

Abstract
Mouse models of type I diabetes offer the potential to combine genetic approaches with other pharmacological or physiological manipulations to investigate the pathophysiology and treatment of diabetic retinopathy. Type I diabetes is induced in mice through chemical toxins or can arise spontaneously from genetic mutations. Both models are associated with retinal vascular and neuronal changes. Retinal transcriptomic responses in C57BL/6J mice treated with streptozotocin and Ins2Akita/+ were compared after 3 months of hyperglycemia. Specific gene expression changes suggest a neurovascular inflammatory response in diabetic retinopathy. Genes common to the two models may represent the response of the retina to hyperglycemia, while changes unique to each model may represent time-dependent disease progression differences in the various models. Further investigation of the commonalities and differences between mouse models of type I diabetes may define cause and effect events in early diabetic retinopathy disease progression.
Electronic supplementary material
The online version of this article (doi:10.1007/s12177-009-9045-3) contains supplementary material, which is available to authorized users.
Keywords: Gene expression, Retina, Type I diabetes, Animal models
Introduction
A variety of rodent models of type I diabetes are used for preclinical investigation of diabetic retinopathy. These models vary according to species (rat or mouse), strain, method of diabetes induction, and duration of diabetes [1]. Depending on the aims of a particular study, a strong rationale exists for any number of combinations of these factors. However, this multiplicity of models complicates comparisons between reports and makes difficult the interpretation of fundamental mechanisms underlying the development of diabetic retinopathy. To assess similarities in the most basic molecular changes, studies have been conducted across species and strains to identify commonalities and differences in retinal gene and protein expression [2, 3].
As part of efforts in preclinical drug development for diabetic retinopathy, we have previously utilized the streptozotocin-treated (STZ) Sprague–Dawley rat model [4–7]. However, mouse models of type I diabetes, permitting ease of genetic manipulation, offer an advantage in disease and therapeutic studies of diabetic retinopathy. STZ-induced diabetic mice have hallmark aspects of retinopathy such as neuronal cell death and vascular dysfunction [8–10]. Recent efforts, lead by the National Institutes of Health-sponsored Animal Models of Diabetic Complications Consortium, have standardized the STZ induction protocol (http://www.amdcc.org). However, the availability of Ins2Akita, a spontaneous, early onset genetic model of type I diabetes with complete penetrance, avoids the effects of STZ on non-insulin producing cell types and provides an attractive alternative.
We have described retinal complications in the Ins2Akita model of type I diabetes [11]. Akita designates a point mutation in the Ins2 gene which leads to accumulation of misfolded Ins2 protein in pancreatic beta cells and ultimately beta cell death [12, 13]. Ins2Akita mice are not obese and are typically hyperglycemic by 5 weeks of age. In contrast to other spontaneous models of type I diabetes in the mouse, Ins2Akita lacks intrinsic immune defects, allowing the retinal inflammatory response to be examined solely in relationship to diabetes-induced metabolic alterations; and it is dominantly inherited on an inbred genetic background, greatly facilitating studies that implement intercrosses with genetically altered mice carrying various mutations that will permit testing of new hypotheses and identification of new drug targets. We have characterized the retinal complications in the Ins2Akita model of type I diabetes and have found increased vascular permeability, leukostasis, and apoptosis, as well as retinal morphological changes [11, 14, 15].
To compare two mouse models of type I diabetes, i.e., STZ-induced and Ins2Akita, a gene expression analysis was conducted examining whole retinas collected after 3 months of hyperglycemia. This time point was chosen as it is the same duration as our previous work with the Sprague–Dawley rat model of type I diabetes [4, 6]. Also, we have demonstrated increased retinal vascular permeability and apoptosis, and decreased ganglion cell density and insulin receptor kinase activity at this time point in the Ins2Akita model [11]. Additionally, decreased ganglion cell density and increased apoptosis are observed at this time point as well in the Ins2Akita model [14]. The results from the present study reveal common aspects of the retinal response to diabetes across mouse (and rat) models with a number of changes also specific to each model. Differences between the models prompting further examination of the time course of retinal dysfunction in these model systems.
Methods
Animal models of type I diabetes
All mice were maintained by the Penn State Diabetic Retinopathy Animal Models Core in accordance with the Institutional Animal Care and Use Committee guidelines under specific pathogen-free conditions and monitored by quarterly sentinel testing. Stock C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME, USA) are replenished twice a year and bred only one generation in house.
C57BL/6J-Ins2Akita/+ mice (The Jackson Laboratory, Bar Harbor, ME, USA) were imported into the colony several years ago and are continuously maintained by crossing with C57BL/6J stock at each generation. Due to the asynchronous replication and resulting inconsistency of the diabetes in Ins2Akita/+ female mice, only male mice were used for these experiments. Ins2Akita/+ males were phenotyped by blood glucose at 5 weeks of age, and assignment was verified by molecular genotyping of ear biopsies taken at the time of retinal harvest (18 weeks).
Diabetes was induced according to the protocol from the Animal Models of Diabetic Complications Consortium http://www.amdcc.org/. Briefly, at 4–5 weeks of age, each day for five consecutive days, male C57BL/6J mice were fasted for approximately 4 h prior to a single intraperitoneal injection of 50 mg/kg streptozotocin (Sigma-Aldrich, St. Louis, MO, USA) in 100 mM sodium citrate buffer, pH 4.5. Blood glucose concentrations were measured after 1 week and each subsequent week. Only mice that had blood glucose concentrations of >250 mg/dL by the end of the second week were included. Control mice were injected with an equal dose of citrate buffer only. STZ and control mice were harvested 12 weeks post-STZ or buffer injection, thus approximately 18 weeks of age. Mice had free access to food and water and were on a 12-h light/dark cycle. No exogenous insulin was delivered. Blood glucose concentrations and percent glycosylated hemoglobin were measured from drops of blood acquired by nicking the tail, using a Lifescan glucose meter and Siemens DCA Analyzer, respectively. Cartridges for the quantitative measurement of hemoglobin A1c are based on specific monoclonal antibody agglutination. At the time of retinal harvest, mice were given a lethal dose of pentobarbital, 100 mg/kg (Ovation Pharmaceuticals Inc., Deerfield, IL, USA) by intraperitoneal injection. Retinas were excised and quickly frozen in liquid nitrogen and stored at −80°C prior to RNA isolation using a protocol to produce standardized whole retinal tissues [6, 11].
