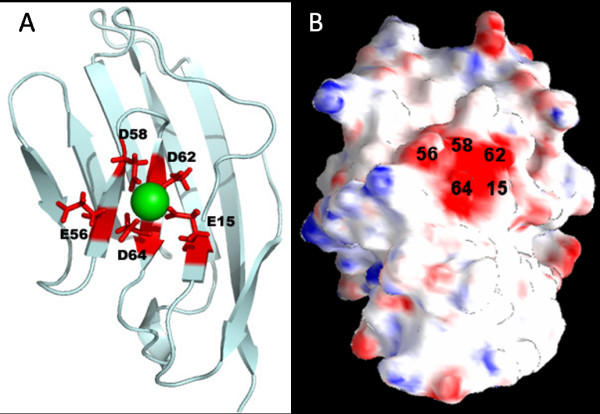
Model structure of designed proteins. (A) Model structure of EEDDD (7E15) and the coordination design of the variants. (B) The surface potential of Ca2+-free EEDDE, calculated using DelPhi[45], shows the highly-charged Ca2+-binding location. The calculation used interior and exterior dielectric constants of 4 and 80, respectively.
2. Methods
2.1. Protein engineering and purification
CD2.7E15 (also referred to as EEDDD in this study) variants were engineered using the classical PCR method from CD2.7E15 DNA in the vector pGEX-2T. The mutations were confirmed using automated DNA sequencing at the Biology Core Facility of Georgia State University. The protein expression, purification, and concentration estimation were carried out as previously reported [32]. The background Ca2+ was minimized by incubating the sample with EGTA first, followed by a pH gradient separation using a HiTrap SP HP column (GE Healthcare) with chelexed (Chelex-100 resin, BioRad) buffers.
2.2. Trp fluorescence
Trp emission spectra over 300-400 nm, with excitation at 282 nm, of proteins (4 μM) in 10 mM Tris, pH 7.3 in the presence of 10 mM Ca2+ or 1 mM EGTA were collected using a PTI fluorimeter and a cuvette with a 1 cm pathlength at room temperature.
2.3. Metal binding
Cation binding was done in 20 mM PIPES-10 mM KCl, pH 6.8. The Tb3+-FRET fluorescence signals were monitored using a PTI fluorimeter following the procedure described previously. The Tb3+ binding affinity was measured by direct Tb3+ titration and the La3+ binding affinity was derived using the competitive binding of La3+ against Tb3+. The Ca2+-binding affinity was measured using 1H-15N HSQC NMR spectra by titrating Ca2+ into the protein sample with 40-50 μM EGTA at the initial point and monitoring the chemical shift changes versus the Ca2+ concentrations. The Kd of each CD2.7E15 variant was calculated using the average of results from multiple resonances and the uncertainty represented the different responses of these resonances to Ca2+ binding.
2.4. Far-UV CD
Far-UV CD spectra were collected on a Jasco-810 spectropolarimeter coupled with a Peltier temperature controller. The signal was monitored over 260-200 nm with four repeat scans. The proteins (25 μM) were in 10 mM Tris-10 mM KCl, pH 7.3 with either 1 mM EGTA, 10 mM Ca2+, or 0.05 mM Tb3+, placed in a 1 mm pathlength cell with a sealed top. Buffer signals were subtracted from the spectra. For thermal titration measurements, CD spectra were taken at 2-5 degree increments with four to six repeat scans over a temperature range from 15 to 85°C. Five minutes were allowed for equilibration at each temperature before the scans were taken. The signal change at 225 nm was plotted using KaleidaGraph and fitted using a two-state transition model,
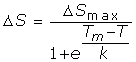 |
(1) |
where ΔS and ΔSmax are the signal changes at temperature T point and the final temperature, respectively; Tm is the transition temperature; and k is a fitting parameter measuring the steepness of the thermal transition. The reported Tm and the uncertainty are the average and standard deviation of three runs, respectively.
2.5. Prediction of mutational effects on folding stability and Ca2+ binding affinity
The model structures of EEDDD with bound Ca2+ were generated by the design program and the structures of the other variants were generated from EEDDD using the program SYBYL (Tripos Co.). Hydrogen atoms were added using SYBYL. The bound Ca2+ was removed for the apo-form structures, and the Ca2+ was replaced by a Tb3+ at the same location to construct the Tb3+-loaded form structures. All Asp and Glu residues were assumed to be unprotonated (and all Lys and Arg protonated); while pKa values are prone to be perturbed when charges are clustered [34], the assumed protonation states seem appropriate for the neutral pH where melting temperatures and ion binding affinities were measured.
