


Abstract
Inhibition of primer extension by ribosome–mRNA complexes (toeprinting) is a proven and powerful technique for studying mechanisms of mRNA translation. Here we have assayed an advanced toeprinting approach that employs fluorescently labeled DNA primers, followed by capillary electrophoresis utilizing standard instruments for sequencing and fragment analysis. We demonstrate that this improved technique is not merely fast and cost-effective, but also brings the primer extension inhibition method up to the next level. The electrophoretic pattern of the primer extension reaction can be characterized with a precision unattainable by the common toeprint analysis utilizing radioactive isotopes. This method allows us to detect and quantify stable ribosomal complexes at all stages of translation, including initiation, elongation and termination, generated during the complete translation process in both the in vitro reconstituted translation system and the cell lysate. We also point out the unique advantages of this new methodology, including the ability to assay sites of the ribosomal complex assembly on several mRNA species in the same reaction mixture.
INTRODUCTION
The introduction of the primer extension inhibition technique, or toeprinting, provided new possibilities in the study of the ribosome–mRNA interactions and general mechanisms of translation. Initially, it was shown that prokaryotic ribosomes located on an RNA template arrested DNA elongation in the polymerase reaction of reverse transcriptase, and from this the positions of ribosomal complexes stalled on the mRNA could be determined quite precisely (1). The real power of this approach was evinced when it was employed to investigate eukaryotic translation initiation at internal ribosome entry sites in both rabbit reticulocyte lysate (RRL) (2) and reconstituted mammalian systems (3). Since then, the method has became a standard tool to study initiation, elongation and termination, i.e. all the stages of eukaryotic mRNA translation (4).
A scheme of a typical toeprinting experiment is shown in Figure 1. The underlying principle of this method is that any stable ribosomal complex stalled on mRNA stops the reverse transcription reaction by blocking polymerase movement along the mRNA. The transcription reaction is initialized with a primer designed to anneal downstream from the expected site of complex assembly and produces a complimentary DNA (cDNA) of characteristic length. The commonly used approach to toeprinting includes the analysis of radioactively labeled reverse transcription reaction products (mainly 32P-labeled) by electrophoretic separation on regular sequencing gel plates. This technique has obvious disadvantages and limitations, including the necessity of handling highly radioactive materials, low reproducibility and intractable problems with quantitative analysis.
Figure 1.

Toeprint analysis of a ribosome–mRNA complex formation. (A) Stable ribosomal (RS) complexes stop reverse transcriptase (RT) at a certain mRNA position generating short cDNA products of specific lengths. Comparison of a (B) classical toeprint experiment—electrophoretic analysis of radioactively labeled products of the primer extension inhibition reaction and (C) alternative approach utilizing fluorescently labeled primers and capillary electrophoresis for detection of the cDNA products. Peaks of fluorescence correspond to the bands on radioautograph.
Alternatively, the DNA primers can be labeled with suitable fluorescent groups and the cDNA molecules that resulted from primer elongation can be separated and detected using modern equipment for fragment analysis or sequencing, first suggested in (5). Separation of these cDNA fragments by capillary electrophoresis gives excellent resolution over a wide range of lengths (from about 20 to over 300 nucleotides), and the relative amount of each fragment can be precisely determined from the integral fluorescent intensity of the corresponding peak. Preliminarily, this fluorescent assay system was tested on initiation ribosome–mRNA complexes formed in the crude cell-free RRL (6).
In this report we describe the methodology and the benefits of capillary toeprinting for studying all main stages of translation, such as initiation, elongation and termination, in cell lysates or in reconstituted eukaryotic translation systems. We demonstrate that this approach is not merely fast and cost-effective, but also increases the precision of the toeprinting technique.
MATERIALS AND METHODS
Recombinant proteins
Plasmids coding for eukaryotic translation factors eIFs 1, 1A, 4A, 4B, 5B, 5 and Escherichia coli Met-tRNA synthetase (7), eRF1 and eRF3c (lacking its N-terminal 138 amino acids) (8) were used for production of the recombinant proteins in E. coli strain BL21DE3. Induction of protein synthesis in the E. coli and subsequent recombinant protein purification procedures generally resembled those described in Refs (7–9).
Non-recombinant proteins and ribosomal subunits
40S and 60S ribosomal subunits, eIFs: 2, 3, 4F and 5B; eEF1H and eEF2 were purified from RRL as described in Refs (7,9).
Messenger RNAs
Plasmids containing firefly luciferase coding region preceded with 5′ untranslated regions (UTRs) of: obelin protein mRNA from Obelia longissima (pObeLucTMV, Obe-Luc mRNA), omega sequence from tobacco mosaic virus 5′ UTR (pTZ10ΩLuc, Ω-Luc mRNA) and 25 nucleotides long poly(A) sequence (pTZA25Luc, A25-Luc mRNA), flanked by T7 RNA polymerase promoter, were described in Refs (10–13), correspondingly.
Plasmid for T7 RNA polymerase transcription of MVHL-stop mRNA encoded four (CAA) repeats at the mRNA 5′-end followed by natural β-globin mRNA 5′ UTR sequence, short open reading frame coding for Met-Val-His-Leu peptide, and UAA stop codon (4).
Plasmids containing firefly luciferase coding region were hydrolyzed with EcoRI endonuclease, the resulting 592 nt of the luciferase open reading frame to be included in the final transcript.
Plasmid coding for MVHL-stop mRNA was hydrolyzed with XhoI endonuclease.
Run-off transcription was carried out using T7 RNA polymerase as it was suggested in the protocols described in (14–16).
Natural capped β-globin mRNA was purified from RRL generally as described (17), the purification procedure was based on the mRNA 3′ poly(A) specific binding to oligo(dT) immobilized matrix.
Aminoacylation of tRNA
Total calf liver tRNA provided by Novagen was separated from high and low molecular weight contaminants by size-exclusion chromatography on Superdex 75 HR column (GE Healthcare).
