

Summary
The formation of platelet-rich thrombi under high shear rates requires both fibrinogen and von Willebrand factor (VWF) as molecular adhesives between platelets. We attempted to describe the role of VWF as a potential substrate and modulator of the fibrinolytic system using binding assays, as well as kinetic measurements on the cleavage of fibrin(ogen) and a synthetic plasmin substrate (Spectrozyme-PL). The similar dissociation constants for the binding of plasminogen, plasmin, and active site-blocked plasmin onto immobilized VWF suggest that the primary binding site in plasmin(ogen) is not the active site. The progressive loss of clottability and generation of degradation products during fibrinogen digestion with plasmin were delayed in the presence of VWF at physiological concentrations, while VWF cleavage was not detectable. Determination of kinetic parameters for fibrinogen degradation by plasmin, miniplasmin and microplasmin showed that VWF did not modify the Km, whereas kcat values decreased with increasing VWF concentrations following the kinetic model of non-competitive inhibition. Inhibitory constants calculated for VWF were in the range of its physiological plasma concentration (5.4 μg/ml, 5.7 μg/ml and 10.0 μg/ml for plasmin, miniplasmin and microplasmin, respectively) and their values suggested a modulating role of the kringle 5 domain in the interaction between VWF and (mini)plasmin. VWF had no effect on the amidolytic activity of plasmin on Spectrozyme-PL, or on fibrin dissolution by (mini)plasmin. Our data suggest that VWF, while a poor plasmin substrate relative to fibrinogen, protects fibrinogen against degradation by plasmin preserving its clottability in plasma and its adhesive role in platelet-rich thrombi.
Keywords: von Willebrand factor, fibrinogen, plasmin, fibrinolysis, thrombolysis
Introduction
Von Willebrand factor (VWF) is a multimeric adhesive protein circulating in the plasma or deposited in the subendothelial layer of the vessel wall (1,2). It is synthesized in the vascular endothelial cells as a monomer of ca 250 kDa, which will form disulfide-bridged dimers, and then, larger multimers ranging from 500 kDa to in excess of 10,000 kDa. Since each monomer possesses binding sites for both vascular collagen types and platelets, the multimeric VWF is able to serve as a multivalent adhesive molecule between platelets and adhesive surfaces, as well as in-between platelets. According to in vitro experimental data gained in various models mimicking thrombus formation under flow conditions, the capture of platelets from flowing blood on thrombogenic surfaces becomes increasingly VWF-dependent with increasing hemodynamic forces. Above ca. 800 s−1 shear rate values VWF seems to be a necessary bridging molecule between the first layer of adhering platelets and their target surfaces (3). The significant role of VWF in the recruitment of further platelets and the vertical propagation of thrombus growth under high shear conditions has also been appreciated (4). Since shear rate values in normal medium-sized arteries are around 500 s−1 and in stenosed arteries are even higher approaching 20,000 s−1, VWF may be an important adhesive glue between platelets in arterial thrombi formed in vivo (1, 5). Fibrinogen is a soluble plasma glycoprotein of 340 kDa that is converted to fibrin by thrombin upon the activation of the blood coagulation cascade. Fibrin forms the insoluble, 3-dimensional matrix of venous thrombi, and both fibrinogen and fibrin serve as multivalent adhesive molecules between platelets in arterial thrombi with nearly 50 % of fibrinogen converted to fibrin (6,7). In addition VWF binds to polymerizing fibrin and this interaction facilitates the binding of VWF to platelets (8). These findings clearly support the concept that both VWF and fibrin(ogen) are necessary for holding platelets together in arterial thrombi (9-11).
The medical treatment of patients with arterial thrombosis has been based on two major approaches, the thrombolytic treatment attempts the enzymatic dissolution of the thrombi, while the invasive cardiologist tries to restore the vessel lumen with mechanical means (12). All presently applied thrombolytic agents activate plasminogen to plasmin, which is considered to be the major fibrinolytic protease cleaving both fibrinogen and fibrin to soluble degradation products (13). In addition to plasmin, elastase and cathepsin G secreted from polymorphonuclear leukocytes may also contribute to the dissolution of thrombi, not only by their direct fibrinolytic activities, but also by modulation of the plasmic system and the formation of miniplasmin, a protease with similar fibrinolytic efficiency, but lacking the first 4 kringle domains of plasmin (14-17). Another plasmin-derivative, microplasmin is an autodigestive product of plasmin sharing the same catalytic domain, but lacking all 5 kringle domains. It is probably formed only under in vitro conditions, and used as a tool for assessing the role of kringle domains in the interactions of plasmin with other molecules (15,18). The efficiency of thrombolysis, in vivo, might be modified by several factors including various components of arterial thrombi that have been shown to interfere with the fibrinolytic system in vitro (19-21). While a number of studies describe the kinetics of fibrin dissolution, data on VWF degradation by plasmin are limited. High enzyme concentrations relative to the effective fibrinolytic concentrations have been investigated, and the affinity of plasmin for VWF has not been determined (22). Considering that fibrinogen is among the most abundant plasma proteins (2-4 g/l in blood), which is well over the Km values of plasmin-derivatives (16,18), whereas VWF concentration is only about 0.01 g/l, it would be difficult to predict the fate of fibrin(ogen) and VWF in the presence of plasmin.
In this paper we describe that at its physiological concentration VWF is able to protect fibrinogen from degradation by plasmin-derivatives, which may help the preservation of fibrinogen clottability during thrombolysis, and stabilize arterial thrombi against thrombolytic treatments.