The Poisson-Boltzmann (PB) equation was solved to calculate the electrostatic free energies of the protein variants in bound- and apo-forms using the UHBD program[35]. A salt concentration of 10 mM in the solution was used. Electrostatic contributions of mutational effects on the Ca2+ binding affinity and folding stability of CD2.7E15 variants were calculated following previously-published protocols [4,36]. The effect of a mutation on the folding stability was calculated as the change in the electrostatic folding free energy:
| (2) |
In our case, the wild-type protein was identified as the original 7E15 (i.e., EEDDD), and the mutant was any of the variants introduced here. The unfolded state of proteins was modeled as individual residues separately dissolved in the solvent. Therefore, in the unfolded state, residues other than the one under mutation make the same contribution to the electrostatic free energies of the wild-type protein and mutant, and do not affect ΔΔGf. Neglecting this contribution, the electrostatic folding free energy of either the wild-type protein or the mutant is:
| (3) |
where Gel(X) is the electrostatic free energy of molecule X; "protein" refers to the folded protein; and "residue" refers to a residue, the one under mutation, that is carved out of the folded structure. Gel has both a Coulombic component and a solvation component.
Calculations of ΔΔGf were done on the apo forms only. A basic assumption underlying the present study is that the metal ions bind to the designed proteins only when they are in the folded state, since only then coordination residues come together to form the binding site. Under this assumption, a state in which the protein is both unfolded and metal ion-loaded does not exist, and it would not be appropriate to apply the procedure for calculating ΔΔGf to the metal ion-loaded forms. We note that preferential binding of the folded state by metal ions shifts the folding-unfolding equilibrium towards the former.
The contribution of a point mutation to metal ion binding affinity can be expressed as the binding free energy difference between the mutant and the wild-type complex:
| (4) |
where ΔGb is the binding free energy, calculated as
| (5) |
The calculated result is to be compared with the counterpart from experimentally determined binding affinity:
| (6) |
where kB is Boltzmann's constant.
3. Results
3.1. Engineering of CD2.7E15 variants
In a previous publication [32], CD2.7E15 referred to a rationally designed Ca2+-binding protein with an engineered Ca2+-binding site at the B, E, and D β-strands of the host protein CD2 (Fig. 1), with coordination residues Glu, Glu, Asp, Asp, and Asp occupying positions 15, 56, 58, 62, and 64, respectively. Here CD2.7E15 is referred to as EEDDD; other variants of EEDDD are referred to using a similar notation, consisting of single-letter codes for the residues occupying the five coordination sites. The EEDDD variant was chosen as the template to investigate the effects of charge and coordination residue type on Ca2+ binding based on the following considerations. First, this metal binding site presented the possibility to study the relationship between Ca2+ binding and protein stability. We have previously shown that the cluster of five negatively charged residues at this location did not unfold the protein and metal binding did not alter the global conformation of the protein [32]. Second, this variant retains two wild-type residues, E56 and D62. Third, using the EEDDD template, a series of variants with local net charges ranging from -5 to -2 by replacing Asp or Glu with Asn or Gln (Table 1) could be generated. Variants with local net charges of -1 or 0 were not generated since they were not expected to bind Ca2+ with a reasonable affinity. Finally, using the EEDDD template, pairs of variants, such as EEDDD/EEDDE or EEDDN/EEDDQ, which possess identical net charges but differ by one methylene group, could also be generated, presenting the opportunity to investigate the size effects of coordination residues.
Table 1.
The cation binding affinities for 7E15 variants
| Protein | Residues | Charge | Kd | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 15 | 56 | 58 | 62 | 64 | Tb3+ (μM)a | La3+ (μM)a | Ca2+ (mM)b | ||
| CD2 | N | E | L | D | K | -1 | NB | NB | NB |
| EEDDD | E | E | D | D | D | -5 | 0.4 ± 0.2 | 0.5 ± 0.1 | 0.10 ± 0.05 |
| EEDDE | E | E | D | D | E | -5 | 0.8 ± 0.2 | 0.7 ± 0.1 | 1.1 ± 0.4 |
| EEDDN | E | E | D | D | N | -4 | 6.3 ± 0.6 | 2.2 ± 0.4 | 2.1 ± 0.5 |
| EEDDQ | E | E | D | D | Q | -4 | 14 ± 3 | 3.2 ± 0.7 | 0.7 ± 0.2 |
| EENDN | E | E | N | D | N | -3 | > 30 | > 15 | > 10 |
| NENDN | N | E | N | D | N | -2 | NB | NB | NB |
Buffer condition: 20 mM PIPES-10 mM KCl, pH 6.8. NB: No observed binding. a: from Tb3+-FRET. b: from NMR HSQC.