RNA isolation
Whole retina tissue was homogenized using a Retsch TissueLyzer II (Qiagen, Valencia, CA, USA) with a 3-mm stainless steel ball in 500 μL TriReagent (Molecular Research Center, Cincinnati, OH, USA) for 30 s at 30 Hz. After homogenization, RNA was isolated following standard Tri Reagent/BCP disruption and phase separation and overnight isopropanol precipitation at −20°C. RNA was further purified using Qiagen RNeasy Mini kit (Qiagen). RNA was assessed for quantity and quality using a NanoDrop ND1000 (Thermo Scientific, Wilmington, DE, USA) and RNA 6000 Nano LabChip with an Agilent 2100 Expert Bioanalyzer (Agilent, Palo Alto, CA, USA), respectively. All RNA used had RNA integrity values greater than 8.
Illumina microarray analysis of mRNA expression
Microarray analysis was performed using Illumina Ref8 microarrays. For the Ins2Akita experiment, n = 8 wild-type samples, and n = 9 Ins2Akita samples were analyzed. For the STZ experiment, n = 8 control and n = 6 STZ-treated animals were analyzed. Samples were labeled according to Illumina TotalPrep RNA Amplification kit (Illumina, San Diego, CA, USA) standard procedures. RNA (250 ng) was reverse transcribed to synthesize first strand cDNA by incubating samples at 42°C for 2 h with T7 Oligo(dT) primer, 10× First Strand buffer, dNTPs, Rnase inhibitor, and ArrayScript. Second strand cDNA was synthesized with 10× second strand buffer, dNTPs, DNA polymerase and Rnase H at 16°C for 2 h. cDNA was purified according to standard procedures. cDNA was eluted in approximately 17.5 μL 55°C nuclease-free water and stored at −20°C overnight. cDNA was in vitro transcribed to synthesize cRNA using MEGAscript (Ambion) kit. Samples were incubated with T7 10× Reaction buffer, T7 Enzyme mix and Biotin-NTP mix at 37°C for 14 h. cRNA was purified according to instructions. cRNA yield was measured using a NanoDrop ND1000 (Thermo Scientific, Wilmington, DE, USA).
Purified cRNA (750 ng) was prepared for hybridization according to instructions for hybridizing to Illumina MouseRef-8 Expression BeadChips. BeadChips were incubated in a hybridization oven for 20 h at 58°C at a rocker speed of 5. After 20 h, BeadChips were disassembled and washed and Streptavadin-Cy3 stained according to Illumina standard procedures. BeadChips were dried by centrifugation at 275 × g for 4 min and subsequently scanned using a BeadArray Reader.
BeadStudio (Illumina) flat files were imported into GeneSpring 7.3 (Agilent) software for data analysis. Each experiment (STZ and Ins2Akita) was treated separately for statistical analyses. Initially probes were filtered to remove those for which no transcript was detected. Using the detection p values initially generated in BeadStudio, probes were required to have detectable signal for six of 14 arrays in the STZ experiment and eight of 17 arrays for the Ins2Akita to be included in the subsequent statistical analyses. The goal of this filtering is to eliminate transcripts not reliably detected from further analysis, while retaining genes potentially expressed in only one group (i.e., expressed only in control or only in diabetic). Therefore, the thresholds were chosen based on the smallest number of samples in a group. The full complement of microarray data is available in the Gene Expression Omnibus [16], accession # GSE19122.
To determine differential expression, a combination of statistical p value (t test, p < 0.05) and fold-change (≥1.4 fold) cutoffs were used in accordance with standards in data analysis [17] and our previously published methods [6].
Quantitative RT-PCR
Quantitative polymerase chain reaction (qPCR) analysis was performed as described previously [4, 6] using the 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA), 384-well optical plates, and Assay-On-Demand (Applied Biosystems) gene specific primers and probes. SDS 2.2.2 software and the 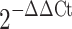 analysis method were used to quantitate relative amounts of product using β-actin as an endogenous control. β-actin levels were determined to be unchanged in an absolute quantification experiment (data not shown). For a full listing of primer/probe sets, see Table 1.
analysis method were used to quantitate relative amounts of product using β-actin as an endogenous control. β-actin levels were determined to be unchanged in an absolute quantification experiment (data not shown). For a full listing of primer/probe sets, see Table 1.
Table 1.