Materials and methods
Streptokinase and plasminogen-depleted human fibrinogen were from Calbiochem (La Jolla, CA). Sepharose CL 4B and Spectrozyme-PL (H-D-norleucyl-hexahydrotyrosyl-lysine-p-nitroanilide) were from GE Healthcare (Uppsala, Sweden) and American Diagnostica (Pfungstadt, Germany), respectively. 4-(2-Aminoethyl)-benzenesulfonyl fluoride (AEBSF, Pefabloc®) and bovine thrombin were purchased from Serva (Heidelberg, Germany), thrombin was further purified by ion-exchange chromatography on sulfopropyl-Sephadex yielding preparation with specific activity of 2100 IU/mg (23). Polyclonal rabbit-anti-human VWF, goat-anti-rabbit IgG peroxidase conjugate, bovine serum albumin (BSA), aprotinin and benzamidine were purchased from Sigma-Aldrich Kft. (Budapest, Hungary).
Human plasminogen was purified by affinity chromatography on Lysine-Sepharose from citrated human plasma containing 10 KIU (kallikrein inhibitor unit)/ml aprotinin and 10 mM benzamidine (24). Miniplasminogen, lacking the first four kringle domains of plasminogen, was prepared by elastase-mediated cleavage of plasminogen followed by affinity chromatography on Lysine-Sepharose (25). Microplasmin, an autocatalytic cleavage product of plasmin was isolated, as described (26). The generation of plasmin and miniplasmin from their zymogens, and determination of the active concentration for the three proteases were performed as previously described (27). All preparations of plasmin(ogen) and its derivatives were examined with gel electrophoresis and silver staining, which substantiated a single band with the expected molecular size under non-reducing denaturing conditions.
Plasmin, plasminogen and plasminPefabloc (Pefabloc active site blocked plasmin) were labeled with Eu-chelate (N1-(p-isothiocyanatobenzyl)-diethylenetriamine-N1,N2,N3,N4-tetraacetic acid chelated with Eu3+) according to the manufacturer’s instructions (Wallac, Turku, Finland) with the efficiency of 1-2 Eu3+-chelate/molecule of protein (determined by using the Europium standard solution from Wallac). Through its isothiocyanate arm the Eu-chelate binds covalently to primary aminogroups of the proteins. Following binding of the Eu-labelled protein to solid phase-immobilized ligand, the unbound protein is washed away and Wallac Enhancement solution® (photon acceptor in acidic detergent solution) is added, which releases the Eu from the protein-bound chelate and forms a new chelate in a micellar environment enhancing the fluorescent signal up to 107-fold. The detection sensitivity of the label is similar to that of radioisotopes and allows detection of attomoles of compound in a 96-well plate format (28,29).
Purification of VWF
Human frozen plasma was thawed in the presence of 6-aminohexanoic acid (0.01 M), benzamidine (10 mM) and aprotinin (10 KIU/ml). Ethanol was added (5 v/v% final concentration) to precipitate VWF, followed by storage at 4°C for 12 h. Thereafter the VWF fraction was separated with 15-min centrifugation at 10,800g and 4°C, the precipitate was resuspended in disodium EDTA (ethylenediamine tetraacetic acid; approx. 5 ml of 0.2 M EDTA added to precipitate from 250 ml plasma, pH 7.4), followed by gel filtration on a Sepharose CL 4B column (1.5 × 40 cm) equilibrated and eluted with 0.025 M citric acid pH 6.15 solution containing 0.05 M NaCl. Finally, fractions containing high molecular weight multimers were dialyzed against HBS buffer (Hepes-Buffered Saline: 0.01 M HEPES, 0.15 M NaCl, pH 7.4) (30). The functional activity of the isolated VWF was characterized by its ability to agglutinate washed, gel-filtered normal human platelets in the presence of ristocetin, defining pooled normal human plasma as 1 U/ml. In parallel the VWF antigen content of the preparations was determined with immunoturbidimetric assay setting the antigen level in pooled normal human plasma to 1 and the ratio of antigen to agglutinating activity was determined for each VWF preparation, which varied in the range 0.8 – 1.5. In all experiments the VWF concentration was adjusted according to the agglutinating activity calibration, but for better interpretation of the mass interrelations results are reported in μg/ml units using conversion based on the antigen/activity ratio and the average VWF concentration in pooled normal human plasma (10 μg/ml) (30). The equivalence of the specific agglutinating activity of the purified VWF preparations and VWF in pooled normal human plasma is an indicator that the multimeric distribution in our preparations corresponds to the multimeric pattern in normal plasma. The purity of the VWF preparations was checked using gel electrophoresis under reducing denaturing conditions with silver staining and immunoblot for VWF antigen, both of which showed a single band with molecular mass of 250 kDa.
Detection of fibrinogen degradation by measuring the clotting time
Plasmin (7.5 nM) was added to fibrinogen (2 g/l in 0.01 M imidazole, 0.15 M NaCl, pH 7.4, containing 2.5 mM CaCl2) in the absence or presence of VWF (10 or 15 μg/ml). After various times of incubation, fibrinogen was clotted with thrombin, and clotting time was measured with a coagulometer KC-1A (Amelung, Lemgo, Germany). Thrombin concentration was set to yield a 10-s clotting time with undegraded fibrinogen. If the clotting time was longer than 120 s, the sample was considered to be non-clottable.