3.2. Conformational analysis of the 7E15 variants
At 25°C the far-UV CD spectra for all of the variants displayed a single negative maximum at ~216 nm, nearly identical to that of wild-type CD2, indicating the maintenance of wild-type β-sheet secondary structure. Moreover, the Trp fluorescence emission spectra of the variants displayed a single maximum at 327 nm, identical to that of the wild-type protein, suggesting that the mutations did not alter the native fold. The CD and the Trp fluorescence spectra remained unchanged with the addition of cations, suggesting the absence of global conformational changes upon metal binding (Fig. 2).
Figure 2.

Conformational analysis by far-UV-CD and Trp fluorescence. (A) The far-UV CD spectra of all 7E15 variants in the presence of 1 mM EGTA or 10 mM Ca2+ in 10 mM Tris, pH 7.4 show a single negative maximum at ~216 nm. CD2 with Ca2+ (open circle), EEDDQ with EGTA (open diamond) or Ca2+ (solid diamond), and EENDN with EGTA (open triangle) or Ca2+ (solid triangle) are shown as examples. (B) The normalized Trp fluorescence spectra for all 7E15 variants almost overlap with a single emission maximum at 327 nm. CD2(open circle), EEDDN with EGTA (open diamond) or Ca2+ (solid diamond), and EEDDE with EGTA (open triangle) or Ca2+ (solid triangle) are shown as examples.
3.3. Metal binding
Taking advantage of aromatic residues such as Trp-32 in the host protein, we used aromatic sensitized fluorescent resonance energy transfer (FRET) to analyze metal binding in the EEDDD variants (Fig. 3). As negative control, the addition of wild-type CD2 into a Tb3+ solution resulted in negligible change in the Tb3+ fluorescence emission at 545 nm. In contrast, the addition of EEDDD, EEDDE, EEDDN, and EEDDQ into the Tb3+ solution resulted in more than 30-fold increases in Tb3+ fluorescence emission. The addition of NENDN only resulted in a slight enhancement relative to CD2, suggesting that the variant bound Tb3+ at most with a weak affinity. The Tb3+-binding affinities were obtained by directly titrating Tb3+ into the protein variants (Table 1). The variants with -5 charges showed the strongest affinities with Kd < 1 μM. The variants with -4 charges show affinities at the μM level. The Tb3+ binding affinity of EENDN was not accurately measured due to the limitation of solubility of the protein in high concentration of Tb3+. La3+ binding affinities were obtained by competitively titrating La3+ into Tb3+-protein mixtures and monitoring the decrease in Tb3+ fluorescence intensity. The trend of La3+-binding affinities for the 7E15 variants was the same as that for Tb3+. That is, the -5-charged variants showed the strongest affinities, followed by the -4, -3 and -2 charged variants. The addition of Ca2+ into Tb3+-protein mixtures led to only a small decrease in the Tb3+ fluorescence, suggesting inefficient competition by Ca2+ due to weaker Ca2+ binding.
Figure 3.

Metal binding studied by Tb3+-FRET. (A) The fluorescence enhancement at 545 nm with increasing concentrations of Tb3+ in 5 μM of EEDDN in 20 mM PIPES, 10 mM KCl pH 6.8. (B) Determination of Tb3+ binding affinity of EEDDN by analyzing the dependence of the fluorescent intensity change on Tb3+ concentration. The solid line is the fitted curve. (C) Decrease in fluorescence by the addition of La3+ into a fixed concentration of Tb3+ and EEDDN. This data was analyzed to obtain Kd for La3+.