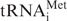 was specifically aminoacylated by methionine using E. coli Met-tRNA synthetase generally as described in Ref. (7). Aminoacylation reaction mixture contained 40 mM Tris–OAc pH 7.5, 10 mM Mg(OAc)2, 4 mM ATP, 0.2 mM l-methionine, 14 KBq/µl [35S] l-methionine (GE Healthcare), 0.25 mAU280nm/µl purified E. coli Met-tRNA synthetase, 0.8 U/µl RNAse inhibitor (RiboLock, Fermentas) and 6–10 µg/ml purified total calf liver tRNA (Novagen). Aminoacylation reaction continued for 25 min at 37°C.
was specifically aminoacylated by methionine using E. coli Met-tRNA synthetase generally as described in Ref. (7). Aminoacylation reaction mixture contained 40 mM Tris–OAc pH 7.5, 10 mM Mg(OAc)2, 4 mM ATP, 0.2 mM l-methionine, 14 KBq/µl [35S] l-methionine (GE Healthcare), 0.25 mAU280nm/µl purified E. coli Met-tRNA synthetase, 0.8 U/µl RNAse inhibitor (RiboLock, Fermentas) and 6–10 µg/ml purified total calf liver tRNA (Novagen). Aminoacylation reaction continued for 25 min at 37°C.
The total calf liver tRNA was aminoacylated by Met, Val, His and Leu using S100 fraction of RRL generally as described in Ref. (7). The aminoacylation reaction mixture contained 40 mM Tris–OAc pH 7.5, 15 mM Mg(OAc)2, 2 mM DTT, 10 mM ATP, 1 mM CTP, 0.05 mM each methionine, valine, histidine, leucine, 14 KBq/µl [35S] l-methionine (GE Healthcare), 1/20 of volume S100 fraction of RRL, 0.8 U/µl RNAse inhibitor (RiboLock, Fermentas) and 3–5 µg/ml purified total calf liver tRNA (Novagen). Aminoacylation reaction continued for 30 min at 37°C.
Formation of translation initiation complexes
Reaction mixtures for initiation complex formation were assembled at 0°C and contained 2.5–100 (normally 15) nM mRNA, 75 nM 40S ribosomal subunits, 60 nM eIF2, 60 nM eIF3, 50 nM eIF3–eIF4F complex, 750 nM eIF1, 750 nM eIF1A, 300 nM eIF4A, 300 nM eIF4B, 60 nM eIF5, 30 nM eIF5B, 75 nM 60S ribosomal subunits and 50 nM Met-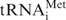 , supplemented with a buffer composed of 40 mM Tris–OAc pH 7.5, 3.7 mM Mg(OAc)2, 2 mM DTT, 0.25 mM spermidine, 2 mM ATP, 0.2 mM GTP (or guanosine 5′-[β,γ-imido]triphosphate, GMP-PNP), 0.1 mM EDTA, 120 mM KCl and 0.3 U/μl RiboLock RNAse inhibitor (Fermentas). The reaction mixtures were assembled in 20 µl, kept at 37°C for 15 min to allow ribosomal–mRNA complex formation, and subsequently used in the toeprint assay.
, supplemented with a buffer composed of 40 mM Tris–OAc pH 7.5, 3.7 mM Mg(OAc)2, 2 mM DTT, 0.25 mM spermidine, 2 mM ATP, 0.2 mM GTP (or guanosine 5′-[β,γ-imido]triphosphate, GMP-PNP), 0.1 mM EDTA, 120 mM KCl and 0.3 U/μl RiboLock RNAse inhibitor (Fermentas). The reaction mixtures were assembled in 20 µl, kept at 37°C for 15 min to allow ribosomal–mRNA complex formation, and subsequently used in the toeprint assay.
Formation of translation initiation complexes in non-fractionated RRL
Reaction mixtures for initiation complex formation were assembled at 0°C and contained 100 nM mRNA, 50% vol/vol nuclease-treated RRL (Flexi RRL, Promega), 20 mM Tris–OAc pH 7.5, 1 mM Mg(OAc)2, 4 mM DTT, 0.5 mM spermidine, 80 mM KCl, 0.3 U/μl RiboLock RNAse inhibitor (Fermentas), 5 mM (ATP + Mg(OAc)2), and were additionally supplemented with 2 mM (GTP + Mg(OAc)2) and 1 mg/ml cycloheximide, or (guanosine 5′-[β,γ-imido]triphosphate + Mg(OAc)2. The reaction mixtures were assembled in 20 µl, kept at 37°C for 5 min to allow ribosomal–mRNA complex formation, and subsequently used in toeprint assay.
Peptide elongation reaction
Peptide elongation reactions resembled those described in Ref. (4). Reaction mixtures contained 75 nM MVHL-stop mRNA, 75 nM 40S ribosomal subunits, 75 nM 60S ribosomal subunits, 200 nM eIF1, 200 nM eIF1A, 200 nM eIF2, 200 nM eIF3, 200 nM eIF4A, 200 nM eIF4B, 200 nM eIF4F, 200 nM eIF5, 200 nM eIF5B, 200 nM eEF1H, 50 nM eEF2 and 25 µg/ml total calf liver tRNA aminoacylated by Met, Val, His, Leu and a buffer composed of 20 mM Tris–OAc pH 7.5, 100 mM KAc, 2.5 mM MgCl2, 2 mM DTT, 1 mM ATP, 0.2 mM GTP, 0.25 mM spermidine and 0.25 U/µl RNAse inhibitor (Fermentas). The reaction mixtures were assembled in 40 µl, incubated at 37°C for 15 min and subsequently used in toeprint assay.