Detection of the degradation of fibrinogen and VWF by SDS-polyacrylamide gel electrophoresis and Western blotting
For fibrinogen degradation, fibrinogen (0.5 g/l in HBS) was incubated with 12.5 nM plasmin at 37 °C in the absence or presence of VWF (10 μg/ml). After various times (30– 120 min) samples were taken into 0.1 M Tris-HCl, 0.1 M NaCl, pH 8.2 buffer containing 2 M urea and 1 w/v% SDS, heated at 100 °C for 1 min and gel electrophoresis was performed on a 7.5 w/v% polyacrylamide gel followed by visualization of protein bands with silver staining. For VWF degradation, samples were prepared similarly, but they were taken in the 1.5 – 24 h time frame and β-mercapto-ethanol (1 v/v%) was also included in the sample buffer to reduce the VWF multimer structure to monomers (250 kDa before proteolysis). Following gel electrophoresis, samples were electroblotted onto nitrocellulose membranes, and VWF and its degradation products were detected with polyclonal rabbit anti-human VWF immunoglobulin G followed by goat-anti-rabbit IgG peroxidase conjugate. The captured secondary antibodies were visualized with the ECL Western blotting analysis system (GE Healthcare, Uppsala, Sweden).
Effect of VWF on kinetic parameters for fibrinogen degradation by plasmin, miniplasmin, and microplasmin
Our previously published models were applied for determination of Km and kcat values for fibrinogen degradation in the absence or presence of VWF (16,18).
A global Km of the proteases for fibrinogen was determined by measurement of their amidolytic activities on the synthetic peptide substrate, Spectrozyme-PL (5-200 μM in HBS) in the absence or presence of 2 g/l fibrinogen competing for the enzyme. The cleavage of the synthetic substrate was followed at 405 nm with a Beckman DU-7500 spectrophotometer, and the initial reaction rate at various Spectrozyme-PL concentrations, v, was calculated by using an extinction coefficient of 8,820 M−1 cm−1 for p-nitroaniline. Km for fibrinogen was calculated using nonlinear curve fitting to the equation v=(vmax · S)/(S + KmSS (1+Fg/KmFg)), where KmSS and KmFg are the Michaelis-Menten constants for Spectrozyme-PL and fibrinogen, and S and Fg are the concentrations of Spectrozyme-PL and fibrinogen. Because VWF at 5 μg/ml concentration did not influence the kinetic parameters for Spectrozyme-PL cleavage (not shown), the same experimental setup could be used for the determination of KmFg in its presence. The addition of VWF did not alter the kinetics of Spectrozyme-PL-cleavage in the presence of fibrinogen either, therefore KmFg values determined in its absence were used for all further calculations.
Determination of a global kcat value for fibrinogen degradation by the proteases was based on the detection of ethanol-soluble fibrinogen degradation products and the application of the integrated form of the Michaelis-Menten equation, as detailed previously (18). Fibrinogen (2 g/l in HBS) was incubated with plasmin (5-20 nM), miniplasmin (10-20 nM) or microplasmin (10-20 nM) in the absence or presence of various concentrations of VWF up to 13.6 μg/ml. At various incubation times, samples were diluted into ice-cold ethanol (20 v/v%) to stop the reaction and precipitate undigested fibrinogen and fibrinogen fragment X. After centrifugation at 20,000g for 5 min, the concentration of ethanol-soluble degradation products was determined from the absorbance of the supernatant measured at 280 nm applying the extinction coefficient of 1.6 for 1 g/l protein. For kcat determinations first fS= S0 − S − KmFg ln(S/S0) was calculated, where S0 and S is the fibrinogen concentration at 0 and t times, respectively, and KmFg is determined independently, as described above. Thereafter, data were fitted to the equation fS=E. kcat .t, where fS is the value at t incubation time with E concentration of the enzyme.
Since the presence of VWF did not alter the KmFg values significantly, but lowered the catalytic rate constants in a concentration dependent way, a non-competitive model was applied to estimate the inhibitory constant for VWF. Values of kcat at various VWF concentrations were expressed in percentage of the value determined on the same day in the absence of VWF, and a 2 parameter nonlinear curve fitting to the equation kcat’ = kcat/(1 + (VWF/KivWf)) was used, where kcat’ is the catalytic rate constant at VWF concentration of VWF, and KivWf is the inhibitory constant.
Measurement of fibrin degradation in the presence of VWF
Formation of fibrin and its degradation by plasmin or miniplasmin was followed by turbidimetry, as described (18). Briefly, fibrinogen (2 g/l in 190 μl of HBS containing 2.5 mM CaCl2) was added to thrombin (30 U/ml in 5 μl) and 5 μl protease (at various final concentrations in the range of 5-20 nM). In the experiments with VWF, it was added to the fibrinogen solution at a final concentration of 10 μg/ml. Turbidity of the fibrin gel was followed at 340 nm with a Dynatech MR 5000 microplate reader (Dynatech Laboratories, Alexandria, VA) at 37 °C, and lysis-time was defined as the time until the turbidity of fibrin decreased to the half-maximal value. Assuming the proteases are saturated with substrate, the lysis-time (t) is a reciprocal function of the enzyme concentration (E) according to the equation t=Ft/(kcat .E), where Ft is the residual substrate concentration at time t, as detailed for kcat determinations in our previous papers (16,18).
In addition to turbidimetry, fibrin degradation was followed by SDS-polyacrylamide gel electrophoresis, as well. Fibrin clots formed from 0.5 g/l fibrinogen in the absence or presence of 10 μg/ml VWF containing 10 nM plasmin were dissolved after various incubation times in reducing or non-reducing sample buffer, and gel electrophoresis was performed as described for fibrinogen degradation above.
