


Abstract
Nrf2 plays pivotal roles in coordinating the antioxidant response and maintaining redox homeostasis. Nrf2 expression is exquisitely regulated; Nrf2 expression is suppressed under unstressed conditions but strikingly induced under oxidative stress. Previous studies showed that stress-induced Nrf2 up-regulation results from both the inhibition of Nrf2 degradation and enhanced Nrf2 translation. In the present study, we elucidate the mechanism underlying translational control of Nrf2. An internal ribosomal entry site (IRES) was identified within the 5′ untranslated region of human Nrf2 mRNA. The IRESNrf2 contains a highly conserved 18S rRNA binding site (RBS) that is required for internal initiation. This IRESNrf2 also contains a hairpin structured inhibitory element (IE) located upstream of the RBS. Deletion of this IE remarkably enhanced translation. Significantly, treatment of cells with hydrogen peroxide (H2O2) and phyto-oxidant sulforaphane further stimulated IRESNrf2-mediated translation initiation despite the attenuation of global protein synthesis. Polyribosomal profile assay confirmed that endogenous Nrf2 mRNAs were recruited into polysomal fractions under oxidative stress conditions. Collectively, these data demonstrate that Nrf2 translation is suppressed under normal conditions and specifically enhanced upon oxidant exposure by internal initiation, and provide a mechanistic explanation for translational control of Nrf2 by oxidative stress.
INTRODUCTION
The initiation of protein translation in eukaryotic cells is mainly regulated via two distinct mechanisms; cap-dependent ribosome scanning and cap-independent internal ribosome entry mediated by internal ribosomal entry sites (IRESs) (1). Normal physiological conditions favor cap-dependent translation (2). The 7-methylguanosine cap (m7GpppN, N is any nucleotide) of mRNA is recognized by eukaryotic initiation factor 4F (eIF4F) complex, consisting of cap-binding protein eIF4E, RNA helicase eIF4A and scaffolding protein eIF4G which recruits 40S ribosome subunit via eIF3. With its associated initiation factors, the 40S ribosome subunit is believed to scan the 5′ untranslated region (UTR) until it finds the initiation codon AUG. Subsequently the 60S ribosome subunit is recruited to assemble the 80S ribosome and polypeptidyl elongation commences (3). Under various cellular and environmental stresses, however, global protein translation declines and the translation of diverse stress-responsive factors driven by IRESs is preferentially upregulated (4,5). The switch from cap-dependent translation to cap-independent translation has been proposed to function as an adaptive response of stress resistance.
Nrf2 (NF-E2 related factor 2) is a basic leucine zipper (bZIP) transcription factor. Nrf2 is the key factor regulating the antioxidant response. Under unstressed conditions, Nrf2 is mainly sequestered in the cytoplasm by a cytoskeleton anchoring protein Keap1 (Kelch-like ECH-associated protein 1) (6). Keap1 is also a substrate adaptor protein for Nrf2 ubiquitination (7,8) that promotes constant degradation of Nrf2. When exposed to oxidative stress, the abundance of stable Nrf2 proteins increases dramatically (9). Nrf2 protein unbound to Keap1 can translocate to cell nucleus in a redox-sensitive manner (10) to orchestrate the transcription of a battery of phase II detoxifying/antioxidant enzymes and phase III efflux transporters (9). As a consequence, these cells can effectively neutralize and remove excess oxidants to restore redox homeostasis. Accumulating evidences show that Nrf2 augmentation may result from redox-sensitive attenuation of Keap1-mediated ubiquitination (11) as well as enhanced translation of Nrf2 mRNA (12). While the regulation of Keap1-mediated Nrf2 ubiquitination has been well elucidated (11), the mechanism underlying Nrf2 translational regulation remains unknown.
In this study, we identified within the 5′-UTR of Nrf2 mRNA a functional IRES. This IRESNrf2 contains a ribosomal binding site (RBS) and a hairpin (HP)-structured inhibitory element (IE). Importantly, the IRESNrf2-driven Nrf2 translation appears to be redox-sensitive. Nrf2 mRNA is selectively recruited into polysomes resulting in enhanced translation of Nrf2 protein. These results reveal a novel mode of regulation of Nrf2 signaling at the level of translation via internal initiation.