The Ca2+-binding affinity of EEDDD was previously determined to be 100 ± 50 μM from the chemical shift changes of the residues proximate to the metal binding position using 1H-15N HSQC spectra of 15N labeled proteins [32]. In this study, similar Ca2+ titrations on the other variants were performed. The resonances in the spectra for the other variants were partially identified by comparing them to the spectrum of EEDDD. For the -4 and -5 charged variants, Ca2+ specifically perturbed several resonances while other resonances maintained their positions throughout the titration process. Fig. 4A shows the 1H-15N HSQC spectra of EEDDQ as an example of data obtained. Perturbed resonances were assigned to coordination residues or their neighbours, including G61, L63, Q64, E58, E56, K66, and Q22. The Ca2+-binding affinities of EEDDE, EEDDQ, and EEDDN were obtained by analyzing the chemical shift perturbations (Fig. 4B and Table 1). Unlike Tb3+ and La3+ binding, the -4 charged EEDDQ showed a stronger Ca2+ binding affinity than the -5 charged EEDDE. Additionally, the chemical shift perturbations induced by Ca2+ binding to EEDDQ were greater than those to EEDDE. The -3 charged variant EENDN displayed small but significant chemical shift perturbations at high Ca2+ concentrations, while the -2 charged variant NENDN did not undergo any significant changes with the addition of up to 13 mM Ca2+ (Fig. 4C). Hence EENDN possessed only weak Ca2+ binding ability, while NENDN did not show observable Ca2+ binding. The null binding results of EENDN and NENDN confirmed that the chemical shift perturbations observed on the other variants were due to specific Ca2+ binding and not to other nonspecific processes such as salt effects.
Figure 4.

Metal binding studied by 2D-NMR. (A) The 1H-15N HSQC spectra of EEDDQ at different Ca2+ concentrations. The Ca2+-induced chemical shift perturbations are indicated by the arrows. Several assigned resonances are labeled. (B) Determination of the Ca2+-binding affinity of EEDDQ by following the chemical shift perturbations as a function of Ca2+ concentration. D62 (solid circle); downfield resonance of Q64 sidechain amide (solid square); upfield resonance of Q64 sidechain amide (solid triangle); K66; ×, Q22 (inverted solid triangle). The solid lines are generated by assuming a 1:1 Ca2+ binding to EEDDQ. The results from different resonances are similar. (C) Overlay of the HSQC spectra of NENDN in the presence of 0.05 mM EGTA (red) or 13.1 mM Ca2+ (green).
3.4. Thermal denaturation of 7E15 variants
The thermal transition temperatures (Tm) of all the variants were obtained using far-UV CD (Fig. 5). At 90°C, EEDDD and the other variants were found to be fully unfolded, just like wild type CD2. Compared with CD2, which possesses a Tm of 61 ± 1°C [19], the clustered negative charges at the β-strands decreased the Tms of EEDDD (41 ± 1°C) and EEDDE (45 ± 3°C) in the absence of metal. Under the same conditions, the Tms of the -4 charged variants were significantly higher, with values of 53 ± 2°C for EEDDN and 56 ± 2°C for EEDDQ. In the absence of metal, Tm values of EENDN (61 ± 1°C) and NENDN (62 ± 1°C) were similar to that of wild-type CD2 (Table 2).
Figure 5.
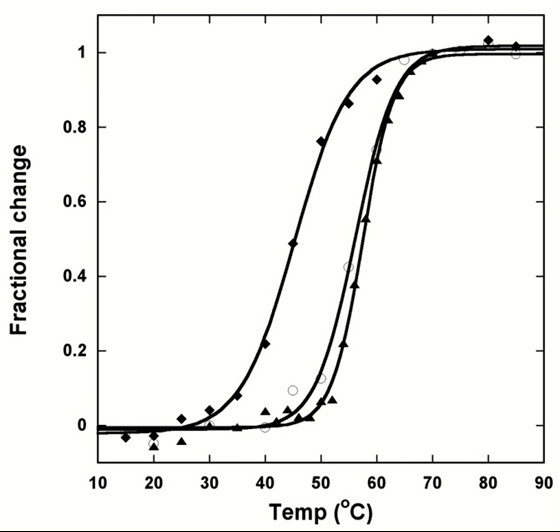
Thermal denaturation. The thermal denaturation of EEDDE in the presence of 1 mM EGTA (solid diamond), 10 mM Ca2+ (open circle), or 0.05 mM Tb3+ (solid triangle) in 10 mM Tris, 10 mM KCl. The solid lines are generated by fitting the data to Equation 1.
Table 2.
