


Abstract
p53 maintains genome integrity by initiating the transcription of genes involved in cell-cycle arrest, senescence, apoptosis and DNA repair. The activity of p53 is regulated by both post-translational modifications and protein–protein interactions. p53 that has been phosphorylated at S366, S378 and T387 binds 14-3-3 proteins in vitro. Here, we show that these sites are potential 14-3-3 binding sites in vivo. Epsilon (ε) and gamma (γ) isoforms required phosphorylation at either of these sites for efficient interaction with p53, while for sigma (σ) and tau (τ) these sites are dispensable. Further, σ and τ bound more weakly to p53 C-terminal phosphopeptides than did ε and γ. However, the four isoforms bound tightly to di-phosphorylated p53 C-terminal peptides than did the mono-phosphorylated counterparts. Interestingly, all the isoforms studied transcriptionally activated wild-type p53. σ and τ stabilized p53 levels in cells, while ε and γ stimulated p53-DNA binding activity in vitro. Overall, the results suggest that structurally and functionally similar 14-3-3 isoforms may exert their regulatory potential on p53 through different mechanisms. We discuss the isoform-specific roles of 14-3-3 in p53 stabilization and activation of specific-DNA binding.
INTRODUCTION
The tumour suppressor p53 plays a vital role in the transcription of target genes that are involved in cell-cycle arrest, DNA repair and apoptosis (1,2). Upon stress stimuli, p53 is activated and stabilized as a sequence-specific transcription factor and its activity is often regulated by various interacting partners (3). The 393 amino acid long protein has an intrinsically disordered N-terminal transactivation domain (p53 TAD), a structured DNA binding core domain (the carrier of hot-spot mutations in cancer), a tetramerization domain (important for cooperative DNA binding) and a C-terminal domain (p53 CTD, negatively regulates specific DNA binding of p53) (4).
The intrinsically unstructured p53 TAD and p53 CTD are target sites for various post-translational modifications such as phosphorylation and acetylation (5,6). p53 CTD is acetylated at K370, K372, K381 and K382 by p300/CBP and these acetylations have been shown to decrease the non-specific DNA binding of p53 CTD in vitro (7). Phosphorylation of p53 at S15, T18, S20 upregulates its transcriptional activity by directly stabilizing it against MDM2-mediated degradation (8,9). Several sites are also phosphorylated in the p53 CTD, particularly, S366, T377, S378, T387 and S392 (10,11). In response to DNA damage, ATM kinase, the gene mutated in ataxia telangiectasia, activates checkpoint kinases Chk1 and Chk2, which phosphorylate p53 at multiple sites, in particular, S313, S314, S366, T377, S378 and T387. Mutation of these phosphorylation sites has differential effects on the acetylation of p53 (10). Phosphorylation at S378 and dephosphorylation at S376 are important for 14-3-3 to activate human p53 (12). However, two additional sites, S366 and T387 have been identified on human p53 CTD in vitro that could be additional binding sites for 14-3-3 proteins (13).
The 14-3-3 family of proteins is involved in diverse functions such as apoptosis, cell-cycle checkpoints and signal transduction pathways (14). The 14-3-3 proteins are dimeric and bind protein targets following their serine/threonine phosphorylation at a consensus motif (15,16). In humans, seven different isoforms have been identified: β, γ, ε, η, τ (also referred as θ), ζ and σ. While most of these isoforms are expressed in all tissues, σ is restricted to epithelial cells (17). The dimeric protein has two binding pockets for phosphoserine- or phosphothreonine- containing motifs. While canonical binding motifs referred to as mode 1 [R(S/Ar)XpS(LEAM)P] and mode 2 [RX(Y/F)XpS[LEAM]P) have been identified for 14-3-3, these proteins interact with targets that deviate significantly from the defined motif (15,18,19). 14-3-3σ has been most widely studied of all, because of its direct linkage to cancer (17). 14-3-3σ is a target gene of p53 and up-regulation of σ leads to cell-cycle arrest (20). A positive feedback loop of p53/14-3-3σ has been proposed, whereby 14-3-3σ stabilizes p53 from MDM2-mediated degradation (21,22).
In the present study, systematic mutational studies were carried out to probe the importance of three p53 phosphorylation sites (S366, S378 and T387) for four isoforms of 14-3-3: γ, ε, τ and σ. Nickel pull-down assays and immunoprecipitation experiments were carried out to identify p53 interactions with different 14-3-3 isoforms. Both the in vitro and the in vivo results agree well while raising the possibility of additional binding sites for 14-3-3 τ and σ. Since the false positive rates are higher in pull-down assays, we carried out fluorescence binding measurements on p53 CTD phosphopeptides for the four isoforms. The experiments confirmed that the isoforms bind p53 CTD but with varying affinities. We discuss the possible modulatory role exerted by 14-3-3 in regulating p53’s transcriptional activity.
MATERIALS AND METHODS
Plasmid construction
For bacterial expression, 14-3-3 isoforms were cloned using BamHI and EcoRI sites into a pET24a vector containing His-lipoyl and Tev protease sites. For transient transfection experiments, cDNAs encoding human p53 and various mutant forms were cloned into pCDNA3.1 (+) (Clontech). cDNAs of human 14-3-3 isoforms were amplified and cloned into pCDNA3.1 (+) vector using BamHI and EcoRI sites. All constructed plasmids were verified by sequencing.
Transfection of cell lines and luciferase assays
H1299 (p53 null) cells stably integrated with the p21-luciferase reporter were grown in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal calf serum and G418. Twenty four hours before transfection, the cells were subcultured from confluence to a 1 : 6 dilution into 6-well plates. For transfections for luciferase assays, each well received 0.1 µg of pCMV-Renilla (Promega), 0.2 µg of pcDNA-p53 and various amounts of pcDNA-14-3-3 plasmids as indicated in the figure legends. The DNA amount was normalized using empty pcDNA3.1 vector. Transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were treated with 0.5 µM CPT or DMSO immediately after transfection. Twenty four hours after drug treatment, cells were washed twice with PBS and lysed using RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 50 mM Tris–HCl at pH 8.0) to which a complete EDTA-free protease inhibitor tablet (Roche) had been added. Luciferase assays were performed with the Dual-Luciferase Reporter Assay system (Promega) in accordance with the manufacturer’s instructions. Wells were prepared in triplicate, and error bars represent 1 SD.
H1299 cells expressing inducible-wild type p53 (a kind gift from Carol Prives, Columbia University, NY, USA) under the control of a tetracycline-regulated promoter were maintained in RPMI 1640 (Invitrogen) containing 10% FCS, 250 µg/ml G418, 2 µg/ml puromycin and 4.5 µg/ml tetracycline (tet) as described (23–25). Transfections were carried out in medium containing tet and cells were induced to express p53 by placing them in tet-free medium. Control cells were maintained in medium containing tet. After 24 h of induction, cells were placed in medium containing tet and CPT (or DMSO). After 24 h of CPT treatment, lysates were prepared for immunoblot or luciferase assays.
Immunoprecipitation and western blots
For immunoprecipitations, transfected cells from three wells of a 6-well plate were washed twice in cold PBS and scraped into 400 µl of RIPA buffer containing a complete EDTA-free protease inhibitor mix. After removing debris by centrifugation, the pre-cleared extract was incubated overnight with 30 µg of 14-3-3 isoform-specific antibodies [σ: ab60311 (Abcam), τ: SC-732, γ: SC-731, ε: SC-1020, (Santa Cruz Biotechnology, Santa Cruz, CA, USA)] and 15 µl of protein G agarose. Beads were collected by centrifugation and washed three times in 10S buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 0.2% NP-40, 0.1% Triton X-100 and 0.01% SDS). Bound proteins were eluted in 60 µl SDS–PAGE sample buffer containing 3 mM DTT. Eluted proteins were separated on 4–12% SDS–PAGE gels and electroblotted onto nitrocellulose membranes. Proteins were detected using mouse anti-p53 (PAb1801) antibodies. Horseradish peroxidase (HRP)-conjugated goat anti-mouse was used at dilution of 1 : 5000. ECL western blotting detection kit (Amersham Pharmacia Biotech, Sweden) was used for protein detection. Immunoprecipitation using phospho-specific antibodies for pS366, pS378 and pT387 (generous gift from Sheau-Yann Shieh, Taiwan) were carried out as described above (10).