Assembly and purification of the pre-TC
Pre-TC was assembled generally as described in Ref. (4). Reaction mixtures contained 92.5 nM MVHL-stop mRNA, 187.5 nM 40S ribosomal subunits, 187.5 nM 60S ribosomal subunits, 312.5 nM eIF1, 312.5 nM eIF1A, 312.5 nM eIF2, 312.5 nM eIF3, 312.5 nM eIF4A, 312.5 nM eIF4B, 312.5 nM eIF4F, 312.5 nM eIF5, 312.5 nM eIF5B, 500 nM eEF1H, 125 nM eEF2 and 187.5 µg/ml total calf liver tRNA aminoacylated by Met, Val, His, Leu and a buffer A composed of 20 mM Tris–OAc pH 7.5, 100 mM KAc, 2.5 mM MgCl2, 2 mM DTT, and supplemented with 1 mM ATP, 0.2 mM GTP, 0.25 mM spermidine and 1 U/µl RNAse inhibitor (Fermentas). The reaction mixtures were assembled in 400 µl, incubated at 37°C for 30 min, loaded onto 10–30% (wt/wt) linear sucrose density gradients formed with buffer A additionally supplemented with 2.5 mM MgCl2 (5 mM MgCl2 final), and centrifuged in a Beckman SW55 rotor at 4°C and 240 000 × g for 95 min. After centrifugation gradients were fractionated, UV absorbance profiles at 260 nm of the fractionated gradients were determined and fractions corresponding to the pre-TC by sedimentation velocity were collected. The collected fractions were thereafter diluted with buffer A containing 1.25 mM MgCl2 to final MgCl2 concentration 2.5 mM, and used for toeprinting analysis or for the translation termination reaction.
Translation termination reaction
The translation termination reaction was performed generally as described in Ref. (4). The resultant solution with purified pre-TC (see above in ‘Assembly and purification of the pre-TC’) was additionally supplemented with 250 nM eRF1 and 250 nM eRF3. The reaction mixtures were assembled in 40 µl, incubated at 37°C for 10 min and subsequently used in the toeprint assay.
Reverse transcription for toeprint assay
The resultant reaction mixtures from ‘Formation of translation initiation complexes’, ‘Formation of translation initiation complexes in non-fractionated RRL’, ‘Peptide elongation reaction’, ‘Assembly and purification of the pre-TC’ or ‘Translation termination reaction’ (see above) were supplemented with 0.5 mM deoxyribonucleoside triphosphate (dATP, dCTP, dGTP, dTTP) each, 2 µM DNA primers with fluorescent labels, and 0.3 U/µl AMV reverse transcriptase (Promega). Simultaneously, the Mg2+ concentration was adjusted to 7 mM. The DNA primers were: 5′ [6-carboxyfluorescein] (FAM)-GGACTCGAAGAACCTCTG 3′ for rabbit β-globin mRNA; 5′ [6-carboxyfluorescein] (FAM)-GATGTTCACCTCGATATG 3′ or 5′ [6-carboxy-4′,5′-dichloro-2′,7′-dimethoxyfluorescein, succinimidyl ester] (JOE)-GATGTTCACCTCGATATG 3′ for mRNAs with luciferase coding region; and 5′ [6-carboxyfluorescein] (FAM)-GCATGTGCAGAGGACAGG 3′ for MHLV-stop mRNA. The reaction mixtures were then incubated at 37°C for 45 min and processed further as described in ‘Analysis of fluorescently labeled cDNA by capillary electrophoresis with fluorescence detection’ below.
Analysis of fluorescently labeled cDNA by capillary electrophoresis with fluorescence detection
The products of the primer extension reactions from ‘Reverse transcription’ or ‘Reverse transcription for toeprint assay’ (see above) were purified with phenol extraction, precipitated in 70% ethanol and 0.7 M NH4OAc and dissolved in 20 µl of 90% formamide with 89 mM Tris–base, 89 mM boric acid and 2 mM EDTA. 0.5 µl aliquots of fluorescent (carboxy-X-rhodamine, CXR) 60–400 bases DNA size standards (Promega) were added to each sample as internal markers for capillary electrophoresis. The cDNAs formed in the primer extension reaction were analyzed by capillary gel electrophoresis in ABI PRISM 3100-Avant Genetic Analyzer (Applera) generally in accordance with the manufacturer manual. The instrument was set up with POP6 (performance optimized polymer 6) and a 50 cm capillary tube and operated with 3100 Data collection software v1.1 (Applera). Run time was about 6000 s. The collected data were processed with GeneMarker 1.5 software (SoftGenetics) running in demonstration mode. Profiles of fluorescent cDNA distribution were attributed to the mRNA sequence using fluorescent (CXR) 60–400 bases DNA size standards (Promega); correction of the size standard calculated lengths to the product length of the actual dye-terminated sequence reactions was done for the first run due to the size discrepancy described in Refs (6,18,19). The plots visualizing distribution of the fluorescence intensities between the cDNA fragments of various lengths were screen-captured from GeneMarker 1.5 Analysis Window. The table with numeric data representing amount of the elongated cDNA oligonucleotide of each specific length as the area occupied by the corresponding cDNA fluorescence peak on electrophoregram was obtained by activating Allele Call and Show Chart/Table functions on the Analysis Window. Fluorescence intensities corresponding to each cDNA peak were measured to determine the amount of reverse transcription products and thus the amount of ribosomal complexes that resulted in the inhibition of primer extension.
RESULTS AND DISCUSSION
Optimization of primer extension reaction
High processivity of reverse transcription ensures that all polymerase molecules that successfully pass the initiation stage will reach the 5′ end of the RNA template. Therefore, the amount of the synthesized full-length cDNA will represent the amount of mRNA. Our first aim was to choose the polymerase, Mg2+ concentration and mode of primer annealing to provide the best processivity of reverse transcription.