Gene symbols, names, alias, assay-on-demand catalogue numbers, and gene IDs for genes examined by quantitative polymerase chain reaction
| Symbol | Name | AOD | Gene ID number | Abbreviations/alias |
|---|---|---|---|---|
| B2M | Beta-2 microglobulin | Mm00437762_m1 | Mm.163 | Ly-m11 |
| C1-Inh | Serine (or cysteine) peptidase inhibitor, clade G, member 1 | Mm00437834_m1 | Mm.38888 | Serping1 |
| C1S | Complement component 1, s subcomponent | Mm00663210_mH | Mm.219527 | C1sa |
| Carhsp1 | Calcium regulated heat stable protein 1 | Mm00787963_s1 | Mm.142095 | Crhsp-24 |
| Ccr5 | Chemokine (C-C motif) receptor 5 | Mm01216171_m1 | Mm.14302 | Cmkbr5 |
| Ch25h | Cholesterol 25-hydroxylase | Mm00515486_s1 | Mm.30824 | m25OH |
| Chi3L1 | Chitinase 3-like 1 | Mm00801477_m1 | Mm.38274 | Brp39 |
| Csrp3 | Cysteine and glycine-rich protein 3 | Mm00443379_m1 | Mm.17235 | CRP3, MLP, MMLP |
| Dcamkl1 | Doublecortin-like kinase 1 | Mm00444950_m1 | Mm.393242 | Dclk |
| Edn2 | Endothelin 2 | Mm00432983_m1 | Mm.284855 | VIC |
| Ednrb | Endothelin receptor type B | Mm00432989_m1 | Mm.229532 | ETR-b |
| Egr1 | Early growth response 1 | Mm00656724_m1 | Mm.181959 | Zenk, Zfp-6, Zif268 |
| Fmod | Fibromodulin | Mm00491215_m1 | Mm.287146 | SLRR2E |
| Gbp2 | Guanylate binding protein 2 | Mm00494575_m1 | Mm.24038 | |
| Gldc | Glycine decarboxylase | Mm00506891_m1 | Mm.274852 | |
| H2-Ab1 | Histocompatibility 2, class II antigen A, beta 1 | Mm00439216_m1 | Mm.254067 | I-Abeta, IAb, Ia-2, Ia2, Rmcs1 |
| Hspb1 | Heat shock protein 1 | Mm00834384_g1 | Mm.13849 | Hsp25 |
| Icam1 | Intercellular adhesion molecule 1 | Mm00516023_m1 | Mm.435508 | MALA-2 |
| IFitm3 | Interferon-induced transmembrane protein 3 | Mm00847057_s1 | Mm.141021 | Cd225, Cdw217, Fgls, IP15, mil-1 |
| Jak3 | Janus kinase 3 | Mm00439962_m1 | Mm.249645 | Fae, wil |
| Kcne2 | Potassium voltage-gated channel, Isk-related subfamily, gene 2 | Mm00506492_m1 | Mm.156736 | MiRP1 |
| Lama5 | Laminin, alpha 5 | Mm01222050_g1 | Mm.4339 | Laminin-511 |
| Lgals3 | Lectin, galactose binding, soluble 3 | Mm00802901_m1 | Mm.248615 | GBP, L-34, Mac-2, gal3 |
| Lgals3bp | Lectin, galactoside-binding, soluble, 3 binding protein | Mm00478303_m1 | Mm.3152 | CyCAP, MAC-2BP, Ppicap |
| Lgals9 | Lectin, galactose binding, soluble 9 | Mm00495295_m1 | Mm.341434 | Gal-9, galectin-9 |
| Mct1 | Solute carrier family 16 (monocarboxylic acid transporters), member 1 | Mm00436566_m1 | Mm.9086 | Slc16a1 |
| Nppa | Natriuretic peptide precursor type A | Mm01255747_g1 | Mm.19961 | ANP, Anf, Pnd |
| Pcgf1 | Polycomb group ring finger 1 | Mm01617932_g1 | Mm.12261 | Nspc1 |
| Pedf | Serine (or cysteine) peptidase inhibitor, clade F, member 1 | Mm00441270_m1 | Mm.2044 | Serpinf1 |
| Slc6a11 (Gat3) | Solute carrier family 6 (neurotransmitter transporter, GABA), member 11 | Mm00556476_m1 | Mm.44683 | Gat3, GAT4, Gabt4 |
| Stat3 | Signal transducer and activator of transcription 3 | Mm00456961_m1 | Mm.249934 | Aprf |
| Timp1 | Tissue inhibitor of metalloproteinase 1 | Mm00441818_m1 | Mm.8245 | Clgi, Timp |
| Tnfrsf12a | Tumor necrosis factor receptor superfamily, member 12a | Mm00489103_m1 | Mm.28518 | Fn14, HPIP, TWEAK-R |
Results
Assessment of diabetes
Mice were harvested after 3 months of hyperglycemia. For the STZ-treated mice, this was 3 months after the last STZ injection. Ins2Akita mice were harvested at 4 months of age. At sample collection, both STZ-induced and Ins2Akita-diabetic mice had significantly elevated blood glucose as compared to the controls (p < 0.001, t test), see Table 2. Hyperglycemic mice were significantly lighter than their respective controls (p < 0.001), but no differences in weight were evident between Ins2Akita and STZ-treated groups, see Table 2.
Table 2.
Animal blood glucose and body weights
| Model | Blood glucose (mg/dL) | Weight (g) | n | ||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | ||
| C57BL/6J-Ins2+/+ | 172 | 41 | 31.4 | 3.3 | 8 |
| C57BL/6J-Ins2Akita/+ | 558 | 38 | 24.5 | 1.8 | 9 |
| C57BL/6J-control | 155 | 26 | 29.4 | 2.2 | 8 |
| C57BL/6J-STZ | 511 | 41 | 23.1 | 2.8 | 6 |
While glycosylated hemoglobin (HbA1c) percentages were not measured in the mice harvested for the gene expression experiments, HbA1c percentages were measured in independent sets of animals after the same duration of diabetes (Table 3). HbA1c percentages do not differ between STZ-induced diabetic mice and Ins2Akita diabetic mice.
Table 3.