Protein expression and purification
Protein expression was carried out as described earlier (13). Briefly, BL21 cells were transformed with 14-3-3 expression plasmids. A single colony was used to inoculate the overnight culture. Three litres of expression cultures at 37°C and 250 r.p.m. were induced with 0.8 mM IPTG at an A600 ∼0.6 and allowed to express for 16 h at 20°C. The cells were harvested and sonicated in cell-cracking buffer of 50 mM sodium phosphate, pH 7.5, 300 mM NaCl, 14 mM β-mercaptoethanol. After centrifugation, the soluble fraction was loaded onto a Hi-trap Ni column and eluted with a 10–250 mM imidazole gradient over 10-column volumes. The pooled fractions were digested with Tev protease overnight at 4°C and then diluted with equilibration buffer followed by loading onto the Ni column. The flow-through was concentrated and further purified on a Superdex 75 26/60 preparative gel filtration (Amersham Biosciences, UK). The proteins were >95% pure as judged by SDS–PAGE analysis.
Nickel pull-down assay
Twenty microlitres of Ni-nitrilotriacetic acid (NTA-Sepharose) beads (Qiagen) were incubated with 5 µg His6, or 5 µg of His6-14-3-3 isoforms in Ni binding buffer (25 mM imidazole, 25 mM Tris–HCl, 150 mM NaCl, 3 mM DTT, pH 7.4, 0.1% NP-40) for 1 h followed by rocking with the cell lysate extract at 4°C for 4 h. After gentle centrifugation, the beads were washed five times with Ni binding buffer. Bound proteins were eluted in 60 µl of SDS–PAGE sample buffer containing 250 mM imidazole and 5 mM DTT and were separated in SDS–PAGE gels. p53 was detected by immunoblotting using PAb1801 (Abcam).
Fluorescence anisotropy and EMSA
Peptides were synthesized on a pioneer peptide synthesizer (Applied Biosystems, UK) as described (13,26). Measurements were recorded on FluoroMax-3 spectrofluorimeter (Jobin Yvon-Horiba) equipped with a Hamilton Microlab Titrator, at 20°C in buffer containing 20 mM HEPES, pH 7.2, 150 mM NaCl and 2 mM DTT. 14-3-3 protein was titrated into a cuvette containing either 1.2-ml fluorescein-labelled peptides (initial concentration 50 nM) as described previously (13). Manual titrations were carried out for mono-phosphorylated p53 CTD peptides, while automatic titrations were performed on all other peptides. Excitation/emission wave-lengths used for fluorescein were 480/530 nm. Data analysis was performed using KaleidaGraph software with a simple one-state binding model (13).
EMSA experiments were carried out as described (13). p21 dsDNA (8 µM) 5′-CGCGAACATGTTCGAACCATGTTCGCG-3′ labelled with fluorescein at the 5′-end was used in experiments. Phosphorylation of p53 using Chk1 and Chk2 was carried out as described earlier (13). Phosphorylated p53 was incubated with different amounts of the 14-3-3 isoforms for 1 h followed by addition of p21 DNA for 30 min in 20 mM HEPES, 150 mM NaCl, 3 mM DTT, 10% glycerol, pH 7.2. Gel images were acquired using a with Typhoon scanner (GE Healthcare) and analysed with ImageQuant TL software (GE Healthcare).
Circular dichroism and DSC measurements
Thermal denaturation of 14-3-3 isoforms was measured by far-UV CD (222 nm) using a Jasco J815 spectropolarimeter and data were fitted to a two-state transition as described. Two point five micromolars of 14-3-3 isoforms were used in a buffer containing 20 mM HEPES, 150 mM NaCl, 3 mM DTE, pH 7.2. Data were recorded in a temperature range of 2°C to 98°C with 1°C/min temperature gradient and 4 s of response. Isosbestic titration was carried out similarly by recording the wavelength scan at various temperatures.
Calorimetric measurements were performed by using a VP-DSC microcalorimeter (MicroCal), equipped with an AutoSampler. Scans were obtained at a protein concentration of 100 µM, in buffer containing 20 mM HEPES, 150 mM NaCl, 3 mM DTE, pH 7.2. A scan rate of 250°C/h was used, over a temperature range from 10°C to 90°C. The reversibility of the transition was checked by cooling and reheating the same sample. Results from the DSC measurements were analysed with the Origin 7.0 software from MicroCal.
RESULTS
Thermodynamic stabilities of 14-3-3 isoforms
Five isoforms of 14-3-3 (γ, β, ε, τ, σ) were recombinantly expressed in Escherichia coli and purified. We measured the equilibrium stabilities before carrying out p53-14-3-3 in vitro binding studies in order to confirm that 14-3-3 is stable during the experiments.
The crystal structures of 14-3-3 proteins are all homodimers (19). Previous reports on dimerization equilibria studies showed ε, τ and γ isoforms are dimeric, while β was found to be in monomer-dimer equilibrium (27). ε has a higher preference to form heterodimers. Each monomer of 14-3-3 consists of a bundle of nine α-helices. Circular dichroism (CD) was used to monitor the thermal unfolding of the helices at 222 nm (Figure 1A). The isoforms exhibited a narrow stability range with melting temperatures (Tm) ranging from 57°C to 64°C. σ was the most stable of the isoforms studied, with an apparent Tm of 64.4°C, while β had the lowest Tm of 57.8°C. Of the five isoforms studied, only the σ isoform exhibited reversible folding behaviour and showed isosbestic point indicative of two-state folding (Figure 1B). We also used differential scanning calorimetry (DSC) to measure the melting temperature of different 14-3-3 isoforms (Figure 1C). A systematic increase in the Tm was observed in DSC as compared to CD, and this is probably due to the oligomeric state of 14-3-3. Oligomeric proteins exhibit concentration dependent Tm shifts in DSC. The 14-3-3 isoforms showed a similar trend in melting temperatures as measured by both CD and DSC. Table 1 lists the Tm values of the 14-3-3 isoforms measured using CD and DSC. The results confirmed that the 14-3-3 isoforms are stable enough for in vitro p53/14-3-3 interactions studies to be carried out.
Figure 1.