Tm values of 7E15 variants
| Protein | Tm (°C) | ||
|---|---|---|---|
| EGTA | Ca2+ | Tb3+ | |
| CD2 | 61 ± 1 | 61 ± 1 | - |
| EEDDD | 41 ± 1 | 51 ± 1 | - |
| EEDDE | 44 ± 2 | 56 ± 1 | 63 ± 2 |
| EEDDN | 52 ± 2 | 58 ± 2 | 54 ± 1 |
| EEDDQ | 56 ± 1 | 61 ± 1 | 60 ± 1 |
| EENDN | 61 ± 2 | 59 ± 2 | 59 ± 2 |
| NENDN | 60 ± 3 | 58 ± 2 | 58 ± 4 |
Using far-UV CD all Tm values were obtained in 10 μM protein solution with 1 mM EGTA, 10 mM Ca2+, and 0.1 mM Tb3+, respectively. The buffer system is 10 mM Tris, 10 mM KCl, and pH 7.4.
A consequence of the assumption that metal ions can only bind to the folded proteins is that the folding stability would be increased by metal binding. Upon binding of Ca2+ or Tb3+, the Tm values of the -4 and -5 charged variants indeed increased significantly (Table 2). Ca2+-binding increased the Tm of -5 variants EEDDD and EEDDE by ~10°C and Tb3+ binding increased the Tm of EEDDE by ~20°C. Additionally, Ca2+ binding increased Tmof -4 variants EEDDN and EEDDQ by ~5°C, and Tb3+ binding increases the Tm of both variants by ~2-4°C. Neither Ca2+ nor Tb3+ was observed to cause significant changes in the Tm values of EENDN and NENDN, consistent with the weak or non-observable metal binding of these variants.
The order of Tms of the variants in the absence of metal binding can be summarized as follows: CD2 (-1) ~NENDN (-2) ~EENDN (-3) > EEDDQ (-4) > EEDDN (-4) > EEDDE (-5) > EEDDD (-5). Upon binding Ca2+, the order Tms was largely the same, except that Tm values of EEDDQ and EENDN were the same. Tb3+ binding introduced another alteration in the ordering of Tms: the Tm of EEDDE now exceeded that of EEDDQ.
3.5. Electrostatics calculations
Upon folding, a cluster of like charges is expected to decrease protein stability due to the individual loss of solvation and the collective repulsion among the charges. Upon metal ion binding, the protein and metal ion simultaneously experience unfavorable desolvation and favorable charge-charge attraction. These electrostatic effects were modeled using the Poisson-Boltzmann (PB) equation, from which the electrostatic contributions to folding stability and binding free energy were calculated. Since the CD, fluorescence, and NMR results all showed that the overall tertiary and secondary structures of the variants were similar to those of wild-type CD2, the structures of the variants on the structure of wild-type CD2 were modeled with minimal changes (i.e., all residues other than the one under mutation were fixed). In addition, it was assumed that the metal binding did not perturb the binding pocket or the global conformation of the protein. The calculations of mutational effects on the folding (binding) free energy ΔΔGf (ΔΔGb) are described in Materials and Methods.
The calculated results for ΔΔGf correlated well with the experimentally observed Tms of the apo forms, with R2 = 0.91 (Fig. 6A). Both the calculations and the experimental data indicated that the folding stability of the variants is ordered in the following way: EEDDE ~EEDDD < EEDDQ ~EEDDN < EENDN ~NENDN. Apparently, the variants with lower net charges experienced less charge repulsion around the binding pocket, thereby increasing folding stability and Tm.
Figure 6.
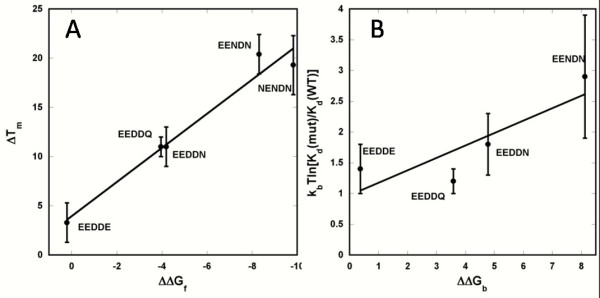
Electrostatics calculations. (A) Correlation of observed Tm values of the apo-7E15 variants and calculated changes in folding energy. (B) Correlation of calculated (ΔΔGb) and experimental results for mutational effects on binding free energy. The error bars are for experimental measurements.
According to our electrostatic calculations of ΔΔGb, the Ca2+ binding affinities of the variants were ordered as follows: EEDDD > EEDDE > EEDDQ > EEDDN > EENDN > NENDN. That is, binding is more favorable at sites with more negative net charge. Compared with the experimental measurements, the order in the Ca2+ binding affinities of EEDDE and EEDDQ was reversed, which may be an indication of non-electrostatic effects not included in our modeling. Nevertheless, there appeared to be good overall agreement between the electrostatic calculations and the experimental measurements regarding the mutational effects on metal-binding affinity (Fig. 6B), suggesting that electrostatic interactions indeed play a major role in the Ca2+ binding affinities of these protein variants.