HbA1c percentages measured in an independent set of animals at the same duration of hyperglycemia
| Model | n | HbA1c (%) | |
|---|---|---|---|
| Mean | SD | ||
| C57BL/6J-Ins2+/+ | 19 | 4.6 | 0.3 |
| C57BL/6J-Ins2Akita/+ | 9 | 12.1 | 1.2 |
| C57BL/6J-Control | 5 | 4.4 | 0.2 |
| C57BL/6J-STZ | 5 | 11.7 | 2.9 |
Microarray profiling of gene expression
Expression profiling of whole retina RNA from both the STZ and Ins2Akita experiments was performed after 3 months of hyperglycemia. Initial filtering for only those probes with detectable signal revealed 16,327 probes detected in six or greater of 14 microarrays in the STZ experiment and 16,165 probes detected in eight or greater of 17 microarrays in the Ins2Akita experiment (Fig. 1a). Of the detected probes, 15,818 (95%) were detected in common between the two experiments.
Fig. 1.

Microarray analysis of retinal gene expression after 3 months of hyperglycemia. a 95% of the probes with detectable signal in the microarray analysis were shared between the two mouse models of type 1 diabetes. b Set analysis of the sets of differentially expressed (p < 0.05, ≥1.4-fold) probes and genes in the two models reveals changes common to both models. The majority of expression changes were specific to one model or the other. c Probes with statistically significant differences in signal (p < 0.05) and ≥1.4-fold change are plotted for each animal in the analysis. The normalized (to a mean control level of 1) intensity is plotted on the y-axis with each individual animal analyzed on the x-axis for both experiments. Each horizontal line is an individual probe. This presentation provides an overview of the interanimal variance as well as differences between the groups
In the STZ experiment, 333 probes, corresponding to 307 genes, were differentially expressed (p < 0.05 and >1.4-fold) in diabetic mice as compared to control mice (Fig. 1b and Supplementary File 1). In the Ins2Akita experiment, 404 probes corresponding to 392 genes were differentially expressed (p < 0.05 and >1.4-fold) in diabetic mice as compared to control mice. When examined, the differentially expressed transcripts in the two experiments were evenly distributed between upregulated and downregulated transcripts in the diabetic mice as compared to their respective controls (Fig. 1c). Some inter-animal variance was observed within groups of diabetic and control mice, but clearly altered levels in expression were observed between control and diabetic animals. In both experiments, 30 probes corresponding to 28 genes were differentially expressed in Ins2Akita and STZ mice (Fig. 1b and Table 4). For all of these commonly regulated transcripts between the two mouse models of type I diabetes, transcripts were either upregulated or downregulated in both models, and none were regulated in opposing directions.
Table 4.
Common changes between the two models identified in the microarray discovery experiments
| Probe number | Mean ratio Ins2/WT | Mean ratio STZ/control | Gene symbol | RefSeq ID |
|---|---|---|---|---|
| 5910072 | 1.55 | 1.73 | 2310016A09RIK | NM_181588.2 |
| 4830543 | 2.28 | 2.07 | 2310061N23RIK | |
| 7400603 | 1.45 | 1.55 | 2810004A10RIK | NM_027265.1 |
| 1470647 | 0.66 | 0.65 | 9030221M09RIK | NM_177298.2 |
| 4730458 | 2.60 | 2.27 | ADAMTS19 | NM_175506.2 |
| 2750209 | 1.58 | 1.80 | AU040576 | NM_178642.3 |
| 5570253 | 1.43 | 1.53 | B2M | NM_009735.2 |
| 3610025 | 1.93 | 1.97 | BC004728 | NM_174992.2 |
| 1510349 | 5.85 | 7.14 | CH25H | NM_009890.1 |
| 1450333 | 4.85 | 3.30 | CLECSF12 | |
| 3170255 | 4.93 | 3.58 | CML4 | NM_023455.2 |
| 1940154 | 1.66 | 1.94 | COBLL1 | |
| 5340722 | 0.70 | 0.70 | CSRP3 | NM_013808.3 |
| 2470133 | 0.64 | 0.61 | CSRP3 | NM_013808.3 |
| 4490730 | 1.54 | 1.69 | D930038M13RIK | NM_178790.2 |
| 4540382 | 1.51 | 1.62 | EGFR | NM_007912.4 |
| 1440307 | 1.66 | 1.90 | H2-AB1 | NM_207105.1 |
| 2570487 | 1.52 | 1.67 | H2-AB1 | NM_207105.1 |
| 840349 | 1.56 | 1.74 | IFITM2 | NM_030694.1 |
| 3840292 | 1.51 | 1.63 | IFITM3 | NM_025378.1 |
| 4880215 | 1.46 | 1.57 | ITPR3 | NM_080553.1 |
| 430132 | 1.97 | 2.01 | LGALS9 | NM_010708.1 |
| 1710768 | 1.97 | 2.04 | LOC56628 | NM_019909.1 |
| 2970598 | 1.47 | 1.61 | MAP3K8 | NM_007746.1 |
| 2490368 | 1.47 | 1.60 | PLSCR2 | NM_008880.2 |
| 6130743 | 1.43 | 1.46 | ROR2 | NM_013846.2 |
| 6840600 | 1.50 | 1.61 | RRBP1 | NM_133626.1 |
| 3830048 | 1.57 | 1.76 | SELENBP1 | NM_009150.2 |
| 380215 | 1.42 | 1.45 | TOR3A | |
| 150424 | 0.55 | 0.55 | XPO1 | NM_001035226.1 |
Mean ratios are presented relative to control values for each of the two experiments
The lists of differentially expressed genes in the two experiments were imported into the Ingenuity Pathway Analysis software for analysis of patterns of gene expression. Network analysis was performed on the results of the two experiments. Network analysis is intended to statistically identify sets of interrelated genes based on their known functions. For the Ins2Akita experiment, the most significantly regulated network of genes related to inflammatory processes (Fig. 2a). For the STZ experiment, the most significantly regulated network centered on genes with known responsiveness to diabetes in either clinical samples or animal models (Fig. 2b). While the two experiments highlighted two different networks, both fit with a hypothesis of diabetic retinopathy as a feed-forward cycle of neuroinflammation [18].