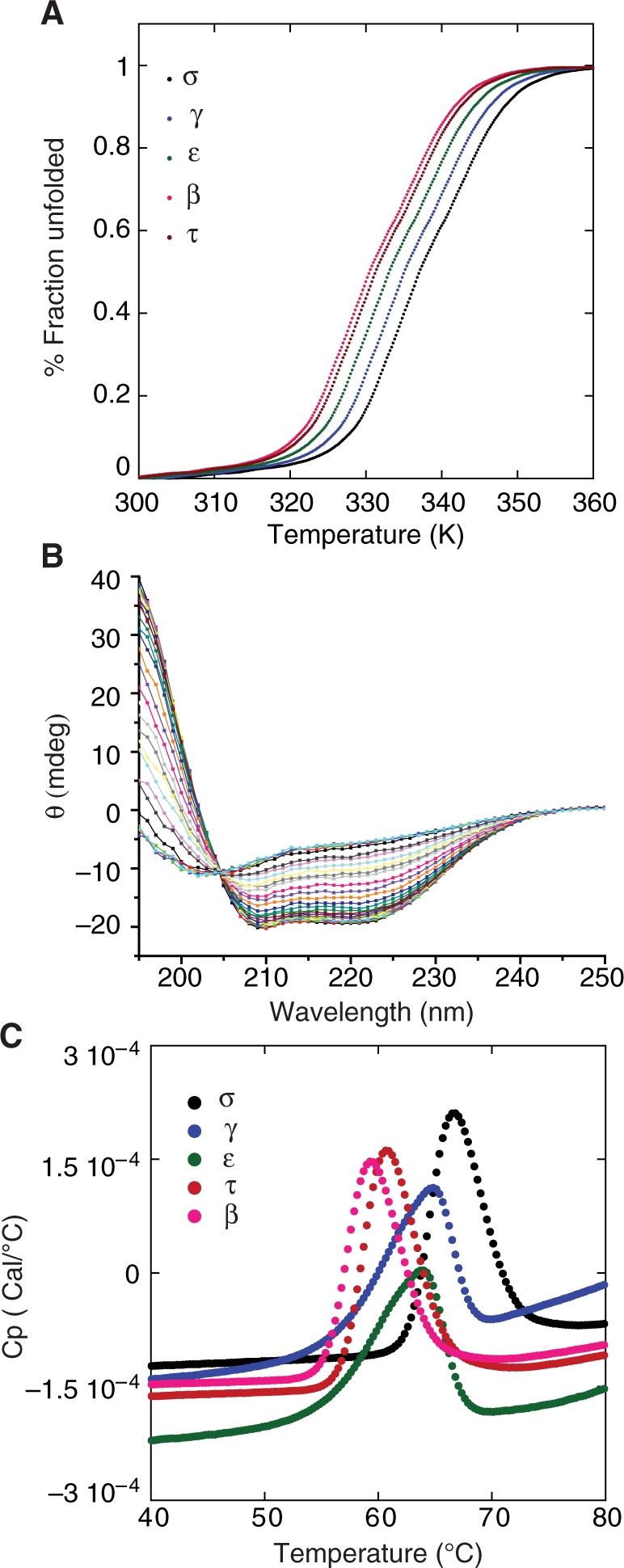
Thermodynamic studies on 14-3-3 isoforms. (A) Thermal unfolding of 14-3-3 isoforms was measured using CD by monitoring the ellipticity at 222 nm. The fraction of unfolded protein is plotted as a function of temperature. Only the σ isoform exhibited reversible folding after denaturation. (B) Isosbestic plot of 14-3-3 σ isoform. Wavelength scans were recorded at different temperatures. A sharp isosbestic point at 205 nm is a clear indication of two-state folding. (C) Thermal denaturation of 14-3-3 isoforms was measured using differential scanning calorimetry in a buffer containing 20 mM HEPES, 150 mM NaCl, 3 mM DTE, pH 7.2 at a heating rate of 250°C/h.
Table 1.
Thermal denaturation of 14-3-3 isoforms
| 14-3-3 isoforms |
Tm (°C) |
|
|---|---|---|
| CD (2.5 µM) | DSC (100 µM) | |
| σ | 64.4 ± 0.4 | 66.5 ± 0.5 |
| γ | 62.4 ± 0.6 | 64.8 ± 0.6 |
| ε | 60.4 ± 0.3 | 63.2 ± 0.4 |
| τ | 58.6 ± 0.4 | 60.5 ± 0.7 |
| β | 57.8 ± 0.7 | 59.4 ± 0.4 |
CD data were recorded in a temperature range of 2°C to 98°C with 1°C/min temperature gradient and 4 s of response time. For DSC experiments, a scan rate of 250°C/h was used, over a temperature range of 10–90°C. Protein samples were measured in a buffer containing 20 mM HEPES, 150 mM NaCl, 3 mM DTE, pH 7.2. The Tm of the 14-3-3 isoforms determined by CD and DSC were compared from each method separately and showed statistically significant differences (P < 0.05) except for τ with β isoform.
Activated p53 interacts with 14-3-3 γ, ε, τ and σ
We previously identified the binding of 14-3-3 γ and ε to p53 phosphorylated at S366, S378 and T387 using peptide screening (13). In order to identify whether 14-3-3 binds to these phosphorylation sites in vitro and if additional binding sites exist in p53, we carried out Nickel pull-down assays. H1299 (p53-null) cell lines were transfected with wild-type (WT) p53 or alanine mutants of the corresponding phosphorylation sites: S366A, S378A, T387A and the triple mutant 3A (S366A/S378A/T387A). Cells were treated with camptothecin (CPT), a topoisomerase inhibitor or DMSO (control) 24 h after transfection. CPT treatment induces double stranded DNA breaks, activating the ATM and Chk1/Chk2 pathway. The Chk1/Chk2 kinases phosphorylate p53 at S366, S378 and T387 (10). In order to confirm that p53 is phosphorylated at these residues, cell lysates were immunoprecipitated with anti- pS366, pS378 and pS387 specific antibodies and immunoblotted for p53 (Figure 2A). Western blots confirmed that p53 is phosphorylated at these residues upon DNA damage. The specificity of the antibodies has been validated elsewhere (10).
Figure 2.

p53/14-3-3 interaction in vitro and in vivo. (A) Phosphorylation status of p53 upon CPT treatment. Immunoprecipitation with pS366-, pS378- and pT387- specific antibodies indicated that these residues are phosphorylated. (B) Ni pull-down of p53 and its Ala mutants by 14-3-3 isoforms. γ, ε, τ and σ bound single Ala mutants of p53: S366A, S378A and T387A while τ and σ but not γ and ε bound the 3A (S366A/S378A/T387A) mutant. (C) H1299 (p53 null) cells were co-transfected with p53 (and different mutants) and 14-3-3 isoforms in seperate experiment, treated with CPT for 24 h, followed by cell lysis and immunoprecipitated with 14-3-3 isoform-specific antibodies. Control experiments carried out on cells co-transfected with p53 (and mutants) and empty (14-3-3 null) vector showed that the interaction of 14-3-3 with p53 is specific. (D) p53 interaction with 14-3-3 isoforms was significantly enhanced upon CPT treatment. A polyclonal antibody that recognizes all 14-3-3 isoforms (K19, Santa Cruz Biotechnology) and a monoclonal antibody for p53 (PAb1801, Abcam) were used.
For the Ni-pull down assays, cell lysates were incubated with His tagged-14-3-3 bound to the Ni-NTA beads and immunoblotted for p53. The isoforms, τ, σ, γ and ε but not β were identified to form a complex with p53. Interestingly, the four 14-3-3 isoforms also bound to all the single Ala mutants. However, τ and σ isoforms bound strongly to the p53 3A mutant, whereas binding by γ and ε was weak or barely detectable. This experiment indicated τ and σ might have additional interaction sites in activated p53 (Figure 2B).
Having identified the isoforms that bind p53 in vitro, we investigated whether these isoforms could also interact with p53 in vivo. Immunoprecipitation results confirmed the observations made from the in vitro pull-down assays, with τ and σ binding to all the mutant forms while γ and ε failed to bind the 3A mutant (Figure 2C). We could not detect significant endogeneous levels of 14-3-3 γ, ε, and τ in H1299 cell lines although the basal level of the σ isoform could be detected on western blots, but upon transfection, both p53 and the 14-3-3 isoforms are over-expressed. We observed similar p53 levels in cells treated with or without CPT (Supplementary Figure S1), but interaction of 14-3-3 with p53 was strongly detectable only upon drug treatment (Figure 2D). This is probably due to the elevation of p53 phosphorylation status in drug-treated cells. However, low levels of p53 could be immunoprecipitated with γ, ε, τ and σ isoform-specific antibodies in the absence of drug treatment suggesting the presence of binding sites in p53 that are constitutively phosphorylated (Figure 2D). The loss of interaction between γ and ε with 3A mutant is not merely due to the replacement to Ala but rather on their inability to be phosphorylated which is supported by the following facts (i) C-terminus of p53 is intrinsically disordered and mutations to Ala would hardly disrupt the structural integrity, (ii) the observation that 14-3-3 interacts with all the single mutants suggests that the Ala replacement at individual position has no effect and hence would be collectively inert. It also indicates that phosphorylation at any one site is sufficient to mediate interaction with 14-3-3 ε and γ, (iii) the observation that ε (and γ) and σ (and τ) interacts differentially with 3A mutant, strongly supports that the C-terminus of p53 is the docking site for ε (and γ) but probably not for σ (and τ). If otherwise, binding of ε (and γ) to 3A mutant, as similar to, σ (and τ) would be observed, and (iv) the transcriptional activity of p53 (in the absence of 14-3-3) is unperturbed with all the mutant proteins, strongly suggesting that the mutations has little effect on the functional aspect of p53 (Figure 4C).