4. Discussion
It is clear that electrostatic interactions play important roles in protein folding and in metal ion binding affinity. As shown in Table 1, trivalent cations bind more strongly to all of the EEDDD variants than divalent Ca2+. Moreover, the variants with five negatively charged residues in the metal binding pocket have higher metal ion binding affinities than variants with fewer negative coordination residues. These observations can be simply explained by charge-charge attraction. However, in addition to the direct charge effect, there are multiple factors contributing to the selectivity of different cations, such as coordination properties, binding pocket size, and solvation energy. For example, Zn2+ and Fe3+ prefer sulfur as coordination atoms instead of oxygen [37,38]. Improper pocket size, in addition to influencing cation selectivity, may also alter the van der Waals interaction energy. Ion solvation energy is also an important factor in cation binding and is responsible for the selectivity of Ca2+ over Mg2+ in metalloproteins [37,38]. We have previously shown that designed Ca2+-binding sites in proteins exhibit strong metal selectivity for Ca2+ and Ln3+ over excess Mg2+, Zn2+, Cu2+, K+, and Na+ [8,19,32,33].
Our highly negative charged designed variants, EEDDD and EEDDE, show the highest binding affinity for metal ions but the lowest folding stability. Binding is more favorable in EEDDD than EEDDE, most likely due to the extra methylene group in the latter, which has the effect of reducing the local charge density, hence decreasing cation binding affinity. This extra methylene group also appears to influence protein stability, as the Tm comparisons of EEDDD/EEDDE and EEDDN/EEDDQ suggest that mutation of Lys 64 by Glu instead of Asp or by Gln instead of Asn increases the thermal stability for both apo and loaded forms (Table 2). It is plausible that the extra methylene group in the carboxylic sidechain reduces the local repulsion among charged coordination residues. A statistical study has shown that Asp is the most preferred coordination residue in naturally-evolved Ca2+ binding sites followed by Glu, Asn, and Gln [16,39]. That is, charged coordination residues are preferred over neutral ones (D and E over N and Q) due to the charged nature of Ca2+. The calculation and experimental results reported here for the EEDDD variants indicate that steric size of the coordination residues is a less important factor for Ca2+ binding.
The introduction of negative charges and the removal of positively charged K64 in all of the EEDDD variants disrupt the local charge balance of CD2 (Fig. 1). Binding of Ln3+ or Ca2+ neutralizes the charge repulsion, resulting in an increase in the thermal transition temperature. The increase in Tm, = 10°C, is more dramatic for the most negatively charged variants (EEDDD and EEDDE). Based on the binding free energy calculations, the effects of cation binding on the Tm can be inferred. Weaker binding, as indicated by a positive ΔΔGb, is expected to correspond to a smaller increase in Tm. The small ΔΔGb value calculated for EEDDE (0.4 kcal/mol) in reference to EEDDD suggests that in the presence of cation binding, EEDDE shows an increase in Tm as significant as EEDDD. The moderate ΔΔGb values calculated for EEDDQ (3.6 kcal/mol) and EEDDN (4.8 kcal/mol) suggest that these two variants show a moderate increase in Tm. On the other hand, the relatively large ΔΔGb values of EENDN (8.2 kcal/mol) and NENDN (10.2 kcal/mol) suggest a relatively small increase in Tm upon cation binding. These predictions agree well with the experimental results.
The predictions of mutational effects by our electrostatic calculations, both on folding stability and on cation binding affinity, achieved good correlations with the experimental results. However, the predictions differed from the experimental results in magnitudes, due to the simplicity of our electrostatic model. First of all, we used fixed charges. In reality, charges will redistribute due to electronic polarization upon folding or upon cation binding; the use of fixed charges will exaggerate electrostatic contributions. Such exaggeration is particularly severe for cation binding, since the residues under mutations are directly coordinated with the bound ion. Second, in modeling mutations, residues other than the one under mutation were fixed. In reality, surrounding residues will readjust; such relaxation will mitigate the mutational effect. Third, the electrostatic calculations were carried out on a single protein conformation. Inclusion of conformational sampling (e.g. by molecular dynamics simulation) as opposed to single conformation calculation, appears to increase the predictive power of our model [40,41]. Finally, we totally neglected non-electrostatic effects, such as hydrophobic and van der Waals interactions.