Binding of labeled plasmin, plasminogen and active-site blocked plasmin to immobilized VWF
96-well microtiter plates were coated with VWF (150 μl 1μg/ml VWF in HBS /well). After 12-hour incubation the VWF solution was discarded, the wells were washed for 5 min with HBS and blocked with 0.5 w/v% BSA in 0.01 M HEPES, 0.1 M NaCl, pH 7.4 for 1 h. After a 5-min wash with HBS, the VWF-coated wells were incubated with plasmin, plasminogen or plasminPefabloc solutions for 10 min (100 μl/well), containing constant amounts of Eu-labeled (0.01 μM) and varying amounts of nonlabeled proteins in a total concentration range of 0.01 to 2 μM. Plasmin(ogen) solutions were then removed and the VWF surface was washed quickly 3 times with HBS. The VWF-bound Eu was released in Enhancement Solution (200 μl/well). The emission fluorescence of Eu was measured with a microplate fluorimeter Victor2 (Wallac; excitation wavelength 340 nm, emission wavelength 615 nm, counting delay 400 μsec). Nonlinear least-squares regression analysis was used to identify the parameters (kNS, VWF and Kd) of a model that assumed a single class of saturable binding sites and a nonsaturable (nonspecific) component according to the equation Pbound=kNS.[P]free+VWF.[P]free/(Kd+[P]free) as described earlier (31). In this equation Pbound is the amount of bound plasmino(gen) and [P]free is the concentration of free plasmin(ogen), VWF is a constant of the experimental system (the amount of surface-immobilized VWF), Kd is the dissociation constant and kNS is a binding constant for the nonsaturable component. The independent variable in the equation, [P]free, was calculated using the conservation equation [P]bound+[P]free=[P]tot, where [P]tot is the nominal plasmin(ogen) concentration applied to the surface. This experimental setup allows the identification of the specific and non-specific component of the interactions (32).
Results
Effects of VWF on the course of fibrinogen degradation by plasmin
When fibrinogen is digested with plasmin, its clottability is gradually impaired, the clotting time with thrombin is prolonged (Fig. 1). Addition of VWF to the fibrinogen counteracts the clotting time-prolonging effect of plasmin, resulting in a longer incubation time needed to reach the unclottable state (Fig. 1). The formation of fibrinogen degradation products by plasmin in the absence or presence of VWF was monitored by SDS-polyacrylamide gel electrophoresis. The generation of unclottable fibrinogen degradation products, fragments Y and D (molecular weights are 150 kDa and 100 kDa, respectively) was markedly delayed in the presence of 10 μg/ml VWF (Fig. 2A). Because of the low concentration of VWF in the samples (10 μg/ml vs 500 μg/ml for fibrinogen), the fate of VWF could be detected only by Western blotting of similar samples. Using the conditions applied in Fig. 2A cleavage of VWF was not observed during the 2 h-incubation period (not shown). However, cleavage of VWF was seen at higher plasmin concentrations, even when fibrinogen was present, although in the absence of fibrinogen the degradation process was faster (Fig. 2B).
Figure 1. Effect of VWF on the clotting of fibrinogen partially digested by plasmin.
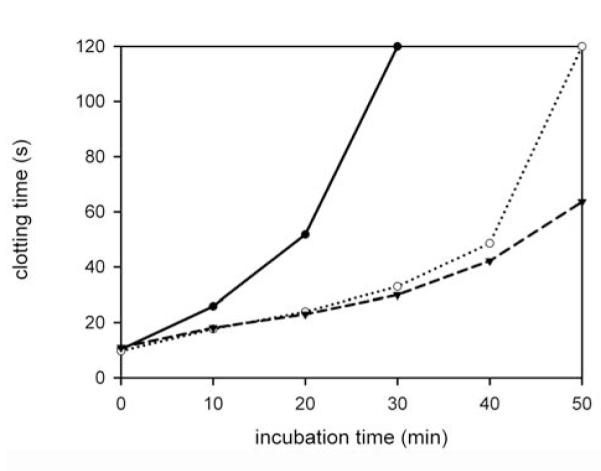
Fibrinogen (2 g/l) was incubated with plasmin (7.5 nM) in the absence (closed circle) or presence of VWF (open circle: 10 μg/ml, triangle down: 15 μg/ml). At the indicated times samples of partially digested fibrinogen were withdrawn and their clotting by thrombin was measured, as described in Materials and methods. Clotting times from a representative experiment of at least 3 are shown, values greater than 120 s are plotted as 120 s.
Figure 2. Degradation patterns of fibrinogen and VWF with plasmin.

A, Fibrinogen (0.5 g/l) was incubated with 12.5 nM plasmin in the absence (lanes 1-4) or presence (lanes 5-8) of 10 μg/ml VWF. Samples were withdrawn at the indicated times, fibrinogen degradation products were separated with electrophoresis on a 7.5 w/v% polyacrylamide gel and visualized with silver staining as described in Materials and methods. B, VWF (10 μg/ml) was incubated with 50 nM plasmin in the presence (lanes 1-4) or absence (lane 5) of fibrinogen (0.5 g/l). Samples were withdrawn at the indicated times, VWF degradation products were separated with electrophoresis on a 7.5 w/v% polyacrylamide gel and visualized with Western blotting, as detailed in Materials and methods.
Effects of VWF on kinetic parameters for fibrinogen degradation by plasmin, miniplasmin and microplasmin
The Michaelis-Menten constants of plasmin and its truncated variants for fibrinogen were determined according to a kinetic model of two competing substrates, fibrinogen and Spectrozyme-PL, and the measurement of the amidolytic cleavage of the latter. In the absence of VWF, fibrinogen did not change the vmax for Spectrozyme-PL, but elevated the Km in a competitive manner, and hence Km for fibrinogen could be computed as detailed in the Methods section. The addition of VWF to Spectrozyme-PL, however, had no influence on the kinetic parameters for the amidolytic reaction, not even when Spectrozyme-PL was added after preincubation of plasmin with VWF (not shown). When VWF was included with fibrinogen and Spectrozyme-PL, the rate of Spectrozyme-PL cleavage by plasmin or microplasmin was not influenced by VWF, therefore Km values for fibrinogen were similar to the ones in the absence of VWF (Table 1). Since these values were in good agreement with our earlier observations, the previously determined Km value for miniplasmin (18) was adopted for further calculations.