Figure 4.

Effect of 14-3-3 isoforms on the transcriptional activity of p53 and various mutants. (A) H1299 cell lines with an integrated p21-luciferase reporter were co-transfected with 14-3-3 isoforms and p53 (and mutants) in separate experiments, followed by CPT treatment. pcDNA 3.1(+) was used as a negative control. All the data shown are for drug treated cells. Error bar represents 1 SD for three independent experiments (*P < 0.0.5). (B) Representative western blot of cell-lysates from H1299 co-transfected with WT p53 or different p53 mutants and 14-3-3 γ, showed similar protein levels. Transfections of ε, τ and σ isoforms also showed similar protein levels (Supplementary Figure S3). (C) Control experiments showing luciferase activity for the wild-type p53 and mutants when co-transfected with empty vectors (in the absence of 14-3-3). (D) Western-blot indicating similar protein levels of 14-3-3 isoforms in separate experiments is shown.
14-3-3 isoforms bind with varying affinities to the p53 CTD
The in-vivo results indicated that while phosphorylation at either S366, S378 or T387 is essential for γ and ε to bind p53, no such prerequisite seems to exist for τ and σ. We previously reported the binding of p53 CTD mono- or di-phosphorylated peptides (pS366, pS378 and pT387) to 14-3-3 isoforms γ and ε. Table 2 lists the peptide sequences used for binding experiments. Using fluorescence titrations, we measured the binding affinities for τ and σ isoforms to these phosphopeptides (Figure 3) and compared them with γ and ε. Table 3 lists the Kds for the four isoforms that were tested for binding to the phosphopeptides. τ and σ isoforms had similar binding affinities to p53 CTD phosphopeptides. However, the affinities were weaker and significantly different from γ and ε. This raises the question of the role of τ and σ isoforms in regulating the activity of p53 by mediating interactions with p53 CTD. All the isoforms showed similar Kds to the mode-2 consensus peptide indicating that the 14-3-3 isoforms used were functionally active. Binding of 14-3-3 to p53 CTD wild-type peptide is not quantifiable since no binding was observed (no increase in polarization) when titrated up to a concentration of 600 µM of 14-3-3.
Table 2.
Peptides tested for binding to 14-3-3 isoforms
| Peptides | Sequence* |
|---|---|
| WT | CRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD |
| pS366 | CRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD |
| pS378 | CRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD |
| pT387 | CRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD |
| pS366/pT387 | CRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD |
| pS378/pT387 | CRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD |
| pS366/pS378 | CRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD |
| Mode 2 binding peptide | ARLYHSLLPAA |
Peptides were labelled using fluorescein 5′ maleimide at the cysteine introduced in the p53CTD (362-393) peptides. S and T indicate phosphorylated serine and threonine, respectively.
Figure 3.

Binding titrations of 14-3-3 τ and σ and fluorescein-labeled peptides. (A) Binding titration of 14-3-3 τ and σ to mode-2 consensus peptide. (B and D) Binding of σ isoform to mono- and di-phosphorylated p53 CTD peptides. (C and E) Binding of τ isoform to mono- and di-phosphorylated p53 CTD peptides. Peptides sequences are listed in Table 2. Experiments were carried out at 20°C in 20 mM HEPES, 150 mM NaCl, 5 mM DTT, pH 7.2. Titrations of γ and ε to p53CTD phosphopeptides were reported earlier (13).
Table 3.
Binding of 14-3-3 isoforms to p53 CTD phosphopeptides
| Peptides |
Kd (µM) |
|||
|---|---|---|---|---|
| γa | εa | τ | σ | |
| pS366 | 17 ± 2 | 16 ± 2 | 28 ± 3 | 24 ± 4 |
| pS378 | 20 ± 2 | 18 ± 3 | 27 ± 3 | 22 ± 3 |
| pT387 | 14 ± 3 | 11 ± 2 | 24 ± 4 | 23 ± 3 |
| pS366/pT387 | 0.14 ± 0.07 | 0.18 ± 0.05 | 6.2 ± 0.3 | 3.5 ± 0.4 |
| pS378 /pT387 | 0.48 ± 0.05 | 0.51 ± 0.03 | 2.1 ± 0.4 | 2.2 ± 0.3 |
| pS378/pT387 | 0.45 ± 0.03 | 0.80 ± 0.06 | 3.3 ± 0.2 | 7.3 ± 0.3 |
| Consensus peptide | 0.43 ± 0.06 | 0.54 ± 0.01 | 0.24 ± 0.02 | 0.12 ± 0.01 |
| WT (362–393) | n.q. | n.q. | n.q. | n.q. |
The peptides were labeled with fluorescein at the free-amino end of p53 CTD (362–393); n.q., not quantifiable
aValues were taken from previously published data (13).
14-3-3 increases the transcriptional activity of p53
We next investigated whether the 14-3-3/p53 interactions plays any role in regulating the transcriptional activity of p53. Luciferase assays were carried out in a H1299 cell line with a stably integrated p21-luciferase reporter gene. Cells were co-transfected with 14-3-3 and p53, followed by CPT treatment. Non-drug treated cells showed only a marginal increase in transcriptional activity, which is probably due to the basal phosphorylation status of p53 (Supplementary Figure S2). But, upon drug treatment, the transcriptional activity of p53 is increased ∼2.5-fold compared with mock-transfected cells. Transfection with each of the 14-3-3 isoform resulted in a significant increase in p53 activity as compared with in the absence of 14-3-3 (P < 0.05). We also co-transfected the p53 mutants S378A, S366A, T387A and 3A mutant with 14-3-3 isoforms in separate experiments. The four isoforms exhibited a similar increase in the transcriptional activity of p53 S366A, S378A and T387A mutants. When compared with WT p53, the difference in transcriptional activity of these mutants was negligible, suggesting that the influence of 14-3-3 in p53 activity was not affected by individual mutations. However, with the 3A mutant, γ and ε had a decreased level of transcriptional activity closer to that of WT p53 in the absence of 14-3-3 (P < 0.05), indicating that these phosphorylation sites are essential for p53 up-regulation by these isoforms. τ and σ had no distinguishable change in the activity level with the 3A mutant, indicating that phosphorylation at these sites may not be necessary for these isoforms to activate p53 (Figure 4A). Control experiments, transfecting with empty vectors, were carried out throughout the study. Cells co-transfected with p53, p53 mutants and empty vector (14-3-3 insert is absent) showed that the proteins are over-expressed. Control experiment carried out in the absence of 14-3-3 showed that the differences in transcriptional activity of WT p53 and the p53 mutants were statistically negligible (Figure 4C). This experiment confirmed that the mutations introduced had little effect on the activity of p53. Overall, the results demonstrated that the presence of 14-3-3 isoforms enhanced the transcriptional activity of p53. Phosphorylation at either of the sites S366, S378 or T387 is necessary and sufficient for γ and ε to activate p53, but is dispensable for τ and σ.
τ and σ isoforms but not γ and ε leads to increased p53 levels
In order to deduce the possible mechanisms behind the observed increase in transcriptional activity, p53 levels have to be monitored inside the cells. To this end, we used H1299 cell lines that can be induced to stably express wild-type p53 by tetracycline regulation. In transient tranfections, the proteins are over-expressed and hence the levels of p53 cannot be monitored accurately. Cells were transfected with 14-3-3 isoforms, followed by induction of p53. The levels of p53 increased in cells transfected with τ and σ isoforms and the effect was manifested only in drug-treated cells. On the other hand, no increase in p53 levels were observed in cells transfected with either γ or ε (Figure 5A).
Figure 5.