Fig. 2.

Network analysis of microarray data for each of the experiments. Network analysis (ingenuity) identifies sets of genes with known relationships that are unlikely (p < 0.05) to occur by random chance. The most significant network for each experiment is shown. In the Ins2Akita experiment, a network with a number of genes related to inflammation (blue borders) was identified, while the STZ experiment network contained a number of genes (blue borders) that have been previously reported to be altered in mRNA or protein expression in animal models or clinical specimens. Red indicates upregulation, and green indicates downregulation of transcript levels. Uncolored genes were not observed to be differentially expressed in the microarray analysis but are integral parts of the network
To confirm differentially expressed transcripts in both models of type I diabetes, focused qPCR validation of microarray experiments was performed. Six genes (B2m; cysteine and glycine-rich protein 3 (Csrp3); interferon-induced transmembrane protein 3 (Ifitm3); Lgals9; H2A histone family, member B3 (H2Ab1); and Ch25h) were chosen for confirmation, of which, four genes demonstrated a statistically significant altered level of expression in both mouse models by qPCR (Fig. 3). These genes were chosen for qPCR confirmation to confirm the validity of the microarray measurements and due to their being some of the largest magnitude changes. Increased expression of B2m, Ifitm3, and Lgals9 and reduced expression of Csrp3 was observed in the diabetic mice as compared to their respective control mice. Significantly, increased H2Ab1 expression was observed only in the STZ-diabetic mice relative to controls. Ch25h expression was not significantly altered in the qPCR confirmation but did trend towards upregulation, as observed in the microarray analysis.
Fig. 3.

Quantitative polymerase chain reaction confirmation of common microarray changes between the two models. *p < 0.05, **p < 0.01, t test, n = 6–9/group, C control, D diabetic
Additionally, three transcripts (Egr1, fibromodulin (Fmod), and glycine dehydrogenase (Gldc)) were examined by qPCR which were observed to be altered in only one mouse model by the microarray analysis (Fig. 4). Egr1 was downregulated in Ins2Akita mice alone, while Fmod and Gldc were upregulated in STZ mice alone in the microarray analysis. qPCR validation confirmed decreased expression of Egr1 in Ins2Akita diabetic mice as compared to controls, but no change was observed in the STZ-diabetic mice as compared to their controls. Increased expression of Fmod, and Gldc was observed in STZ diabetic mice relative to control mice in qPCR validation. No change was observed in Ins2Akita diabetic animals as compared to controls in the qPCR measurements. In total, eight of the nine genes examined by qPCR validated the microarray results an indication of the reliability of the microarray data generated.
Fig. 4.

Quantitative polymerase chain reaction confirmation of microarray changes specific to one model. *p < 0.05, **p < 0.01, t test, n = 6–9/group, C control, D diabetic
Previously, we examined whole retina mRNA expression changes in the STZ-induced Sprague–Dawley rat model of type I diabetes at 3 months of hyperglycemia [6]. We also demonstrated that a set of retinal genes, a biomarker panel, are accurate classifiers of disease state and may be used to gauge the efficacy of therapies in preclinical drug development using that model system [4]. This biomarker panel of 24 genes was also developed for 3 months of hyperglycemia time point. The genes in the biomarker panel were also examined in the retinal mRNA from the STZ and Ins2Akita experiments by qPCR for potential commonalities in gene expression changes (Table 5). Some common changes were observed with increased expression of C1Inh and decreased expression of Kcne2 in STZ and Ins2Akita diabetic mice as compared to their respective controls. Additionally, Gbp2 and Lgals3bp expression was evident in the STZ diabetic mouse and increased laminin alpha 5 (Lama5) in the Ins2Akita diabetic mouse. While only a minority of the rat mRNA biomarkers were observed to change in these mouse models, it is of interest to note that all of those common changes were of the same direction and magnitude.
Table 5.
Previously reported retinal gene expression changes in the rat STZ model at 3 months diabetes examined in the STZ mouse and Ins2Akita mouse models by quantitative polymerase chain reaction
| Gene | Model | ||
|---|---|---|---|
| Rat STZa | Ins2Akita | Mouse STZ | |
| C1INH (Serping1) | 2.6** | 1.8* | 2.1* |
| C1S | 2.8** | – | – |
| Carhsp1 | 2.6*** | – | – |
| Ccr5 | 1.6** | – | – |
| Chi3L1 | 2.7** | – | – |
| Dcamkl1 | 0.8** | – | – |
| Edn2 | 3.7* | – | – |
| Ednrb | 1.6** | – | – |
| Slc6a11 (Gat3) | 0.6*** | – | – |
| Gbp2 | 2.4* | – | 2.8*** |
| Hspb1 | 14.2** | – | – |
| Icam1 | 1.7** | – | – |
| Jak3 | 1.9** | – | – |
| Kcne2 | 0.5*** | 0.8 ** | 0.8* |
| Lama5 | 1.8*** | 1.4 ** | – |
| Lgals3 | 8.0*** | – | – |
| Lgals3bp | 1.7* | – | 2.0* |
| Mct1 | 0.6*** | – | – |
| Nppa | 0.5*** | – | – |
| Pcgf1 | 0.7*** | – | – |
| Pedf | 1.9* | – | – |
| Stat3 | 1.4** | – | – |
| Timp1 | 4.0** | – | – |
| Tnfrsf12a | 2.2*** | – | – |
– not changed
*p < 0.05 (t test)
**p < 0.01 (t test)
***p < 0.001 (t test)
aPrimary quantitative polymerase chain reaction data for STZ rats was previously reported [4]
Discussion
The present study examined diabetes-responsive retinal gene expression to advance three related goals: (1) provide targets for future biomarker development, (2) increase knowledge of the retinopathological process in diabetes though comparison of a toxin-induced and a spontaneous genetic form of type I diabetes, and (3) compare the similarity of retinal gene expression responses between mouse and rat models of type I diabetes.