Table 1. Kinetic parameters for fibrinogen degradation in the absence or presence of VWF.
Km, kcat and KivWf values (mean ± standard errors) were determined as detailed in Materials and methods.
| additive | Plasmin | Miniplasmin | Microplasmin | |
|---|---|---|---|---|
| Km (μM) | none | 1.7 ± 0.3 | 8.1a | 10.2 ± 2.9 |
| VWF 5 μg/ml |
1.5 ± 0.3 | n.d. | 8.1 ± 2.2 | |
|
| ||||
| kcat (min−1) | none | 7.2 ± 0.8 | 10.9 ± 1.8 | 4.8 ± 0.4 |
| VWF 10 μg/ml |
2.5 ± 0.4 | 4.5 ± 1.2 | 2.5 ± 0.1 | |
|
| ||||
| KivWf ( μg/ml) | 5.4 ± 0.6 | 5.7 ± 1.2 | 10.0 ± 2.7 | |
from Ref.18
n.d.: not determined
Determination of catalytic rate constants for fibrinogen degradation by plasmin, miniplasmin, and microplasmin were based on the measurement of ethanol-soluble fibrinogen degradation products (representative curves for microplasmin with and without VWF are presented in Fig. 3, Inset). When VWF was present in the course of fibrinogen degradation, kcat values decreased with rising VWF concentrations, and the experimental data followed the kinetic model of non-competitive inhibition (Fig. 3). Inhibitory constants calculated for plasmin and miniplasmin (a plasmin derivative lacking the first 4 kringle domains) were similar, whereas that for microplasmin (lacking all 5 kringle domains) was higher (Table 1.)
Figure 3. Effect of VWF on the catalytic rate constants for fibrinogen degradation by microplasmin.
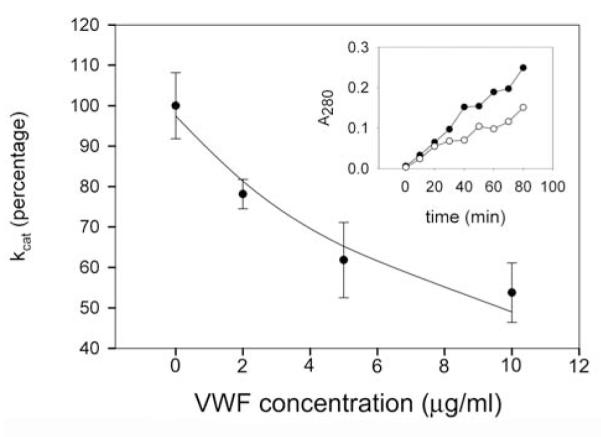
Fibrinogen (2 g/l) was incubated with microplasmin in the absence or presence of VWF and catalytic rate constants (kcat) for fibrinogen degradation were determined from experiments illustrated in the Inset, as detailed in Materials and methods. Values of kcat are expressed as percentage of the value in the absence of VWF, mean ± SEM are shown. Line shows regression values fitted to the equation kcat’ = kcat/ (1 + (VWF/KivWf)). Inset: Representative experiment on the kinetics of fibrinogen degradation by 32 nM microplasmin in the absence (closed circle) or presence (open circle) of 10 μg/ml VWF. Symbols present the absorbance at 280 nm of ethanol-soluble fibrinogen degradation products.
Dissolution of fibrin in the presence of VWF
Turbidity of the fibrin gel was followed in the course of fibrin formation and its solubilization by plasmin and miniplasmin at various enzyme concentrations was characterized with the turbidimetric lysis-time values (Fig. 4). Both fibrin and VWF were applied in the physiological concentration range (2 g/l for fibrin, 10 μg/ml for VWF), with no biologically significant alteration in the rate of fibrin dissolution found in the presence of VWF (the computed kcat values for plasmin are 4.5±0.2 min−1 in the presence of VWF and 5.1±0.4 min−1 in its absence, whereas the same values for miniplasmin are 3.5±0.1 min−1 and 4.2±0.2 min−1, respectively). The kinetic pattern of fibrin degradation product generation by plasmin, as observed with SDS-polyacrylamide gel electrophoresis, was not significantly altered by the presence of VWF, either (Fig. 4, Inset).
Figure 4. Effect of VWF on fibrin dissolution by plasmin and miniplasmin.
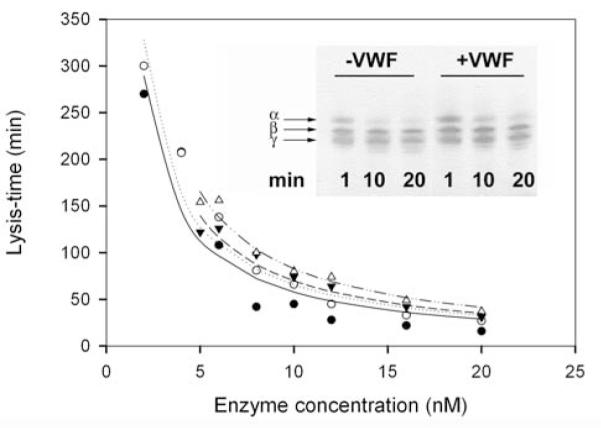
Lysis-times for fibrin dissolution with various concentrations of plasmin (circles) or miniplasmin (triangles) in the absence (closed symbols) or presence (open symbols) of 10 μg/ml VWF were determined from turbidimetric fibrinolytic assay, as detailed in Materials and methods, and plotted against the enzyme concentration applied. Lines show regression curves fitted to the equation t=Ft/(kcat .E). Inset: Fibrin degradation monitored by SDS-polyacrylamide gel electrophoresis. Fibrin clots formed from 0.5 g/l fibrinogen in the absence or presence of 10 μg/ml VWF containing 10 nM plasmin were incubated for the indicated time at 37 °C. Thereafter the clots were dissolved in 0.1 M Tris-HCl, 0.1 M NaCl, pH 8.2 buffer containing 2 M urea, 1 v/v% β-mercapto-ethanol and 1 w/v% SDS and gel electrophoresis was performed on a 12.5 w/v% polyacrylamide gel followed by visualization of protein bands with silver staining (α, β and γ indicate the three polypeptide chains of fibrin).