14-3-3 τ and σ stabilize p53. (A) H1299 cells expressing inducible p53 were transfected with 14-3-3 isoforms, followed by induction of p53 for 24 h (in tet-free medium). p53 induction was stopped by placing the cells in tet-containing medium and cells were treated with 0.5 µM CPT or DMSO (control) for an additional 24 h. Cell extracts were prepared and resolved by SDS–PAGE followed by immunoblotting for p53 using PAb1801 antibody. γ and ε had no effect on the p53 levels while τ and σ increased p53 levels. (B) Dose–response luciferase assay for 14-3-3 τ and σ is shown. No stimulation of luciferase activity was observed when p53 is not induced. (C) H1299 cells were transfected with increasing amounts of 14-3-3 τ and σ. Twenty-four hours post-transfection, cells were lysed and immunoblotted with anti-τ, anti-σ and anti-p53. A notable increase in p53 level was observed. (D) Dose–response luciferase assay for 14-3-3 γ and ε is shown. No stimulation of luciferase activity was observed when p53 is not induced. (E) Immunoblotting of cell lysates from H1299 cells transfected with increasing amounts of 14-3-3 γ and ε showed no pronounced effect on the p53 levels.
We further validated this observation by transfecting different amounts of 14-3-3 isoforms and looking at the transcriptional activity and p53 levels. In support of our earlier observations, p53 levels and hence transcriptional activity increased with increasing amounts of τ and σ isoforms while the transcriptional activity but not p53 levels increased with higher amounts of γ and ε isoforms (Figure 5B–D). Although the increase in p53 levels was not dramatic with τ and σ isoforms, it is still evident when compared with γ and ε which showed constant p53 levels (Figure 5E). In order to rule out the possibility that τ and σ isoforms might introduce any artifactual effect by interacting with components of transcriptional machinery, H1299 cell-lines that express stable GFP upon induction was used as a negative control. These cell-lines are tetracycline-regulated, similar to the H1299 cell-lines expressing p53 used in this study. The GFP expressing cell-lines were transfected with all the four isoforms with and without CPT treatement. From the western blot, it is evident that 14-3-3 τ and σ had little effect on GFP levels indicating that the increase in p53 level observed is 14-3-3 τ- and σ- specific (Supplementary Figure S4). The observation that 14-3-3 τ and σ stabilize p53 in cells is further reinforced by the previous report, wherein 14-3-3σ was shown to stabilize p53 by inhibiting its turnover and is mediated by blocking the activity of MDM2 towards p53. Hence, in the present study, τ and σ isoforms might stabilize p53 either by directly binding to p53 and preventing degradation or through other mechanisms. Transfection of empty vectors at increasing amounts showed no stabilization of p53, further confirming the observation that the p53 stabilization observed when transfected with τ and σ isoforms is τ and σ-mediated (data not shown).
γ and ε isoforms but not τ and σ enhance specific-DNA binding of p53 in vitro
We previously showed that the γ isoform enhanced the DNA binding activity of p53 in vitro (13). Here we examined whether ε, τ and σ isoforms had any similar effects on the DNA binding properties of p53. First, electrophoretic mobility shift assays (EMSA) were carried out on full-length p53 phosphorylated using Chk1 and Chk2 kinases and increasing amounts of 14-3-3 isoforms: γ, ε, τ and σ and p21 response element (13). In addition to the previously identified γ isoform, ε also enhanced the specific-DNA binding of p53 (Figure 6A). The increase in binding was sequence specific across different lengths of DNA (Figure 6B). In contrast, τ and σ isoforms had no effects on the DNA binding activities of p53 (Figure 6A). However, the p53-DNA band is super-shifted in the presence of τ and σ isoforms indicating ternary complex formation. Further, the presence of p53 and 14-3-3 in the complex were confirmed by in-gel digestion followed by mass-spectrometry (Supplementary Figure S5). No complex formation of 14-3-3 and DNA, in the absence of p53 was observed, confirming that the stimulation observed is mediated by activating p53 (Figure 6C).
Figure 6.

Effect of 14-3-3 on DNA binding of p53. (A) EMSA experiments carried out with the 14-3-3 isoforms are shown in separate panels. Lane 1: only DNA (8 µM), lane 2-8: p53 (8 µM) and increasing amounts of 14-3-3 (0, 2, 4, 8, 10, 15, 20 µM, respectively). γ and ε isoforms showed enhancement of the p53-DNA band with the free DNA band decreasing in intensity. A 27 bp containing p21 response element was used. (B) Sequence-specific DNA binding was observed across 40 bp and 55 bp DNA containing p53 response element. Lanes 1, 3, 5, 7, 9 and lanes 2, 4, 6, 8, 10 were loaded with 40 bp and 55 bp, respectively. In the presence of 14-3-3 ε and γ (20 µM: Lanes 7–10), the band is supershifted, and the free DNA band has disappeared. (C) Control experiment showing no complex formation of 14-3-3 and DNA occurs in the absence of p53. When p53 is present, ε and γ but not τ and σ enhance the sequence-specific DNA binding of p53. (D) Binding titration profiles of p53 to p21 response element labeled with fluorescein at 5′-end in the absence and presence of 14-3-3 respectively. The data fitting was done using Hill equation, [p53]h / (kdh/ [p53]h). ε and γ showed a decrease in cooperative DNA binding of p53, while τ and σ had no pronounced effect.
To determine the DNA binding enhancement quantitatively, we used our previous approach of florescence titration using fluorescein-labeled p21 response element (13). The ε isoform increased the DNA binding affinity of p53 to DNA from 48 nM to 25 nM with the Hill Constant dropping from ∼2.1 to 1.2 indicating the decrease in cooperativity of DNA binding of p53 (Figure 6D). This is similar to the γ isoform which we reported earlier (13). However, in the presence of τ and σ isoforms, the Kd of p53 to DNA increased only slightly to 42 nM and the Hill Constant dropped to ∼1.9 indicating that these isoforms had little effect on the DNA binding properties of p53 (Figure 6D).
DISCUSSION
We previously identified that, in addition to the documented pS378, pS366 and pT387 could serve as additional interaction sites on p53 with 14-3-3 (13). Here, we report that (i) 14-3-3 ε and γ interact with p53, phosphoyrlated at S366, S378 and T387 in vitro and in vivo, (ii) 14-3-3 τ and σ isoforms have additional binding sites in p53, (iii) all the four isoforms enhances the transcriptional activity of p53, iv) 14-3-3 τ and σ increase p53 levels in cells by stabilizing p53 and (v) 14-3-3 ε and γ activated p53 for sequence-specific DNA binding by stabilizing the tetramer formation.
First, we measured the thermodynamic stability of the 14-3-3 isoforms to confirm they are properly folded and stable, before proceeding with further experiments. All five isoforms had high values of Tm (Table 1). The σ isoform showed higher thermal stability than other isoforms.
Interaction sites of 14-3-3 on p53
p53 is phosphorylated at S376 and S378 under normal conditions. Upon IR-treatment, S376 is dephosphorylated, creating a 14-3-3 consensus binding site (12). The 14-3-3 family of proteins interacts with various partners with considerable deviation from the defined canonical motif (19). Recently, additional 14-3-3 binding sites on p53 have been proposed based on peptide screening, where p53 CTD phosphorylated at S366 and T387 bound with similar or higher affinities to 14-3-3 γ and ε (13). In the present study, Ni-pull-down assays with 14-3-3 isoforms showed that while σ and τ interacted with p53 mutated to Ala at either 366, 378, 387 or the triple mutant 3A (366A/378A/387A), γ and ε failed to interact with 3A mutant. This observation indicates the possible existence of other sites of interaction for σ and τ isoforms. We also tested the β isoform but it did not show any detectable binding to wild-type p53 (data not shown). The in vitro results agreed well with the immunoprecipitation results, further confirming that phosphorylation at S378 is dispensable for 14-3-3 interactions. Lee et al. (21) have shown that the phosphorylation status of mouse p53 at S373 and S375 (human equivalents S376 and S378) is not essential for 14-3-3 binding. The existence of multiple binding sites for target binding might act as a fail-safe mechanism whereby the failure to phosphorylate one residue is compensated for by phosphorylation at other sites. A similar mechanism exists for the phosphorylation sites of the p53 transactivation domain, where multiple phosphorylations at residues S15, T18, S20 stabilize p53 in cells and prevent MDM2-mediated degradation (5,28). A 24-fold reduction in MDM2 binding is observed when the p53 trans-activation domain is phosphorylated at multiple sites (29).