Biomarker development
As part of a translational diabetic retinopathy drug development center, one of our goals is to develop and optimize tools for enhancing the efficacy of preclinical drug development. Previously, we have proposed a process for the development and use of mRNA biomarkers in preclinical drug development [4]. This biomarker discovery and validation process was then used to create a diabetic retinopathy mRNA biomarker panel for use with the STZ rat model of type I diabetes [4]. The initial aspect of this development process is biomarker target discovery, and one of the goals of the present study was to conduct biomarker target discovery in the STZ and Ins2Akita mouse models of type I diabetes. The expression changes identified through microarray screening in the present study can serve as biomarker targets for future validation and classification analysis studies. The successful qPCR confirmation of targets from the microarray analysis suggests that additional confirmations of microarray targets will be successful.
Effects of specific genes
A total of 13 genes were demonstrated by qPCR to be differentially expressed with diabetes in this study. These genes were selected to (1) confirm the validity of the array measurements, (2) confirm genes identified in common between the two models, and (3) examine specific changes we have previously observed in the STZ rat model. These confirmations are limited in number, and additional targets from the microarray analysis can be examined in future studies. Each of these genes has potential actions in the pathophysiology of diabetic retinopathy, and several has been previously reported to be altered in expression in other animal models of diabetes. A brief description of each of the validated genes provides insight into how they may contribute to the growing hypothesis of diabetic retinopathy as a feed forward cycle of inflammation, neuronal damage, and microvascular dysfunction [18]. While diabetic retinopathy research in the past has primarily focused on vascular dysfunction, this hypothesis suggests that neuronal, inflammatory, and vascular changes are interrelated and that neuronal damage can induce inflammation and vascular permeability.
Genes related to inflammatory process validated in this study include C1-Inh, Gbp2, Ifitm3, and B2m. C1-Inh (also known as serpin protease inhibitor, clade g (Serping1)) has recently been described in a number of clinical and animal model reports on diabetic retinopathy. C1-Inh, induced in both the STZ and Ins2Akita mouse models, inhibits acute phase response signaling and the classical complement systems through inhibition of C1r, C1s, MASP-1, or MASP-2 [19]. Increased C1-Inh protein expression has been observed in the vitreous of diabetic patients [20, 21] as well as increased mRNA expression in the retina of STZ-induced diabetic rats [4, 6, 22]. As an inhibitor of complement activation, increased levels of C1-Inh may be a compensatory reaction to limit the inflammatory response. In the retina, C1-Inh expression has been localized to photoreceptors and inner nuclear layer [23]. As the role of the complement system in the inflammatory aspects of diabetic retinopathy is becoming better understood [24], this particular gene is an attractive focus for further examination in mechanistic studies.
The role of Gbp2, induced in the STZ model (guanylate binding protein 2, interferon-inducible) is poorly understood in the retina, but Gbp2 has previously been demonstrated to be induced in the retina of rat models of type I diabetes [6, 22]. Gbp2 encodes a GTPase and is induced in a number of extraretinal inflammatory responses. Ifitm3, induced in both the STZ and Ins2Akita mouse models, is present in both neurons and endothelial cells in brain [25] and has been previously reported to be induced in the retina of diabetic rats [22]. Beta-2-microglobulin (B2m), induced in both the STZ and Ins2Akita mouse models, is primarily known for its role in antigen presentation but in type I diabetes is associated with nephropathy and vascular complications [26]. B2m is widely expressed in the retina [27] and induced levels of B2m in the retina may play a similar role in vascular dysfunction.
Several genes (Cspr3, Fmod, and Lama5) related to tissue structure were observed to increase in the present study. The function of Csrp3, reduced in expression in both the STZ and Ins2Akita mouse models, is unknown in the retina, but the protein contains two LIM domains for protein–protein interactions. The role of cysteine and glycine-rich protein family genes in actin cytoskeleton organization may be related to diabetes-induced alterations in the retinal neuron morphology [14]. Fmod, induced in only the STZ model, is a proteoglycan component of the extracellular matrix. While the functional outcomes of increased Fmod are unknown, its role in the extracellular matrix may be of importance in diabetes-induced alterations in retinal thickness [11]. Lama5, induced only in the Ins2Akita mice, is a member of the large laminin gene family which are major components of basement membranes. Specifically, Lama5 is present in the interphotoreceptor matrices and outer plexiform layer and associates with the retinal vasculature [28]. Previous reports have demonstrated diabetes-induced increases in other laminin genes [29, 30], and we have observed increased Lama5 mRNA expression in the Sprague–Dawley, STZ-induced diabetic rat after 3 months of diabetes [4].
Lgals3bp and Lgals9, increased in expression in both models, are members of the galectin family of beta-galactoside-binding proteins induced by a number of inflammatory molecules and infections. While Lgals3bp and Lgals9 are uncharacterized in the retina, they may interact with Lgals3 in diabetes-induced blood-retinal-barrier breakdown and accumulation of advanced glycation end products [31]. Lgals3 null mice have been shown to have less disruption of the blood-retina-barrier with diabetes than wild-type mice [32].