In summary, using de novo designed Ca2+ binding proteins, which circumvent complications related to cooperative binding and Ca2+-induced conformational change in natural proteins, enabled us to reach several conclusions. By increasing the number of negatively charged coordination residues from 2 to 5 in a relatively restricted Ca2+-binding site, the Ca2+ binding affinities were increased by more than 3 orders of magnitude. The metal selectivity for trivalent Tb3+ over divalent Ca2+ was also increased by more than 100 fold. On the other hand, increasing the number of negatively charged coordination residues decreased the thermal transition temperatures of the apo-proteins due to the repulsion between the negatively charged residues in the apo-form. The thermal stability of the proteins was regained upon Ca2+ and Ln3+ binding to the designed Ca2+ binding pocket. Our study thus show that the charged coordination residues increase Ca2+ and Ln3+ affinity at the expense of decreased protein stability due to the charge repulsion of the Ca2+-free form. Furthermore, charge numbers of -3 or -4 for Ca2+ binding favor protein stability as well as Ca2+ binding. The steric size of the coordination residues is not crucial as long as the Ca2+-binding pocket is properly formed. In addition to revealing key factors involved in Ca2+ binding affinity and Ca2+-conferred thermal stabilization in natural Ca2+ binding proteins, our results regarding the net charge and coordination type provides important insights into engineering proteins such as thermoenzymes with enhanced stability [42-44].
Contributor Information
Anna Wilkins Maniccia, Email: alwilkins@gmail.com.
Wei Yang, Email: yangwei@ciac.jl.cn.
Julian A Johnson, Email: juju10683@yahoo.com.
Shunyi Li, Email: lisy@hubu.edu.cn.
Harianto Tjong, Email: harianto@mailer.sb.fsu.edu.
Huan-Xiang Zhou, Email: zhou4@fsu.edu.
Lev A Shaket, Email: levshak88@yahoo.com.
Jenny J Yang, Email: chejjy@langate.gsu.edu.
Acknowledgements
We thank Jin Zou, Dan Adams, and Michael Kirberger for their critical reading of this manuscript and helpful discussions. This work is supported in part by the following sponsors: NIH GM070555, NIH GM 62999 and NSF MCB-0092486 to JJY; NIH GM058187 to HXZ; NIH predoctoral fellowship to AWM and NIH supplemental fellowship to JAJ; CNSF grant 30870491 to WY.
References
- Honig B, Nicholls A. Science. 1995. pp. 1144–9. [DOI] [PubMed]
- Xiao L, Honig B. J Mol Biol. 1999. pp. 1435–44. [DOI] [PubMed]
- Perl D, Schmid FX. J Mol Biol. 2001. pp. 343–57. [DOI] [PubMed]
- Dong F, Vijayakumar M, Zhou HX. Biophys J. 2003. pp. 49–60. [DOI] [PMC free article] [PubMed]
- Wunderlich M, Martin A, Schmid FX. J Mol Biol. 2005. pp. 1063–76. [DOI] [PubMed]
- Henzl MT, Graham JS. FEBS Lett. 1999. pp. 241–5. [DOI] [PubMed]
- Henzl MT, Larson JD, Agah S. Biochemistry. 2000. pp. 5859–67. [DOI] [PubMed]
- Yang JJ, Gawthrop A, Ye Y. Protein Pept Lett. 2003. pp. 331–45. [DOI] [PubMed]
- Protasevich I, Ranjbar B, Lobachov V, Makarov A, Gilli R, Briand C, Lafitte D, Haiech J. Biochemistry. 1997. pp. 2017–24. [DOI] [PubMed]
- Tsalkova TN, Privalov PL. J Mol Biol. 1985. pp. 533–44. [DOI] [PubMed]
- Prasad A, Pedigo S. Biochemistry. 2005. pp. 13692–701. [DOI] [PubMed]