The interaction of p53 with 14-3-3 isoforms is more pronounced upon drug (CPT) treatment, indicating that 14-3-3 binding sites (phosphorylated residues) are created upon DNA damage. CPT induces double-stranded DNA breaks and activates ATM, which acts upstream in the activation of the Chk1/Chk2 pathway (30,31). ATM-dependent dephosphorylation of p53 at S376 and Chk1/Chk2 dependent phosphorylation of p53 at S378, S366 and T387 generate 14-3-3 binding sites in vivo (10,12). Inhibition of these kinases affects the phosphorylation levels of S366 and T387 (10). While CPT has been used throughout this study to induce DNA damage, use of other chemotherapeutic agents such as nutlin, cisplatin, actinomycin D or ionizing radiations might provide detailed insight into the need of phosphorylation for efficient 14-3-3 interaction at the molecular level.
The in vitro peptide affinity studies carried out in the present study showed that di-phosphorylated peptides bound with higher affinity than their mono-phosphorylated counterparts in all four 14-3-3 isoforms studied, suggesting that di-phosphorylated forms of p53 (pS366/pS378, pS378/pS387, pS366/pT387) may be of biological importance. This is most likely because 14-3-3 is a dimeric protein and can simultaneously accommodate two phospho groups in the binding cavities resulting in a greater specificity and higher affinity (16,19); We previously showed that one 14-3-3 dimer binds one di-phosphorylated peptide (13). Comparison of the binding affinities of γ and ε with τ and σ to p53 CTD phoshopeptides revealed that γ and ε had higher affinities for mono- and di-phosphopepides. τ and σ isoforms showed a 1.7- fold decrease in binding to mono-phosphorylated peptides when compared with γ and ε. The effect is more pronounced with a 20-fold decrease in binding to di-phosphorylated peptides. This raised the question of whether σ and τ isoforms are directly involved in regulating p53.
Stavridi et al. (32) have shown that γ, ε and τ isoforms show IR-specific interaction with wild-type p53 but σ failed to interact, while Yang et al. (22) reported that 14-3-3 σ interacts with p53 and positively regulates the transcriptional activity of p53. However, we observed here that all four isoforms interact with p53 but the interaction sites are seemingly different. The interaction site for 14-3-3 γ and ε reside in p53 CTD, consistent with the previous reports (32). σ and τ isoforms interact with p53 CTD, albeit with weak affinity as compared to γ and ε. Furthermore, the finding that σ and τ isoforms interact with p53 3A mutant in drug-treated cells, strongly indicates the existence of additional binding sites in p53. The transactivation domain of p53 (p53 TAD) is extensively phosphorylated upon DNA damage (10 phosphorylation sites) and plays a crucial role in modulating interactions with various targets such as MDM2, p300 and PC4 (26,29). p53 TAD is the target for a number of kinases and might create additional binding sites for σ and τ isoforms. Using in-vitro peptide screening, S100 proteins, which were previously known to interact with the C-terminal domain of p53, has been shown to interact with p53 TAD (33). Hence, a systematic screening of various mono-phosphorylated and combinations of di-phosphorylated peptides would probably enable the identification of interaction sites of σ and τ, if any, in the p53 transactivation domain. However, it must be noted that 14-3-3 isoform specificity is low for the optimal motifs but much more pronounced towards sub-optimal motifs, resulting in functional specificity and hence, identifying the σ and τ interaction sites on p53 is more challenging and a formidable task, as p53 lacks optimal binding motif of 14-3-3 (34,35).
Transcriptional activation of p53 by 14-3-3 isoforms
We carried out transcriptional activity assays to study the effect of 14-3-3 isoforms in regulating the transcriptional activity of p53. The results clearly demonstrated that all four isoforms activate p53 transcriptionally. The single Ala p53 mutants showed a similar activity to wild type p53 when co-transfected with 14-3-3 γ, ε, σ and τ, while with the 3A mutant, γ and ε induced a reduction in transcriptional level close to wild-type p53 only, but σ and τ had no such detrimental effect. This observation further confirmed that phosphorylations at S366, S378 and T387 are dispensable for σ and τ to activate p53 transcriptionally.
Stabilization of p53 by 14-3-3 σ and τ
Transcriptional activity of p53 is mainly regulated by two mechanisms: stabilization leading to increased p53 levels and activation of p53 for sequence-specific DNA binding. Hence, we investigated the mechanism behind 14-3-3-mediated transcriptional activation of p53. Transcriptional assays were carried out in H1299 cell-lines that stably express wild-type p53 under the control of a tetracycline-regulated promoter. The results clearly demonstrated that σ and τ isoforms enhanced the transcriptional activity of p53, by increasing p53 levels, probably through stabilization. However, no increase in p53 levels was observed with γ and ε indicating the stabilization mechanism is isoform-specific.
Stabilization of p53 in response to DNA damage is associated primarily with inhibition of MDM2-mediated degradation. This inhibition is achieved through different routes such as phosphorylation of p53 resulting in reduced affinity of MDM2 to p53, activation of p19ARF leading to inhibition of ubiquitin-ligase activity and regulation of subcellular localization of MDM2 (36). In an interesting study, Yang et al. (22) have reported that 14-3-3 enhances the transcriptional activity of p53 by increasing its half-life. Further, 14-3-3 σ decreases the half-life of MDM2 and blocks MDM2-mediated p53 degradation by inhibiting MDM2’s ubiquitin ligase activity and also interferes with the nuclear-export activity of MDM2 towards p53 (22). Our results, in general, are consistent with these observations and, further suggest that in addition to the previously identified σ isoform, τ might exert a similar regulatory mechanism towards p53. However, from our results, (i) it is possible that p53 might have additional σ and τ binding sites, which could directly interfere with MDM2-mediated degradation, (ii) these sites are created only upon DNA damage. This hypothesis is supported by previous report that upon DNA damage, 14-3-3σ stabilize p53 partly by direct binding and that this characteristic was not observed in p19ARF (22). It must be noted that 14-3-3 proteins can also bind unphosphorylated targets and these targets bind to 14-3-3 proteins in the same location as phosphorylated targets (37). Moreover, 14-3-3 σ has been shown to have different substrate specificity in comparison to other isoforms. Hence, if p53 TAD anchors additional binding sites for σ and τ that are created upon DNA damage, it is possible that binding of σ and τ to p53 TAD could competitively inhibit the binding of MDM2 to p53 TAD, resulting in increased p53 levels, similar to competitive binding of p300 and MDM2 to p53 TAD (29). While this is speculative at the moment, further experiments to identify σ and τ interaction sites on p53 would unravel the stabilization mechanism behind direct 14-3-3 and p53 interaction. Perhaps there exist parallel pathways that could also lead to stabilization of p53 in the presence of 14-3-3. For instance, 14-3-3 τ stabilizes E2F-1 and E2F-1, in turn, stabilizes p53 by direct binding. Further, E2F-1 upregulates p14ARF, which negatively regulates MDM2 (38,39). In a different pathway, Akt (protein kinase B) activation enhances p53 degradation. 14-3-3 σ binds and inhibits Akt, resulting in p53 stabilization (40). Hence, stabilization of p53 by 14-3-3 could be achieved through different pathways. Perhaps, several other mechanisms of 14-3-3 mediated p53 stabilization remain unexplored.