Plasmin, plasminogen and active-site blocked plasmin binding to immobilized VWF
The molecular interaction of VWF and plasmin was characterized on immobilized VWF surface with Eu-labeled and non-labeled ligands using a bimolecular binding model and a nonlinear regression analysis of the experimental data (Fig. 5). Evaluation of the equilibrium binding data for VWF gave a dissociation constant of Kd = 0.16±0.006 μM for plasmin, Kd = 0.10±0.002 μM for plasminogen and Kd = 0.27±0.007 μM for active-site blocked plasmin.
Figure 5. Equilibrium binding of plasmin(ogen) to VWF.
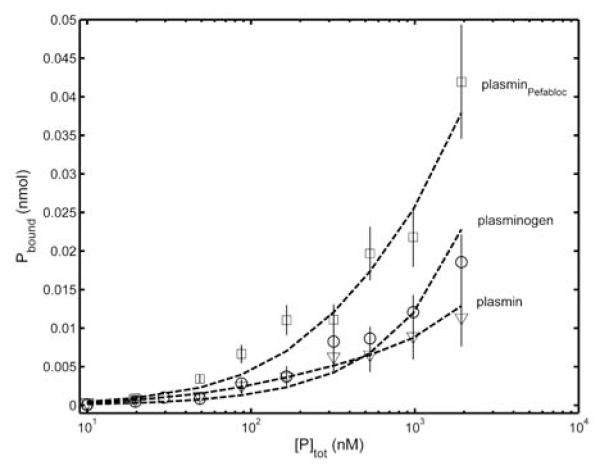
Mixtures of Eu-plasmin(ogen) or Eu-plasminPefabloc (10 nM) and varying amounts of the respective non-labeled ligand (0 - 2 μM) were incubated in VWF-coated microtiter plates for 10 min. After washing the VWF-bound Eu was measured, as described in Materials and methods. The data of five independent experiments are evaluated in a single procedure for each ligand and results are presented as mean (symbols), its standard deviation (vertical lines) and the best fit (dashed line), which is obtained for a single class of binding sites according to the equation Pbound=kNS.[P]free+VWF.[P]free/(Kd+[P]free).
Discussion
The formation of platelet-rich thrombi is important for physiological hemostasis, as well as for pathological intravascular thrombosis, both events occurring at hemodynamic conditions characterized by high shear stress. According to data gained from in vitro experimental models, as well as from histological analysis of ex vivo thrombi, such thrombi are glued by VWF and fibrinogen, 50 % of which is converted to fibrin (4-11). For the development of thrombolytic strategies, the process cannot be simulated in simplified fibrinolytic models, as several thrombus components have been shown to influence various steps of plasmin-dependent fibrinolysis (19-21). Since the role of VWF as an adhesive molecule in arterial thrombi is widely appreciated, and data on its degradation in the course of physiological or therapeutical thrombolysis are sparse, we initiated a study on its potential interaction with the plasmin-mediated fibrinolytic system.
For quantitative characterization of their possible physical interaction, binding experiments were performed on an immobilized VWF surface (Fig. 5). We have found, that the dissociation constants for plasmin-, or plasminogen-binding to VWF are in the same order of magnitude as the values reported in the literature for the plasminogen – fibrin binding (0.4 – 25 μM according to Refs. 33, 34), thus, these molecular interactions are not negligible. Considering that similar values were gained for plasmin, plasminogen and active site-blocked plasmin, an allosteric site could be the primary site of their interaction.
One synthetic peptide (Spectrozyme-PL) and two natural substrates (fibrinogen and fibrin) were applied to investigate the influence of VWF on the enzymatic activity of plasmin and its des-kringle derivatives. According to spectrophotometric measurements VWF does not alter the amidolytic cleveage of Spectrozyme-PL, a low molecular-weight peptide substrate (not shown), which also indicates that the primary interaction of VWF with plasmin does not involve its active site. Degradation of fibrinogen by plasmin and its des-kringle derivatives, however, was significantly delayed in the presence of physiological concentrations of VWF. Analysis of the formation of fibrinogen degradation products by SDS-PAGE, follow-up of fibrinogen clottability, and measurements on the generation of ethanol-soluble fibrinogen degradation products showed equivocally that VWF protects fibrinogen from degradation by plasmin and its des-kringle derivatives (Figs 1-3). Kinetic parameters estimated for fibrinogen degradation by the enzymes followed the model of non-competitive inhibition by VWF with an inhibitory constant of 5-10 μg/ml, which falls into the physiological plasma concentration range (Table 1). The higher inhibitory constant in the case of microplasmin (lacking all 5 kringle domains) compared to plasmin and miniplasmin suggests that kringle 5 might play a significant modulating role in the interaction between an allosteric site in the catalytic domain of the enzymes and VWF. While generation of fibrinogen degradation products was notably delayed in the presence of VWF at 3-fold molar excess of VWF monomers over plasmin (Fig. 2A), cleavage of VWF was not observable under the same circumstances, only at approximately equimolar VWF monomer and enzyme concentrations and after longer incubation times (Fig 2B). This is in accordance with earlier data on VWF cleavage by plasmin, where plasmin was applied at 200 nM concentration, a relatively high value compared to its effective fibrinolytic concentrations (22). Compared to fibrinogen, plasmin degradation of fibrin was not significantly inhibited by VWF, probably because of the known much higher enzyme affinity for fibrin than for fibrinogen (16,18). Thus, the overall functional consequence of plasmin binding to VWF is to prevent the digestion of large molecular substrates (fibrinogen) allowing free access of small substrates (Spectrozyme-PL) to the active site, but this sequestration of the protease is overcome by substrates with higher affinity (fibrin).