We set up primer extension reactions directly in the reaction mixture used for ribosome–mRNA complex assembly by the addition of four deoxynucleotide triphosphates, extra Mg2+ and certain reverse transcriptase. We chose avian mieloblastosis virus (AMV-RT) and moloney murine leukemia virus (MMLV-RT) reverse transcriptases that are known to be successfully used for toeprinting. Both reverse transcriptases provided excellent processivity and yield at the final Mg2+ concentration of 4–8 mM (Supplementary Figure S1). We used two different approaches to primer hybridization, either pre-annealing with mRNA (1,20) or addition of the excess primer to the reaction mixture upon the assembly of the ribosome–mRNA complexes (3,21). In the latter case the yield of cDNA products was higher, probably because of re-initiation and multiple rounds of mRNA transcription. While the reverse transcription background generated by MMLV-RT was significantly lower when a primer was pre-annealed to mRNA after high-temperature melting (Supplementary Figure S1A versus S1B), the processivity of AMV-RT was high regardless of the method of primer addition. Moreover, only the AMV-RT allowed low reverse transcription background, substantial cDNA yield, and high specificity of primer hybridization for the simultaneous analysis of two different mRNAs in the same reaction mixture (Supplementary Figure S1D). We conclude that AMV-RT has advantages for any toeprinting analyses, while MMLV-RT is more useful with primer pre-annealing techniques.
Ribosomal complexes to be analyzed by primer extension method
To provide a comprehensive snapshot view of a whole translation process we performed a sequential mixing of model mRNAs with specific translation factors, aminoacylated tRNAs and ribosomal subunits, generally as described in (4) for eukaryotic translation system reconstitution.
The initiation 48S ribosomal complex is the first stable intermediate in translation. To assemble the 48S complex, we added translation initiation factors eIF1, eIF1A, eIF2, eIF3, eIF4A, eIF4B and eIF4F together with mRNA, Met-tRNAi and purified small (40S) ribosomal subunits to the reaction mixture. In the presence of ATP this led to the recruitment of 40S ribosomal subunits at the 5′ mRNA end with further 5′ UTR scanning (Figure 2A). As a result, the initiating ribosome recognized the start codon in a proper nucleotide context and formed a stable complex (Figure 2B). The 48S complex formed in this way is presumed to include initiation factors eIF1A, eIF3, eIF2:GTP with Met-tRNAi located in the P-site of the 40S ribosomal subunit. Formation of the 48S complex usually serves as a probe for the efficiency of initiation and can be detected by fluorescent toeprint analysis. The calculated relative yield of the complex can be used to estimate the effect of specific factors on the efficiency of translation initiation.
Figure 2.

Schematic depiction of ribosome–mRNA complexes representing different stages of the translation process. (A) mRNA 5′-end recognition and 5′ UTR scanning. Necessary initiation factors, 40S ribosomal subunit and initiator aminoacyl tRNA are indicated. (B–E) Arrows indicate the factors required for the transition to the consequent stable ribosome–mRNA complex.
To complete the initiation process and assemble full-sized initiation 80S ribosome on the mRNA we added initiation factors eIF5, eIF5B and 60S ribosomal subunits to the 48S complex. This led to the eIF5-induced hydrolysis of the eIF2-bound GTP and the release of eIF2:GDP, the attachment of eIF5B:GTP to the complex and 60S ribosomal subunit joining followed by the hydrolysis of eIF5B-bound GTP and the release of eIF5B:GDP. The resultant stable 80S complex is considered to contain no initiation factors and is capable of further translation stages (Figure 2C). It can be clearly discriminated from the 48S complex by toeprint analysis (2,22).
To switch on the elongation process we added elongation factors eEF1H, eEF2 and elongator aminoacyl tRNAs. In the system we used, the elongating ribosomes can stall at either the codon with a missing cognate aminoacyl tRNA, or at the first nonsense (stop) codon, depending on the set of aminoacyl tRNAs added. The specific length of cDNA products reflected the position of such ribosomes on the mRNA in primer extension analysis. The ribosomal complex with the stop codon in the A-site (pre-termination complex) signified the completion of the elongation reaction and became capable of the further nascent peptide release (Figure 2D).
Finally, we added termination factors eRF1 and eRF3 to the pre-termination complex that induced peptidyl-tRNA hydrolysis, the release of nascent peptide and the formation of termination complex (TC) (Figure 2E). Toeprint analysis revealed a shift of the ribosome on the mRNA due to the termination complex formation.
We used the fluorescent assay system to detect, discriminate and quantify the abovementioned stable ribosome–mRNA complexes.
Detection of ribosomal complexes on mRNA
Stable ribosomal complexes assembled on mRNA block reverse transcription and thus result in primer extension products shorter than the full-length cDNA. The size of these short products (toeprints) is strictly determined by the position of the complex on the mRNA. The extension reaction is arrested at a certain distance from the actual complex position on the mRNA and the halt of reverse transcription does not result in homogeneous toeprint cDNA. The appearance of three major cDNA peaks on electrophoregram is typical of toeprint signals from all ribosome–mRNA complexes—initiation, elongation and termination. These peaks correspond to +16th, +17th and +18th nucleotides reckoning downstream from the first nucleotide in the P-site of the ribosome (Figure 1B).
Reverse transcriptase can be stopped not only by ribosomal complexes, but also by other effectors including stable elements of mRNA structure, incidental mRNA chain breaks, tightly bound ligands, or even some template sequences. Specific stop signals corresponding to the ribosomal complexes can be deduced by setting up a control experiment where complex formation is not allowed. In all cases, except a few viral RNAs with unique IRESes, eIF2 is strictly required for 48S complex assembly. Therefore, to perform a negative control experiment we excluded eIF2 from the reaction mixture. As shown in Figure 3A, the presence of eIF2 in the reaction mixture affected mainly the major complex-specific peaks, while unspecific signals remained unchanged. In some special cases [e.g. IRESes of cricket paralysis virus (23) and hepatitis C virus (24)] where eIF2 is not mandatory for initiation, some alternative control reaction can be used.
Figure 3.