Additional genes confirmed to be altered in expression by qPCR (Egr1, Kcne2, Gldc, and H2Ab1) have less clear relationships to the known pathophysiological processes of diabetic retinopathy. The transcription factor Egr1, suppressed in the Ins2Akita mice, is best known for its regulatory role in a large set of genes involved in neuronal plasticity [33]. A potential role for increased Egr1 expression has been suggested in retinal microglial activation [34], but this role has yet to be fully elucidated, and the functional outcome of the reduced Egr1 expression observed in the Ins2Akita diabetic mouse remains to be determined. The potassium voltage-gated channel, Isk-related family, member 2 gene, Kcne2, which is decreased in expression in both models, is most characterized for its role in heart disease but has been previously demonstrated to be reduced in the retina in a rat model of type I diabetes [6]. Gldc and H2Ab1 are both increased in only the STZ model. Gldc catalyzes glycine into glyoxylate. H2Ab1 is a histone family gene and an abundant protein in chromatin. Alterations in histone expression could potentially have roles in epigenetic control of gene expression and present another example of an attractive focus for further experimentation.
Relationship between the animal models
This report is, to our knowledge, the first direct comparison of retinal gene expression changes in the STZ and Ins2Akita mouse models of type I diabetes and demonstrates that while there are commonly regulated genes, there are also substantial differences in the gene expression response at 3 months of hyperglycemia. Previous studies have compared the gene and protein expression changes with diabetes (e.g., [3]) and found significant differences between rodent strains, most likely attributable to genetic background. To eliminate any contributing variables from the genetic background in the comparison of the two mouse models, both experimental groups in this study shared the C57BL/6J genetic background, making the single nucleotide change in the Ins2 gene the only genetic difference between the two models. Both models had similar levels of glycemic insult (blood glucose and HbA1c at sacrifice) and have been demonstrated to show increased vascular permeability and neuronal death/dysfunction [9–11, 14, 15, 27]. A potential cause of the differences between the two models could arise from the initiating event, for example, non-beta cell toxicity with STZ, or from varied progression of the diabetes process, as the percent beta cell loss and concomitant decrease in circulating insulin has not been quantified in either model at comparable time points. Further characterization of functional and molecular events in the context of specific intermediate durations of diabetes will be required to interpret the differences observed between the STZ-induced and the Ins2Akita retinal transcriptomes, as well as the differences between the two mouse models and the STZ-induced rat model.
These findings add to the knowledge of the retinal molecular alterations in response to diabetes in two widely used mouse models. The ultimate goal of this research is to improve clinical care of diabetic patients, and these models should share important commonalities with the human condition. While the mouse models examined here share key features with the human disease state (e.g., vascular leakage, apoptosis, and neuronal dysfunction), our knowledge of the time course of molecular changes in the human retina with diabetes remains limited due to the difficulties in obtaining samples. Comprehensive studies from human (or non-human primate) tissue are critically needed to provide a comparator for data sets from other animal models. This would allow for careful comparison of various animal models to the human condition and rationale choices of models for both drug development and basic science studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(XLS 86 kb)
Acknowledgements
This work was supported by a grant from the Juvenile Diabetes Research Foundation (4-2002-455) and a grant from Pennsylvania Department of Health using Tobacco Settlement Funds. The Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Willard M. Freeman and Georgina V. Bixler contributed equally to this work.
References
- 1.Chatzigeorgiou A, Halapas A, Kalafatakis K, Kamper E. The use of animal models in the study of diabetes mellitus. In Vivo. 2009;23:245–258. [PubMed] [Google Scholar]
- 2.Obrosova IG, Drel VR, Kumagai AK, Szabo C, Pacher P, Stevens MJ. Early diabetes-induced biochemical changes in the retina: comparison of rat and mouse models. Diabetologia. 2006;49:2525–2533. doi: 10.1007/s00125-006-0356-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirwin SJ, Kanaly ST, Linke NA, Edelman JL. Strain-dependent increases in retinal inflammatory proteins and photoreceptor FGF-2 expression in streptozotocin-induced diabetic rats. Invest Ophthalmol Vis Sci. 2009;50:5396–5404. doi: 10.1167/iovs.09-3474. [DOI] [PubMed] [Google Scholar]
- 4.Freeman WM, Bixler GV, Brucklacher RM, Lin C-M, Patel KM, VanGuilder HD, et al. A novel multi-step evaluation process of preclinical drug development biomarkers. The Pharmacogenomics J. 2009; In press. [DOI] [PubMed]
- 5.Fort PE, Freeman WM, Losiewicz MK, Singh RS, Gardner TW. The retinal proteome in experimental diabetic retinopathy: Up-regulation of crystallins and reversal by systemic and periocular insulin. Mol Cell Proteomics. 2008;8:767–779. doi: 10.1074/mcp.M800326-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brucklacher RM, Patel KM, Vanguilder HD, Bixler GV, Barber AJ, Antonetti DA, et al. Whole genome assessment of the retinal response to diabetes reveals a progressive neurovascular inflammatory response. BMC Med Genomics. 2008;1:26. doi: 10.1186/1755-8794-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanguilder HD, Brucklacher RM, Patel K, Ellis RW, Freeman WM, Barber AJ. Diabetes downregulates presynaptic proteins and reduces basal synapsin I phosphorylation in rat retina. Eur J NeuroSci. 2008;28:1–11. doi: 10.1111/j.1460-9568.2008.06322.x. [DOI] [PubMed] [Google Scholar]