Cleavage of fibrinogen in the course of thrombolytic therapy is clinically important for both therapy efficacy and potential hemorrhagic side effects (37). One could speculate that VWF present in arterial thrombi may play not only a role as a platelet-platelet adhesive, but can also contribute to the resistance of thrombi against fibrinolysis. On the other hand, VWF circulating in plasma may help to preserve fibrinogen clottability in the course of systemic thrombolysis. The failure of this fibrinogen sparing mechanism may contribute to the disturbances in hemostasis and the bleeding disorder in von Willebrand disease. When both fibrinogen and VWF are presented at their physiological plasma concentrations for cleavage by plasmin, the enzyme prefers fibrinogen as a substrate, however, VWF cleavage is also a possible event at sites of arterial thrombi where enzymes may be present at high local concentrations (19) and thus plasmin can facilitate the clearance of VWF. Because plasmin interacts with both chains of factor VIII (35,36), which circulates in complex with VWF, the presence of factor VIII together with VWF in vivo may further divert plasmin from action on fibrinogen. The appearance of plasmic degradation products of VWF has been suggested as an indicator of VWF-related hemostatic abnormalities contributing to the bleeding complications occurring after thrombolytic therapy, even though the overall functional VWF tests did not indicate a major drop of VWF activities below the normal levels (37). Our present data suggests that the presence of VWF in plasma, as well as in arterial thrombi is worth of considering not only as a molecular glue for the formation of platelet-rich plugs, but also as a potential target and modulator of thrombolysis.
Acknowledgements
We acknowledge the technical expertise of Ida Horváth and Györgyi Oravecz. The authors are grateful to Dr. Colin Longstaff (National Institute for Biological Standards and Control, South Mimms, UK) for helpful discussion and language editing of the manuscript.
Appendix
| 1. | What is known on this topic
|
| 2. | What this paper adds
|
Footnotes
This work was supported by the Hungarian Scientific Research Fund [OTKA-F42475], [OTKA 75430], [OTKA K60123], Medical Scientific Council [ETT 005/2009] and the Wellcome Trust [083174/B/07/Z];.
References
- 1.Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemostasis. 2003;1:1335–42. doi: 10.1046/j.1538-7836.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- 2.Ruggeri ZM. Von Willebrand factor: Looking back and looking forward. Thromb Haemost. 2007;98:55–62. [PubMed] [Google Scholar]
- 3.Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–97. doi: 10.1016/s0092-8674(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 4.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–66. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 5.Ruggeri ZM, Savage B. Hemostasis and thrombosis. Basic principles and clinical practice. 5th ed Lippincott Williams and Wilkins; 2006. Platelet-vessel wall interactions in flowing blood; pp. 723–35. [Google Scholar]
- 6.Weiss HJ, Turitto VT, Baumgartner HR. Role of shear rate and platelets in promoting fibrin formation on rabbit subendothelium. Studies utilizing patients with quantitative and qualitative platelet defects. J Clin Invest. 1986;78:1072–82. doi: 10.1172/JCI112663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McBane RD, II, Ford MAP, Karnicki K, et al. Fibrinogen, fibrin and crosslinking in aging arterial thrombi. Thromb Haemost. 2000;84:83–7. [PubMed] [Google Scholar]
- 8.Loscalzo J, Inbal A, Handin RI. von Willebrand protein facilitates platelet incorporation in polymerizing fibrin. J Clin Invest. 1986;78:1112–9. doi: 10.1172/JCI112668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goto S, Ikeda Y, Saldivar E, Ruggeri ZM. Distinct mechanisms of platelet aggregation as a consequence of different shearing flow conditions. J Clin Invest. 1998;101:479–486. doi: 10.1172/JCI973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruggeri ZM, Dent JA, Saldivar E. Contribution of distinct adhesive interactions to platelet aggregation in flowing blood. Blood. 1999;94:172–8. [PubMed] [Google Scholar]
- 11.Tsuji S, Sugimoto M, Miyata S, et al. Real-time analysis of mural thrombus formation in various platelet aggregation disorders: distinct shear-dependent roles of platelet receptors and adhesive proteins under flow. Blood. 1999;94:968–75. [PubMed] [Google Scholar]
- 12.Gelfand EV, Cannon CP. Hemostasis and thrombosis. Basic principles and clinical practice. 5th ed Lippincott Williams and Wilkins; 2006. Acute coronary syndromes; pp. 1387–404. [Google Scholar]
- 13.Collen D, Lijnen HR. Basic and clinical aspects of fibrinolysis and thrombolysis. Blood. 1991;78:3114–24. [PubMed] [Google Scholar]
- 14.Machovich R, Owen WG. The elastase-mediated pathway of fibrinolysis. Blood Coagul Fibrinolysis. 1990;1:79–90. doi: 10.1097/00001721-199003000-00011. [DOI] [PubMed] [Google Scholar]