Detection of the initiation ribosome–mRNA 48S complex on natural β-globin mRNA. Lower curves show the case where 40S ribosomal subunits, eIFs: 1, 1A, 2, 3, 4A, 4B, 4F and Met-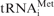 were present in the reaction mixture. Upper curves are the result of a control reaction where eIF2 was excluded. (A) The original fluorescence readout of the capillary electrophoresis. (B) Same as (A), but the cDNA peaks of fluorescence (blue line) were aligned with the mRNA sequence using Promega CXR size standards (red line). (C) Scaled region of (B) between the 5′-end and toeprint signals, normalized by the integral fluorescence intensity. The aligned β-globin mRNA sequence is shown in the bottom. Three main electrophoregram zones are indicated (see text).
were present in the reaction mixture. Upper curves are the result of a control reaction where eIF2 was excluded. (A) The original fluorescence readout of the capillary electrophoresis. (B) Same as (A), but the cDNA peaks of fluorescence (blue line) were aligned with the mRNA sequence using Promega CXR size standards (red line). (C) Scaled region of (B) between the 5′-end and toeprint signals, normalized by the integral fluorescence intensity. The aligned β-globin mRNA sequence is shown in the bottom. Three main electrophoregram zones are indicated (see text).
Analysis of electrophoregrams
To determine the lengths of the cDNA products, i.e. the position of the ribosomal complex on mRNA we aligned the electrophoregram with the nucleotide sequence of the mRNA. First of all, the dye-terminated sequencing reaction was set up for the mRNA to be analyzed. Products of this reaction were used to properly align commercially available fluorescent size standards with each cDNA sequence (6,18,19). The calibrated markers were added to the mixture of cDNA oligonucleotides derived from the toeprint reaction and loaded on the capillary electrophoresis. Fluorescences of the cDNA and the size standards were detected simultaneously through different channels. Using these data, peaks of cDNA fluorescence were attributed to the proper position of the mRNA sequence (Figure 3B).
A direct interpretation and comparison of the electrophoregrams may be incorrect. The absolute amplitude of a fluorescence peak depends not only on the efficiency of reverse transcription arrest, but also on the general efficiency of the transcription reaction, on unsystematic losses of cDNA during sample preparation and on the non-repeatability of capillary tube loading. Thus, we normalized the fluorescence readouts of the gels uniformly. We used the integral fluorescence intensity calculated from the electrophoregrams (excluding fluorescence of the free primer) as a normalizing factor. Indeed, it directly reflected the total molar amount of cDNAs synthesized or, in other words, the number of processive reverse transcriptase runs. In an electrophoregram normalized by this factor, the peak area indicated the percent of reverse transcriptase stops at given position of mRNA (Figure 3C). Such normalization made it possible to compare the results of independent toeprint experiments directly.
To simplify the calculations, we divided toeprint electrophoregrams into three main zones. First, the 5′-end zone corresponding to the full-sized reverse transcription product, which is normally represented not by a single peak as might be expected, but by the group of peaks that can be explained by the artifacts of mRNA transcription initiation and of reverse transcription termination. For example, the so-called slippage of T7 RNA polymerase is prominent on templates with homopolynucleotide stretches near the promoter region, such as poly(A) mRNA leaders (Figure 9). We determined the 5′-end zone as a group of adjacent peaks located close to the estimated position of the full-size cDNA and exceeding the local noise (Figure 3C). The other important zone covers three peaks corresponding to the complex-specific (toeprint) cDNA. Peaks of cDNA fluorescence between these two zones represented the unspecific stops of reverse transcriptase. This ‘unspecific’ zone serves as an intrinsic indicator of the quality of the experiment: if its fluorescence is low it can be accounted simply as an addition to the 5′-end zone, if the fluorescence is significant, the interpretation of the results could be ambiguous.
Figure 9.

Detection of the 48S complex assembly sites in the mixture of natural β-globin mRNA (FAM fluorescence, blue line) and synthetic A25-Luc mRNA (JOE fluorescence, red line). Both fluorescence readouts from FAM and JOE are normalized by the integral fluorescence intensity of the corresponding wavelength. The aligned β-globin and A25-Luc mRNA sequences are shown in the bottom. Complex-specific electrophoregram zones are indicated, and relative yield of the 48S complex assembled on the corresponding mRNA is shown.
We determined the efficiency of ribosome–mRNA complex assembly as the integral fluorescence of the complex-specific zone divided by the total fluorescence of all three zones (5′-end, unspecific and complex-specific). This gave us the percent of mRNAs with bound ribosomal complexes.
Yield of the ribosomal initiation complexes
The concentrations of ribosomal subunits and translation factors we used were similar to those described in Ref. (25), although slightly corrected to better conform to the component ratio in vivo (26). Because the concentration of mRNA in vivo is volatile, we used it as a convenient factor for system optimization. Figure 4 represents the dependence of 48S complex formation efficiency on the concentration of β-globin mRNA. This mRNA was previously characterized as strictly cap- and scanning-dependent and requires the full canonical set of initiation factors (eIF1, eIF1A, eIF2, eIF3, eIF4A, eIF4B and eIF4F) to assemble the 48S complex (27,28). As might be expected, the percent of mRNA with ribosomal complexes continuously decreased with the increase of mRNA concentration (Figure 4B). However, the absolute amount of the 48S complex increased linearly at low mRNA concentrations and reached maximum at about 50 nM mRNA. This value was in agreement with the concentration of the limiting initiation factor eIF4F. Further addition of mRNA caused inhibition of complex assembly that can be explained by the partial sequestration of protein factors by excess mRNA [mRNA self inhibition effect, see Refs (11,13)]. At the same time, at very low mRNA concentrations, some impurities like cross-contamination of the factors may have a significant effect on complex formation. Therefore, we chose the working mRNA concentration of 15 nM, because it fell in the range where a high complex formation efficiency coincided with the linear response of the complex yield to mRNA concentration. The adequacy of the selected concentration was examined using four different model mRNAs; the level of complex yield was almost identical for all of them (Figure 4C).
Figure 4.