- Synowiecki J, Grzybowska B, Zdzieblo A. Crit Rev Food Sci Nutr. 2006. pp. 197–205. [DOI] [PubMed]
- Frommel C, Hohne WE. Biochim Biophys Acta. 1981. pp. 25–31. [DOI] [PubMed]
- Wenk M, Mayr EM. Eur J Biochem. 1998. pp. 604–10. [DOI] [PubMed]
- D'Auria S, Ausili A, Marabotti A, Varriale A, Scognamiglio V, Staiano M, Bertoli E, Rossi M, Tanfani F. J Biochem (Tokyo) 2006. pp. 213–21. [DOI] [PubMed]
- Linse S, Forsen S. Adv Second Messenger Phosphoprotein Res. 1995. pp. 89–151. [DOI] [PubMed]
- Falke JJ, Drake SK, Hazard AL, Peersen OB. Q Rev Biophys. 1994. pp. 219–90. [DOI] [PubMed]
- Reid RE, Hodges RS. J Theor Biol. 1980. pp. 401–44. [DOI] [PubMed]
- Wilkins AL, Ye Y, Yang W, Lee HW, Liu ZR, Yang JJ. Protein Eng. 2002. pp. 571–4. [DOI] [PubMed]
- Marsden BJ, Shaw GS, Sykes BD. Biochem Cell Biol. 1990. pp. 587–601. [DOI] [PubMed]
- Kirberger M, Wang X, Deng H, Yang W, Chen G, Yang JJ. J Biol Inorg Chem. 2008. pp. 1169–81. [DOI] [PubMed]
- Black DJ, Selfridge JE, Persechini A. Biochemistry. 2007. pp. 13415–24. [DOI] [PMC free article] [PubMed]
- Wu X, Reid RE. Biochemistry. 1997. pp. 8649–56. [DOI] [PubMed]
- Starovasnik MA, Su DR, Beckingham K, Klevit RE. Protein Sci. 1992. pp. 245–53. [DOI] [PMC free article] [PubMed]
- Waltersson Y, Linse S, Brodin P, Grundstrom T. Biochemistry. 1993. pp. 7866–71. [DOI] [PubMed]
- Henzl MT, Agah S, Larson JD. Biochemistry. 2004. pp. 10906–17. [DOI] [PubMed]
- Henzl MT, Hapak RC, Goodpasture EA. Biochemistry. 1996. pp. 5856–69. [DOI] [PubMed]
- Falke JJ, Snyder EE, Thatcher KC, Voertler CS. Biochemistry. 1991. pp. 8690–7. [DOI] [PubMed]
- Reid RE, Gariepy J, Saund AK, Hodges RS. J Biol Chem. 1981. pp. 2742–51. [PubMed]
- Gariepy J, Sykes BD, Reid RE, Hodges RS. Biochemistry. 1982. pp. 1506–12. [DOI] [PubMed]
- Yang W, Tsai T, Kats M, Yang JJ. J Pept Res. 2000. pp. 203–15. [DOI] [PubMed]
- Maniccia AW, Yang W, Li SY, Johnson JA, Yang JJ. Biochemistry. 2006. pp. 5848–56. [DOI] [PubMed]
- Jones LM, Yang W, Maniccia AW, Harrison A, Merwe PA van der, Yang JJ. Protein Sci. 2008. pp. 439–49. [DOI] [PMC free article] [PubMed]
- Isvoran A, Craescu CT, Alexov E. Eur Biophys J. 2007. pp. 225–37. [DOI] [PubMed]
- Madura JD, Briggs JM, Wade RC, Davis ME, Luty BA, Ilin A, Antosiewicz J, Gilson MK, Bagheri B, Scott LR, Mccammon JA. Computer Physics Communications. 1995. pp. 57–95. [DOI]
- Dong F, Zhou HX. Biophys J. 2002. pp. 1341–7. [DOI] [PMC free article] [PubMed]
- Dudev T, Chang LY, Lim C. J Am Chem Soc. 2005. pp. 4091–103. [DOI] [PubMed]
- Dudev T, Lim C. Chem Rev. 2003. pp. 773–88. [DOI] [PubMed]
- Pidcock E, Moore GR. J Biol Inorg Chem. 2001. pp. 479–89. [DOI] [PubMed]
- Tjong H, Zhou HX. Biophys J. 2008. pp. 2601–9. [DOI] [PMC free article] [PubMed]
- Tjong H, Zhou HX. J Chem Theory Comput. 2008. pp. 1733–44. [DOI] [PMC free article] [PubMed]
- Looger LL, Dwyer MA, Smith JJ, Hellinga HW. Nature. 2003. pp. 185–90. [DOI] [PubMed]
- Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Science. 2003. pp. 1364–8. [DOI] [PubMed]
- Lawrence MS, Phillips KJ, Liu DR. J Am Chem Soc. 2007. pp. 10110–2. [DOI] [PMC free article] [PubMed]
- Rocchia W, Alexov E, Honig B. Journal of Physical Chemistry B. 2001. pp. 6507–6514. [DOI]