- 15.Kolev K, Tenekedjiev K, Komorowicz E, et al. Functional evaluation of the structural features of proteases and their substrate in fibrin surface degradation. J Biol Chem. 1997;272:13666–75. doi: 10.1074/jbc.272.21.13666. [DOI] [PubMed] [Google Scholar]
- 16.Kolev K, Komorowicz E, Owen WG, et al. Quantitative comparison of fibrin degradation with plasmin, miniplasmin, neutrophil leukocyte elastase and cathepsin G. Thromb Haemost. 1996;75:140–6. [PubMed] [Google Scholar]
- 17.Komorowicz E, Kolev K, Lerant I, et al. Flow rate-modulated dissolution of fibrin with clot-embedded and circulating proteases. Circ Res. 1998;82:1102–8. doi: 10.1161/01.res.82.10.1102. [DOI] [PubMed] [Google Scholar]
- 18.Komorowicz E, Kolev K, Machovich R. Fibrinolysis with des-kringle derivatives of plasmin and its modulation by plasma protease inhibitors. Biochemistry. 1998;37:9112–8. doi: 10.1021/bi980180d. [DOI] [PubMed] [Google Scholar]
- 19.Kolev K, Machovich R. Molecular and cellular modulation of fibrinolysis. Thromb Haemost. 2003;89:610–21. [PubMed] [Google Scholar]
- 20.Tanka-Salamon A, Tenekedjiev K, Machovich R, et al. Suppressed catalytic efficiency of plasmin in the presence of long-chain fatty acids. Identification of kinetic parameters from continuous enzymatic assay with Monte Carlo simulation. FEBS Journal. 2008;275:1274–82. doi: 10.1111/j.1742-4658.2008.06288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gombas J, Tanka-Salamon A, Skopal J, et al. Modulation of fibrinolysis by the combined action of phospholipids and immunoglobulins. Blood Coagul Fibrinolysis. 2008;19:82–8. doi: 10.1097/MBC.0b013e3282f38c6f. [DOI] [PubMed] [Google Scholar]
- 22.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res. 2000;98:323–32. doi: 10.1016/s0049-3848(99)00242-x. [DOI] [PubMed] [Google Scholar]
- 23.Lundblad RL, Kingdon HS, Mann KG. Thrombin. Meth Enzymol. 1976;45:156–76. doi: 10.1016/s0076-6879(76)45017-6. [DOI] [PubMed] [Google Scholar]
- 24.Deutsch DG, Mertz ET. Plasminogen purification from human plasma by affinity chromatography. Science. 1970;170:1095–6. doi: 10.1126/science.170.3962.1095. [DOI] [PubMed] [Google Scholar]
- 25.Machovich R, Owen WG. An elastase-dependent pathway of plasminogen activation. Biochemistry. 1989;28:4517–2. doi: 10.1021/bi00436a059. [DOI] [PubMed] [Google Scholar]
- 26.Wu HL, Shi GY, Bender ML. Preparation and purification of microplasmin. Proc Natl Acad Sci USA. 1987;84:8292–5. doi: 10.1073/pnas.84.23.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolev K, Léránt I, Tenekedjiev K, et al. Regulation of fibrinolytic activity of neutrophil leukocyte elastase, plasmin and miniplasmin by plasma protease inhibitors. J Biol Chem. 1994;269:17030–4. [PubMed] [Google Scholar]
- 28.Inglese J, Samama P, Patel S, et al. Chemokine receptor-ligand interactions measured using time-resolved fluorescence. Biochemistry. 1998;37:2372–7. doi: 10.1021/bi972161u. [DOI] [PubMed] [Google Scholar]
- 29.Handl HL, Vagner J, Yamamura HI, et al. Lanthanide-based time-resolved fluorescence of in cyto ligand-receptor interactions. Anal Biochem. 2004;330:242–50. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 30.Thorell L, Blomback B. Purification of the factor VIII complex. Thromb Res. 1984;35:431–50. doi: 10.1016/0049-3848(84)90235-4. [DOI] [PubMed] [Google Scholar]
- 31.Kolev K, Tenekedjiev K, Ajtai K, et al. Myosin: a non-covalent stabilizer of fibrin in the process of clot dissolution. Blood. 2003;101:4380–6. doi: 10.1182/blood-2002-10-3227. [DOI] [PubMed] [Google Scholar]
- 32.Johnson ML, Frasier SG. Nonlinear least-squares analysis. Meth Enzymol. 1985;117:301–42. [Google Scholar]
- 33.Fleury V, Anglés-Cano E. Characterization of the binding of plasminogen to fibrin surfaces: the role of carboxy-terminal lysines. Biochemistry. 1991;30:7630–8. doi: 10.1021/bi00244a035. [DOI] [PubMed] [Google Scholar]
- 34.Bok RA, Mangel WF. Quantitative characterization of the binding of plasminogen to intact fibrin clots, lysine-sepharose, and fibrin cleaved by plasmin. Biochemistry. 1985;24:3279–86. doi: 10.1021/bi00334a031. [DOI] [PubMed] [Google Scholar]
- 35.Nogami K, Nishiya K, Saenko EL, et al. Identification of a plasmin-interactive site within the A2 domain of the factor VIII heavy chain. Biochim Biophys Acta. 2008;1784:753–63. doi: 10.1016/j.bbapap.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 36.Nogami K, Nishiya K, Saenko EL, et al. Identification of plasmin-interactive sites in the light chain of factor VIII responsible for proteolytic cleavage at Lys36. J Biol Chem. 2009;284:6934–45. doi: 10.1074/jbc.M802224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Federici AB, Berkowitz SD, Zimmerman TS, et al. Proteolysis of von Willebrand factor after thrombolytic therapy in patients with acute myocardial infarction. Blood. 1992;79:38–44. [PubMed] [Google Scholar]