The dependency of 48S complex yield on the concentration of mRNA in the reconstituted translation system. 40S ribosomal subunits, eIFs: 1, 1A, 2, 3, 4A, 4B, 4F and Met-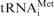 where present in the reaction mixture. (A) Aligned and normalized electrophoregrams of 48S complex toeprint analysis at indicated β-globin mRNA concentrations. (B) Dependency of the 48S complex concentration (solid line) and relative yield (dashed line) on the β-globin mRNA concentration in the reaction mixture. (C) Comparison of 48S complex formation on different messages. Indicated mRNAs were present in the reaction mixture at the same concentration of 15 nM.
where present in the reaction mixture. (A) Aligned and normalized electrophoregrams of 48S complex toeprint analysis at indicated β-globin mRNA concentrations. (B) Dependency of the 48S complex concentration (solid line) and relative yield (dashed line) on the β-globin mRNA concentration in the reaction mixture. (C) Comparison of 48S complex formation on different messages. Indicated mRNAs were present in the reaction mixture at the same concentration of 15 nM.
Discrimination of the ribosome–mRNA complexes
The mutual distribution of the fluorescence intensities between +16, +17 and +18 toeprint peaks depended on the type of the ribosome–mRNA complex (22) (Figure 5). The high resolution of capillary electrophoresis facilitated visualization of this distribution. In the case of 48S complex assembled on the start codon of model mRNA, the +16 stop signal was equal to or slightly exceeded the +17 signal, with a third toeprint on +18 being minor (Figures 5 and 6).
Figure 5.

Specific toeprint patterns of initiation 48S, initiation 80S (elongation-capable), stalled elongation (pre-TC) and termination (TC) ribosomal–mRNA complexes. Numbering of the cDNA peaks corresponds to the position of mRNA nucleotide relative to the triplet located in the P-site of the stalled ribosome.
Figure 6.

Comparison of the toeprint patterns generated by the 48S complexes assembled on different mRNAs (natural β-globin mRNA, non-capped synthetic luciferase-coding mRNAs with A25, Ω TMV and Obe-Luc leaders) at equal reaction conditions. Peaks numbered as in Figure 5.
Change in structural organization of the ribosomal complex on 60S subunit recruitment and formation of the initiating 80S ribosome was reflected in alteration of the toeprint peak distribution. Here the +17 toeprint signal became predominant while peaks +16 and +18 turned minor and were equally represented (Figure 5). This effect was robust and reproducible on different mRNAs and we used it for relative quantitation of the 48S and 80S initiation ribosomal complexes.
The elongation (pre-termination) complex (pre-TC) with the last sense codon located in the P-site and the stop codon—in the A site of the ribosome also resulted in one major cDNA peak located at +17 nucleotide and minor +16, +18 toeprint signals (Figure 5). The overall toeprint pattern of Pre-TC closely resembled that of the 80S initiation complex.
On transition to the termination complex the major cDNA peak shifted from position +17 to +19, thus appearing beyond the initial pre-TC three-peak pattern (Figure 5). Although such substantial change could be the result of dramatic conformational rearrangements of the ribosomal complex itself, it can also be explained by the alteration of the ribosome position on the mRNA. In the latter case enumeration of the toeprint peak positions from the last sense codon became arbitrary since another nucleotide triplet entered P-site.
Although the structural differences between the ribosome–mRNA complexes described can be detected by a conventional toeprint assay with the use of radioactively labeled cDNA (4,7,22), fluorescent toeprinting offers means of their quantitative description. The precise position of the ribosomal complex on mRNA and distribution of fluorescence intensities between toeprint peaks provided sufficient data for us to discriminate between different initiation, elongation and termination complexes without any a priori knowledge of the type of complex formed. This feature can be extremely useful in the studies of translation in partially fractionated or non-fractionated translation systems.
Quantitation of translation initiation, elongation and termination reactions
Capillary electrophoresis of fluorescent toeprint cDNAs offers high sensitivity. As little as 15 fmol of each mRNA can be analyzed in a single reaction. The high dynamic range and linear response of the fluorescent signal to the amount of relevant cDNA makes calculation of the yield of the particular ribosome–mRNA complex an easy task. To perform such quantitation, we attributed the peaks on the electrophoregram to the corresponding mRNA sequence and normalized them by the total fluorescence of the sample (as described above). As an example, the initiation complexes were assembled on model mRNA with poly(A) 5′ UTR (A25-Luc) in a translation system reconstituted in vitro and on a natural β-globin mRNA in the crude nuclease-treated RRL (Figure 7A and B, respectively). In the latter case, 2 mM GMP-PNP was applied to stall translation initiation at the stage of the 48S complex assembly, and 1 mg/ml cycloheximide was added to prevent the 80S initiation complex from entering the elongation process [as described in Ref. (22)]. The calculated yield of the 48S initiation complex was about 40% on A25-Luc mRNA and 60% on β-globin mRNA. The 48S-characteristic distribution of the fluorescence intensities was well developed: the +16 and +17 toeprint signals were major, both providing about 90% of the total amount of complex-specific reverse polymerase stalls while the residual 10% fell onto +18 toeprint peak.
Figure 7.

Quantitation of translation initiation complexes assembled on β-globin mRNA in non-fractionated RRL (A), and on A25-Luc mRNA in reconstituted eukaryotic initiation system (B). Components of the reaction mixtures used for complex assembly are briefly described on the right. Components required for the assembly of certain complexes are indicated in the bottom of the plots. Only complex-specific zones of the electrophoregrams are shown. Relative distribution of the total complex-specific fluorescence between the peaks is indicated above the plots. Peaks numbered as in Figure 5.
A significant change of the toeprint pattern (Figure 7) resulted from an accumulation of the 80S initiation complexes induced by the addition of certain components (eIFs 5, 5B; 60S ribosomal subunits) to the reconstituted system or by the addition of cycloheximide instead of GMP-PNP to the RRL system. In both cases the +16 toeprint peak underwent the maximum change and therefore represented the 48S to 80S transition most reliably: it decreased almost 2-fold for A25-Luc mRNA (Figure 7B) and almost 4-fold for β-globin mRNA (Figure 7A). The extent of +16 peak decrease upon 80S complex assembly served us as a measure of the subunit joining reaction efficiency. For example when eIF5—a catalyst of the ribosome subunits joining reaction—was not included in the reaction mixture with A25-Luc mRNA the +16 signal decreased only 1.4-fold instead of 2-fold after addition of eIF5B and 60S ribosomal subunits. It should be noticed also, that different mRNAs and translation systems provided slightly different distribution between the toeprint peaks (Figures 6 and 7). Therefore, we can compare toeprint patterns only of the complexes formed on the same mRNA and in the same type of translation system.