Activation of specific DNA binding of p53 by 14-3-3 γ and ε
The role of p53 CTD in regulating the sequence-specific DNA binding of p53 has been controversial. While negative regulatory role for p53 CTD has long been proposed, it has been questioned in the recent years (41,42). Further, a positive regulatory role of p53 CTD has been shown for DNA binding and transactivation of target promoters in vivo. p53 CTD is essential for efficient sliding along DNA. Importantly, phosphorylation of p53 CTD has little effect on DNA sliding (41). In the context of short DNA, the stability of sequence-specific complex is decreased due to rapid linear diffusion of p53 across the ends. However, in the present study, any influence of DNA sliding can be ruled out by the fact the stimulation of specific DNA binding of p53 by 14-3-3 was observed across different lengths of DNA (Figure 6B). A positive regulatory role of phosphorylated p53 CTD can be derived by its interactions with cofactors such as 14-3-3. It has been shown that the DNA binding of p53 can be modulated by co-factors that perturb conformational equilibrium between dimers and tetramers (43). Invoking a similar mechanism, activation of specific DNA binding can be achieved by regulating the tetramerization of p53.
In our previous study, we proposed a model whereby 14-3-3 γ sequesters dimeric p53 to the tetrameric active DNA binding form (13). p53 exists in equilibrium between dimers and tetramers at low concentrations. Binding of two dimers facilitates the sequence specific DNA binding of p53 in a co-operative manner (44). Cofactors like 14-3-3 stabilize the tetrameric form of p53 by shifting the dimer-tetramer equilibrium in favour of active tetrameric form, resulting in increased DNA binding. To our knowledge, we were the first to demonstrate previously, using fluorescence aided-Analytical ultracentrifugation, that 14-3-3 sequesters dimeric p53 to form tetramers, resulting in reduced co-operativitiy and increased DNA binding (13). The increase in DNA binding of p53 (from 44 to 22 nM) with a concomitant reduction in Hill Constant (from 2.1 to 1.1) observed here, in the presence of 14-3-3 γ and ε, results from stable tetramer formation of p53. In other words, the dissociation constant of p53 to dimers and tetramers is decreased in the presence of 14-3-3.
Previous reports have also made similar observations on the influence of tetramerization in activating the sequence-specific DNA binding of p53. Deppert and co-workers (45) showed that Bi-functional redox factor 1 (Ref-1) enhances the specific DNA binding of p53 by promoting association of dimers into tetramers and de-stacking of higher order oligomers into the tetrameric form. Acetylation of p53 activates sequence-specific DNA binding of p53 and is mediated by promoting tetramerization (46). In addition, p53β isoform hetero-tetramerize with wild-type p53 and stimulates it for bax expression but not p21 (47). BCCIP (BRCA2 and CDKN1A-interacting protein) enhances transactivation of p53-target genes by facilitating stable tetramer formation (48). In an elegant study, c-Abl tyrosine kinase was shown to interact with p53 CTD and activate DNA binding by decreasing the dissociation rate of p53–DNA complex. This activation is independent of its kinase activity. Interestingly, the interaction of c-Abl with p53 stabilized the tetrameric conformation of p53, leading to selective stimulation and transactivation of p53 to p21 but not bax response element (49). While in the present study, p21 response element was used to study the effect of 14-3-3 on p53, both in vivo and in vitro, it might be interesting to see if 14-3-3 can exert promoter selectivity for activation of specific p53 target genes involved in either growth arrest or apoptosis, similar to c-Abl.
Role of tetramerization in functional regulation of p53
Tetramerization leads to the most active DNA binding form of p53 (50). Dimerization of p53 occurs co-translationally in polysomes while tetramerization is mediated at post-translational level in solution. It is proposed that tetramerization of p53 could represent a form of functional regulation (51). For instance, S100 proteins modulate p53’s response by binding to monomeric or tetrameric p53, resulting in inhibitory or stimulatory effect (33). In unstressed cells, the cellular concentration of p53 is very low (1–10 nM) and may exist predominantly in monomeric or dimeric forms (49). It is likely that in response to DNA damage, tetramerization of p53 is promoted directly by (i) interaction with co-factors such as BCCIP, c-Abl or Ref-1 and (ii) phoshorylation of S392 or (iii) in a sequential manner of phosphorylation of p53 at S366, S378 or T387 followed by 14-3-3-mediated stable tetramer formation (45,49,52). In this regard, it might be interesting to see if co-factors such as BCCIP, c-Abl and Ref-1 regulate tetramerization in a similar manner as 14-3-3 by reducing the co-operativity of DNA binding.
CONCLUSIONS
Upon genotoxic stress, two independent events take place. First, p53 is induced and stabilized, and second, p53 is activated to its functional form leading to cell-cycle arrest or apoptosis. These events are shown to be independent and separable as either stabilization of p53 through oncogene expression or by the proteosome inhibitor LLnL, is insufficient to activate p53-dependent cell-cycle arrest or apoptosis (53,54). In our study, γ and ε isoforms but not σ and τ activated the DNA-binding ability of p53 in vitro. We and others have shown this effect and a model has been proposed, whereby 14-3-3 associates with phosphorylated p53 shifting the dimer-tetramer equilibrium of p53 to the DNA-binding tetrameric form, leading to a high affinity complex (12,13). The augmentation of transcriptional activity of p53 by σ and τ is probably mediated by stabilizing p53 levels in cells. We propose that 14-3-3 isoforms behave in a functionally similar way toward p53 but through diverse mechanisms (Figure 7). One possible reason could be that the 14-3-3 levels are cell-specific due to its multi-functional roles and hence different mechanisms are exerted towards different targets. The findings here lead to important questions such as why 14-3-3 employs different regulatory mechanisms towards p53, although the function is similar, is this behaviour p53-specific and more importantly can the isoforms be swapped for the mechanisms and what are the determining factors in choosing different mechanisms. The solutions to these questions could potentially shed more light on the mechanistic preference of other regulators of p53.
Figure 7.

Possible role of 14-3-3 in the p53 pathway. Upon stress signal, kinases are activated that phosphorylate p53 at multiple sites creating 14-3-3 binding motifs in vivo. p53 is stabilized against MDM2-mediated degradation by 14-3-3 τ and σ. Isoforms ε and γ modulate the transcriptional activity of p53 by stimulating sequence-specific DNA binding of p53. p53 is stabilized and activated for the transcription of target genes involved in cell-cycle arrest or apoptotic pathways. 14-3-3 σ is a target gene of p53 and is directly involved in cell-cycle arrest, inhibiting G2-M progression. Further, a positive feedback loop has been proposed for p53 and 14-3-3 σ (21,22).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Cambridge Commonwealth Trusts; Medical Research Council (to S.R.). Funding for open access charge: Medical Research Council (MRC).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Sheau-Yann Shieh, National Tsing Hua University, Taiwan and Dr Carol Prives, Columbia University, NY for generous gifts of p53 phosphospecific antibodies and H1299 cell lines with tetracycline-regulated inducible p53, respectively. We thank Drs TS Wong and JL Kaar for critical reading of the article and valuable suggestions.