- 8.Martin PM, Roon P, Van Ells TK, Ganapathy V, Smith SB. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci. 2004;45:3330–3336. doi: 10.1167/iovs.04-0247. [DOI] [PubMed] [Google Scholar]
- 9.Feit-Leichman RA, Kinouchi R, Takeda M, Fan Z, Mohr S, Kern TS, et al. Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Invest Ophthalmol Vis Sci. 2005;46:4281–4287. doi: 10.1167/iovs.04-1361. [DOI] [PubMed] [Google Scholar]
- 10.Nishikiori N, Osanai M, Chiba H, Kojima T, Inatomi S, Ohguro H, et al. Experimental effect of retinoic acids on apoptosis during the development of diabetic retinopathy. Clin Ophthalmol. 2008;2:233–235. doi: 10.2147/opth.s2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barber AJ, Antonetti DA, Kern TS, Reiter CE, Soans RS, Krady JK, et al. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005;46:2210–2218. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA, Takeuchi T. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes. 2003;52:409–416. doi: 10.2337/diabetes.52.2.409. [DOI] [PubMed] [Google Scholar]
- 14.Gastinger MJ, Kunselman AR, Conboy EE, Bronson SK, Barber AJ. Dendrite remodeling and other abnormalities in the retinal ganglion cells of Ins2 Akita diabetic mice. Invest Ophthalmol Vis Sci. 2008;49:2635–2642. doi: 10.1167/iovs.07-0683. [DOI] [PubMed] [Google Scholar]
- 15.Gastinger MJ, Singh RS, Barber AJ. Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita-diabetic mouse retina. Invest Ophthalmol Vis Sci. 2006;47:3143–3150. doi: 10.1167/iovs.05-1376. [DOI] [PubMed] [Google Scholar]
- 16.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 18.Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55:2401–2411. doi: 10.2337/db05-1635. [DOI] [PubMed] [Google Scholar]
- 19.Beinrohr L, Dobo J, Zavodszky P, Gal P. C1, MBL-MASPs and C1-inhibitor: novel approaches for targeting complement-mediated inflammation. Trends Mol Med. 2008;14:511–521. doi: 10.1016/j.molmed.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 20.Gao BB, Clermont A, Rook S, Fonda SJ, Srinivasan VJ, Wojtkowski M, et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med. 2007;13:181–188. doi: 10.1038/nm1534. [DOI] [PubMed] [Google Scholar]
- 21.Gao BB, Chen X, Timothy N, Aiello LP, Feener EP. Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J Proteome Res. 2008;7:2516–2525. doi: 10.1021/pr800112g. [DOI] [PubMed] [Google Scholar]
- 22.Gerhardinger C, Costa MB, Coulombe MC, Toth I, Hoehn T, Grosu P. Expression of acute-phase response proteins in retinal Muller cells in diabetes. Invest Ophthalmol Vis Sci. 2005;46:349–357. doi: 10.1167/iovs.04-0860. [DOI] [PubMed] [Google Scholar]
- 23.Mullins RF, Faidley EA, Daggett HT, Jomary C, Lotery AJ, Stone EM. Localization of complement 1 inhibitor (C1INH/SERPING1) in human eyes with age-related macular degeneration. Exp Eye Res. 2009;89:767–773. doi: 10.1016/j.exer.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jha P, Bora PS, Bora NS. The role of complement system in ocular diseases including uveitis and macular degeneration. Mol Immunol. 2007;44:3901–3908. doi: 10.1016/j.molimm.2007.06.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry. 2007;7:46. doi: 10.1186/1471-244X-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong CY, Chia KS, Ling SL. Urinary protein excretion in Type 2 diabetes with complications. J Diab Complications. 2000;14:259–265. doi: 10.1016/S1056-8727(00)00119-7. [DOI] [PubMed] [Google Scholar]
- 27.Martin PM, Gnana-Prakasam JP, Roon P, Smith RG, Smith SB, Ganapathy V. Expression and polarized localization of the hemochromatosis gene product HFE in retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2006;47:4238–4244. doi: 10.1167/iovs.06-0026. [DOI] [PubMed] [Google Scholar]
- 28.Libby RT, Champliaud MF, Claudepierre T, Xu Y, Gibbons EP, Koch M, et al. Laminin expression in adult and developing retinae: evidence of two novel CNS laminins. J Neurosci. 2000;20:6517–6528. doi: 10.1523/JNEUROSCI.20-17-06517.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishikawa T, Giardino I, Edelstein D, Brownlee M. Changes in diabetic retinal matrix protein mRNA levels in a common transgenic mouse strain. Curr Eye Res. 2000;21:581–587. [PubMed] [Google Scholar]
- 30.Stitt A, Gardiner TA, Alderson NL, Canning P, Frizzell N, Duffy N, et al. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes. 2002;51:2826–2832. doi: 10.2337/diabetes.51.9.2826. [DOI] [PubMed] [Google Scholar]
- 31.Stitt AW, McGoldrick C, Rice-McCaldin A, McCance DR, Glenn JV, Hsu DK, et al. Impaired retinal angiogenesis in diabetes: role of advanced glycation end products and galectin-3. Diabetes. 2005;54:785–794. doi: 10.2337/diabetes.54.3.785. [DOI] [PubMed] [Google Scholar]
- 32.Canning P, Glenn JV, Hsu DK, Liu FT, Gardiner TA, Stitt AW. Inhibition of advanced glycation and absence of galectin-3 prevent blood-retinal barrier dysfunction during short-term diabetes. Exp Diabetes Res. 2007;2007:51837. doi: 10.1155/2007/51837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knapska E, Kaczmarek L. A gene for neuronal plasticity in the mammalian brain: Zif268/Egr-1/NGFI-A/Krox-24/TIS8/ZENK? Prog Neurobiol. 2004;74:183–211. doi: 10.1016/j.pneurobio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Langmann T, Ebert S, Walczak Y, Weigelt K, Ehrengruber MU, Stiewe T, et al. Induction of early growth response-1 mediates microglia activation in vitro but is dispensable in vivo . Neuromolecular Med. 2009;11:87–96. doi: 10.1007/s12017-009-8061-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS 86 kb)