Transition of the initiation 80S complex to pre-TC occurred when the set of elongation factors (eEF1H, eEF2) and required aminoacyl tRNAs were included in the reaction. When all of these factors were in excess over the mRNA, two ribosome–mRNA complexes became dominant—the 80S initiation complex on the start AUG codon and pre-TC stalled immediately before the stop codon. For a reliable assay of the subsequent termination reaction, we had to block the transition between these two complexes. This was achieved by the removal of the initiation and elongation factors from the reaction mixture using sucrose density gradient centrifugation. Fractions corresponding to 80S sedimentation coefficient were collected and used in termination experiments. This procedure removed free mRNA from the sample and made standard normalization by the total mRNA amount impossible. Therefore, we used integral fluorescence specific to the 80S initiation complex as a normalizing factor. It was correct in this specific case, because termination factors did not affect the initiation complex (4).
The termination reaction was induced by the addition of either eRF1 and eRF3 with GTP, or eRF1 alone. In both cases the most prominent change between the pre- and post-termination toeprint patterns were the decrease of the +17 peak and a proportional increase of the +19 peak (Figure 8). Here the peak +17 to peak +19 ratio changed from 10:1 to 1:10 upon addition of both termination factors (Figure 8C). In contrast, when eRF3 was omitted these ratios changed to 7:1 and 1:5, respectively, indicating that the termination reaction was less efficient (Figure 8B).
Figure 8.

Quantitation of translation termination reaction on MVHL-STOP mRNA. Plot (A) depicts toeprint pattern of the purified pre-TC, which was used both as a control and as a termination reaction substrate. Only complex-specific zone of the electrophoregram is shown. Plot (B) shows the toeprint pattern after addition of termination factor eRF1 to the pre-TC. Plot (C) is a result of addition of both eRF1 and eRF3.
To summarize, the fluorescent toeprint assay allowed us to calculate the yield of each stable ribosome–mRNA complex and even to estimate the relative efficiency of translation stages under different conditions.
Detection of multiple sites of complex assembly in a single reaction
Standard equipment for capillary electrophoresis allows detection of fluorescence at various wavelengths, providing the ability to discriminate cDNAs generated from primers labeled with different fluorophores. Thus, we were able to analyze the ribosomal complex assembly simultaneously on two or more mRNAs (or at distinct sites of a single mRNA in the same reaction mixture). Using primer oligonucleotides with different sequences we performed their hybridization with mRNA in situ after completion of the initiation complex assembly. An example of such an approach is illustrated in Figure 9, where 48S complexes formed on β-globin and A25-Luc mRNAs were analyzed simultaneously in the same reaction mixture. When the use of sequence-specific primers is not possible, e.g. if mRNAs with different 5′ UTRs and the same coding region are analyzed, primers of the same sequence but bearing different fluorophores may be pre-annealed with the corresponding mRNAs.
This multi-primer approach opens a new way to study competition of different mRNAs for translation initiation factors and to estimate the relative efficiency of initiation of different mRNAs directly in non-fractionated cell lysates. Moreover, the method allows comparison of the intercistronic IRES activity with the efficiency of 5′ end-based initiation on the same mRNA.
CONCLUSIONS
The toeprinting technique using fluorescently labeled primers followed by capillary electrophoresis provides a unique ability to locate, identify and quantify any stable ribosome–mRNA complexes. The type and yield of the complexes can be easily determined based only on toeprinting data. This is a convenient, fast and reliable technique that brings the primer extension inhibition method up to a new level of precision. It potentiates the toeprint method with capabilities that are inaccessible by routine radioisotope approaches.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Foundation for Basic Research (06-04-48964-а, 09-04-01729-а to A.S.S., 09-04-00537-а to N.E.S., 09-04-01726-a to Z.A.A. and 08-04-01091-а to E.Z.A.); State Support of Leading Scientific Groups of the Russian Federation (NSh-4610.2008.4 to A.S.S.); Program for basic researches on Molecular and Cellular Biology of the Presidium of Russian Academy of Sciences (to A.S.S. and L.L.K.). Funding for open access charge: Institute of Protein Research.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Andrey Poltaraus for providing them indispensable methodological and instrumental help. They are very grateful to Tatyana Pestova and Christopher Hellen for providing them with expression vectors for recombinant mammalian initiation factors.