REFERENCES
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH. p53: death star. Cell. 2000;103:691–694. doi: 10.1016/s0092-8674(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 3.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 4.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 5.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 6.Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 7.Friedler A, Veprintsev DB, Freund SM, von Glos KI, Fersht AR. Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure. 2005;13:629–636. doi: 10.1016/j.str.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 8.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 9.Unger T, Juven-Gershon T, Moallem E, Berger M, Vogt Sionov R, Lozano G, Oren M, Haupt Y. Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J. 1999;18:1805–1814. doi: 10.1093/emboj/18.7.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ou YH, Chung PH, Sun TP, Shieh SY. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol. Biol. Cell. 2005;16:1684–1695. doi: 10.1091/mbc.E04-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yap DB, Hsieh JK, Zhong S, Heath V, Gusterson B, Crook T, Lu X. Ser392 phosphorylation regulates the oncogenic function of mutant p53. Cancer Res. 2004;64:4749–4754. doi: 10.1158/0008-5472.CAN-1305-2. [DOI] [PubMed] [Google Scholar]
- 12.Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat. Genet. 1998;19:175–178. doi: 10.1038/542. [DOI] [PubMed] [Google Scholar]
- 13.Rajagopalan S, Jaulent AM, Wells M, Veprintsev DB, Fersht AR. 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 2008;36:5983–5991. doi: 10.1093/nar/gkn598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackintosh C. Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem. J. 2004;381:329–342. doi: 10.1042/BJ20031332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 16.Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 17.Hermeking H. The 14-3-3 cancer connection. Nat. Rev. Cancer. 2003;3:931–943. doi: 10.1038/nrc1230. [DOI] [PubMed] [Google Scholar]
- 18.Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffe MB. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol. Cell. 1999;4:153–166. doi: 10.1016/s1097-2765(00)80363-9. [DOI] [PubMed] [Google Scholar]
- 19.Gardino AK, Smerdon SJ, Yaffe MB. Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin. Cancer Biol. 2006;16:173–182. doi: 10.1016/j.semcancer.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 21.Lee MH, Lozano G. Regulation of the p53-MDM2 pathway by 14-3-3 sigma and other proteins. Semin. Cancer Biol. 2006;16:225–234. doi: 10.1016/j.semcancer.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 22.Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol. Cell Biol. 2003;23:7096–7107. doi: 10.1128/MCB.23.20.7096-7107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 1996;10:2438–2451. doi: 10.1101/gad.10.19.2438. [DOI] [PubMed] [Google Scholar]
- 24.Baptiste-Okoh N, Barsotti AM, Prives C. A role for caspase 2 and PIDD in the process of p53-mediated apoptosis. Proc. Natl Acad. Sci. USA. 2008;105:1937–1942. doi: 10.1073/pnas.0711800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baptiste N, Friedlander P, Chen X, Prives C. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene. 2002;21:9–21. doi: 10.1038/sj.onc.1205015. [DOI] [PubMed] [Google Scholar]
- 26.Rajagopalan S, Andreeva A, Teufel DP, Freund SM, Fersht AR. Interaction between the Transactivation Domain of p53 and PC4 exemplifies acidic activation domains as single-stranded DNA mimics. J. Biol. Chem. 2009;284:21728–21737. doi: 10.1074/jbc.M109.006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, Lee WH, Sobott F, Papagrigoriou E, Robinson CV, Grossmann JG, Sundstrom M, Doyle DA, Elkins JM. Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc. Natl Acad. Sci. USA. 2006;103:17237–17242. doi: 10.1073/pnas.0605779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meek DW. Multisite phosphorylation and the integration of stress signals at p53. Cell Signal. 1998;10:159–166. doi: 10.1016/s0898-6568(97)00119-8. [DOI] [PubMed] [Google Scholar]
- 29.Teufel DP, Bycroft M, Fersht AR. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene. 2009;28:2112–2118. doi: 10.1038/onc.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian B, Yang Q, Mao Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat. Cell Biol. 2009;11:211–218. doi: 10.1038/ncb1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen Q, Scorah J, Phear G, Rodgers G, Rodgers S, Meuth M. A mutant allele of MRE11 found in mismatch repair-deficient tumor cells suppresses the cellular response to DNA replication fork stress in a dominant negative manner. Mol. Biol. Cell. 2008;19:1693–1705. doi: 10.1091/mbc.E07-09-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stavridi ES, Chehab NH, Malikzay A, Halazonetis TD. Substitutions that compromise the ionizing radiation-induced association of p53 with 14-3-3 proteins also compromise the ability of p53 to induce cell cycle arrest. Cancer Res. 2001;61:7030–7033. [PubMed] [Google Scholar]
- 33.van Dieck J, Fernandez-Fernandez MR, Veprintsev DB, Fersht AR. Modulation of the oligomerization state of p53 by differential binding of proteins of the S100 family to p53 monomers and tetramers. J. Biol. Chem. 2009;284:13804–13811. doi: 10.1074/jbc.M901351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bridges D, Moorhead GB. 14-3-3 proteins: a number of functions for a numbered protein. Sci. STKE. 2005;2005:re10. doi: 10.1126/stke.2962005re10. [DOI] [PubMed] [Google Scholar]
- 35.Rosenquist M, Sehnke P, Ferl RJ, Sommarin M, Larsson C. Evolution of the 14-3-3 protein family: does the large number of isoforms in multicellular organisms reflect functional specificity? J. Mol. Evol. 2000;51:446–458. doi: 10.1007/s002390010107. [DOI] [PubMed] [Google Scholar]
- 36.Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol. Cell Biol. 2000;20:3224–3233. doi: 10.1128/mcb.20.9.3224-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petosa C, Masters SC, Bankston LA, Pohl J, Wang B, Fu H, Liddington RC. 14-3-3zeta binds a phosphorylated Raf peptide and an unphosphorylated peptide via its conserved amphipathic groove. J. Biol. Chem. 1998;273:16305–16310. doi: 10.1074/jbc.273.26.16305. [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Liu K, Lin FT, Lin WC. A role for 14-3-3 tau in E2F1 stabilization and DNA damage-induced apoptosis. J. Biol. Chem. 2004;279:54140–54152. doi: 10.1074/jbc.M410493200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindstrom MS, Wiman KG. Myc and E2F1 induce p53 through p14ARF-independent mechanisms in human fibroblasts. Oncogene. 2003;22:4993–5005. doi: 10.1038/sj.onc.1206659. [DOI] [PubMed] [Google Scholar]
- 40.Yang H, Wen YY, Zhao R, Lin YL, Fournier K, Yang HY, Qiu Y, Diaz J, Laronga C, Lee MH. DNA damage-induced protein 14-3-3 sigma inhibits protein kinase B/Akt activation and suppresses Akt-activated cancer. Cancer Res. 2006;66:3096–3105. doi: 10.1158/0008-5472.CAN-05-3620. [DOI] [PubMed] [Google Scholar]
- 41.McKinney K, Mattia M, Gottifredi V, Prives C. p53 linear diffusion along DNA requires its C terminus. Mol. Cell. 2004;16:413–424. doi: 10.1016/j.molcel.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 42.Sauer M, Bretz AC, Beinoraviciute-Kellner R, Beitzinger M, Burek C, Rosenwald A, Harms GS, Stiewe T. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic Acids Res. 2008;36:1900–1912. doi: 10.1093/nar/gkn044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLure KG, Lee PW. p53 DNA binding can be modulated by factors that alter the conformational equilibrium. EMBO J. 1999;18:763–770. doi: 10.1093/emboj/18.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinberg RL, Veprintsev DB, Fersht AR. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004;341:1145–1159. doi: 10.1016/j.jmb.2004.06.071. [DOI] [PubMed] [Google Scholar]
- 45.Hanson S, Kim E, Deppert W. Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene. 2005;24:1641–1647. doi: 10.1038/sj.onc.1208351. [DOI] [PubMed] [Google Scholar]
- 46.Li AG, Piluso LG, Cai X, Wei G, Sellers WR, Liu X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol. Cell. 2006;23:575–587. doi: 10.1016/j.molcel.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 47.Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–2137. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meng X, Yue J, Liu Z, Shen Z. Abrogation of the transactivation activity of p53 by BCCIP down-regulation. J. Biol. Chem. 2007;282:1570–1576. doi: 10.1074/jbc.M607520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei G, Li AG, Liu X. Insights into selective activation of p53 DNA binding by c-Abl. J. Biol. Chem. 2005;280:12271–12278. doi: 10.1074/jbc.M409522200. [DOI] [PubMed] [Google Scholar]
- 50.Chene P. The role of tetramerization in p53 function. Oncogene. 2001;20:2611–2617. doi: 10.1038/sj.onc.1204373. [DOI] [PubMed] [Google Scholar]
- 51.Nicholls CD, McLure KG, Shields MA, Lee PW. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J. Biol. Chem. 2002;277:12937–12945. doi: 10.1074/jbc.M108815200. [DOI] [PubMed] [Google Scholar]
- 52.Sakaguchi K, Sakamoto H, Lewis MS, Anderson CW, Erickson JW, Appella E, Xie D. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry. 1997;36:10117–10124. doi: 10.1021/bi970759w. [DOI] [PubMed] [Google Scholar]
- 53.Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53(1) Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- 54.Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.