REFERENCES
- 1.Hartz D, McPheeters DS, Traut R, Gold L. Extension inhibition analysis of translation initiation complexes. Methods Enzymol. 1988;164:419–425. doi: 10.1016/s0076-6879(88)64058-4. [DOI] [PubMed] [Google Scholar]
- 2.Anthony DD, Merrick WC. Analysis of 40 S and 80 S complexes with mRNA as measured by sucrose density gradients and primer extension inhibition. J. Biol. Chem. 1992;267:1554–1562. [PubMed] [Google Scholar]
- 3.Pestova TV, Hellen CU, Shatsky IN. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol. 1996;16:6859–6869. doi: 10.1128/mcb.16.12.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell. 2006;125:1125–1136. doi: 10.1016/j.cell.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 5.Sachs MS, Wang Z, Gaba A, Fang P, Belk J, Ganesan R, Amrani N, Jacobson A. Toeprint analysis of the positioning of translation apparatus components at initiation and termination codons of fungal mRNAs. Methods. 2002;26:105–114. doi: 10.1016/S1046-2023(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 6.Gould PS, Bird H, Easton AJ. Translation toeprinting assays using fluorescently labeled primers and capillary electrophoresis. Biotechniques. 2005;38:397–400. doi: 10.2144/05383ST02. [DOI] [PubMed] [Google Scholar]
- 7.Pisarev AV, Unbehaun A, Hellen CU, Pestova TV. Assembly and analysis of eukaryotic translation initiation complexes. Methods Enzymol. 2007;430:147–177. doi: 10.1016/S0076-6879(07)30007-4. [DOI] [PubMed] [Google Scholar]
- 8.Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, Kisselev LL. Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA. 1999;5:1014–1020. doi: 10.1017/s135583829999043x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shirokikh NE, Spirin AS. Poly(A) leader of eukaryotic mRNA bypasses the dependence of translation on initiation factors. Proc. Natl Acad. Sci. USA. 2008;105:10738–10743. doi: 10.1073/pnas.0804940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kopeina GS, Afonina ZA, Gromova KV, Shirokov VA, Vasiliev VD, Spirin AS. Step-wise formation of eukaryotic double-row polyribosomes and circular translation of polysomal mRNA. Nucleic Acids Res. 2008;36:2476–2488. doi: 10.1093/nar/gkm1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaloiko LA, Granovsky IE, Ivashina TV, Ksenzenko VN, Shirokov VA, Spirin AS. Effective non-viral leader for cap-independent translation in a eukaryotic cell-free system. Biotechnol. Bioeng. 2004;88:730–739. doi: 10.1002/bit.20267. [DOI] [PubMed] [Google Scholar]
- 12.Alekhina OM, Vassilenko KS, Spirin AS. Translation of non-capped mRNAs in a eukaryotic cell-free system: acceleration of initiation rate in the course of polysome formation. Nucleic Acids Res. 2007;35:6547–6559. doi: 10.1093/nar/gkm725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gudkov AT, Ozerova MV, Shiryaev VM, Spirin AS. 5′-poly(A) sequence as an effective leader for translation in eukaryotic cell-free systems. Biotechnol. Bioeng. 2005;91:468–473. doi: 10.1002/bit.20525. [DOI] [PubMed] [Google Scholar]
- 14.Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 15.Pokrovskaya ID, Gurevich VV. In vitro transcription: preparative RNA yields in analytical scale reactions. Anal. Biochem. 1994;220:420–423. doi: 10.1006/abio.1994.1360. [DOI] [PubMed] [Google Scholar]
- 16.Gurevich VV, Pokrovskaya ID, Obukhova TA, Zozulya SA. Preparative in vitro mRNA synthesis using SP6 and T7 RNA polymerases. Anal. Biochem. 1991;195:207–213. doi: 10.1016/0003-2697(91)90318-n. [DOI] [PubMed] [Google Scholar]
- 17.Pisarev AV, Skabkin MA, Thomas AA, Merrick WC, Ovchinnikov LP, Shatsky IN. Positive and negative effects of the major mammalian messenger ribonucleoprotein p50 on binding of 40 S ribosomal subunits to the initiation codon of beta-globin mRNA. J. Biol. Chem. 2002;277:15445–15451. doi: 10.1074/jbc.M111954200. [DOI] [PubMed] [Google Scholar]
- 18.Fekete RA, Miller MJ, Chattoraj DK. Fluorescently labeled oligonucleotide extension: a rapid and quantitative protocol for primer extension. Biotechniques. 2003;35:90–94, 97–98. doi: 10.2144/03351rr01. [DOI] [PubMed] [Google Scholar]
- 19.Yindeeyoungyeon W, Schell MA. Footprinting with an automated capillary DNA sequencer. Biotechniques. 2000;29:1034–1036. doi: 10.2144/00295st05. 1038, 1040–1041. [DOI] [PubMed] [Google Scholar]
- 20.Kozak M. Primer extension analysis of eukaryotic ribosome-mRNA complexes. Nucleic Acids Res. 1998;26:4853–4859. doi: 10.1093/nar/26.21.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du H, Tarpey R, Babitzke P. The trp RNA-binding attenuation protein regulates TrpG synthesis by binding to the trpG ribosome binding site of Bacillus subtilis. J. Bacteriol. 1997;179:2582–2586. doi: 10.1128/jb.179.8.2582-2586.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dmitriev SE, Pisarev AV, Rubtsova MP, Dunaevsky YE, Shatsky IN. Conversion of 48S translation preinitiation complexes into 80S initiation complexes as revealed by toeprinting. FEBS Lett. 2003;533:99–104. doi: 10.1016/s0014-5793(02)03776-6. [DOI] [PubMed] [Google Scholar]
- 23.Pestova TV, Lomakin IB, Hellen CU. Position of the CrPV IRES on the 40S subunit and factor dependence of IRES/80S ribosome assembly. EMBO Rep. 2004;5:906–913. doi: 10.1038/sj.embor.7400240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terenin IM, Dmitriev SE, Andreev DE, Shatsky IN. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nat. Struct. Mol. Biol. 2008;15:836–841. doi: 10.1038/nsmb.1445. [DOI] [PubMed] [Google Scholar]
- 25.Lomakin IB, Shirokikh NE, Yusupov MM, Hellen CU, Pestova TV. The fidelity of translation initiation: reciprocal activities of eIF1, IF3 and YciH. EMBO J. 2006;25:196–210. doi: 10.1038/sj.emboj.7600904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von der Haar T, McCarthy JE. Intracellular translation initiation factor levels in Saccharomyces cerevisiae and their role in cap-complex function. Mol. Microbiol. 2002;46:531–544. doi: 10.1046/j.1365-2958.2002.03172.x. [DOI] [PubMed] [Google Scholar]
- 27.Pestova TV, Borukhov SI, Hellen CU. Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature. 1998;394:854–859. doi: 10.1038/29703. [DOI] [PubMed] [Google Scholar]
- 28.Pestova TV, Kolupaeva VG. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002;16:2906–2922. doi: 10.1101/gad.1020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.