MATERIALS AND METHODS
Cell culture and chemicals
Human cervical squamous cancerous HeLa cells and human embryonic kidney (HEK) cells were obtained from ATCC (Manassas, VA, USA). HeLa and HEK cells were cultured as monolayer using minimum essential medium (MEM) supplemented with 10% fetal bovine serum, 2.2 mg/ml sodium bicarbonate, 100 U/ml penicillin and 100 µg/ml streptomycin. Human hepatoma G2 cell (HepG2) was also obtained from ATCC. HepG2 cells were cultured as monolayer using F-12 medium supplemented with 10% fetal bovine serum, 1.7 mg/ml sodium bicarbonate, 0.1 unit/ml insulin, 0.5 × minimal essential amino acids, 100 U/ml penicillin and 100 U/ml streptomycin. Reducing glutathione (GSH), N-acetyl cysteine (NAC) and hydroperoxide (H2O2) were purchased from Sigma (St Louis, MO, USA). Sulforaphane (SFN) was purchased from Lkt Lab (St Paul, MN, USA).
Plasmid construction
A segment of 5′-UTR and part of open reading frame (ORF) of hNrf2 mRNA (−83 to +45, with translation start codon as +1) was PCR amplified from genomic DNA extracted from HeLa cells using the QIAamp DNA mini kit (Qiagen). This Nrf2 segment was subcloned into the intercistronic region of a bicistronic vector (13). As a positive control, the 5′-UTR of p53 (−134 to −1) (14,15) was also amplified from genomic DNA and subcloned into the pRluc-Fluc vector. To rule out the possibility of ribosomal read through, we added a HP structure (16) upstream of the Nrf2 5′-UTR with minor modification. The internal screening KpnI site was replaced by a XhoI site. To rule out the possibility of promoter activities of the Nrf2 5′-UTR, we subcloned Nrf2 5′-UTR into the pGL3 basic and control vector (Promega) and analyzed the expression of its downstream Fluc activities. As a negative control, a short linker sequence (ATAAATAAA) was inserted into the intercistroic region, creating a pRluc-null-Fluc plasmid. All constructs were verified by sequencing.
Dual luciferase assay
Dual Luciferase Reporter Assay (Promega, Madison, WI, USA) was used to measure the activities of Renilla (Renilla reniformis) luciferase (Rluc) and firefly (Photinus pyralis) luciferase (Fluc) according to the manufacturer’s instruction. Briefly, 24 h after transfection, cells were harvested in 0.3 ml passive lysis buffer solution and rocked gently at room temperature for 20 min. Cell debris was discarded after 10 min centrifugation at 12 000 r.p.m. Sirius luminometer (Berthold Detection System) was used to measure the Fluc and Rluc intensities. To measure the Fluc activities, 10 µl lysate was mixed with 50 µl Luciferase Assay Reagent II to generate a stabilized luminescent signal. After the Fluc luminescence was quantified, the Fluc reaction was quenched and the Rluc was simultaneously initiated by adding 50 µl Stop & Glo Reagent. Rluc/Fluc activities were normalized by total protein concentration.
Western blotting
Rabbit anti-eIF4E (FL-217), anti-phospho(S209)-eIF4E, anti-Nrf2 (H-300) and anti-GAPDH (FL-335) antibodies were purchased from St Cruz Biotechnology Inc. Rabbit anti-eIF2α and anti-Ser51-phospho-eIF2α (119A11) were purchased from Cell Signaling (Danvers, MA, USA). Two hours after H2O2 and SFN treatments, cells were harvested in TGN buffer [50 mM Tris, pH 7.5, 150 mM NaCl, 1% Tween-20, 0.3% NP-40, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 1X protease inhibitor mixture (Roche), 100 µg/ml Leupeptin, 10 µg/ml aprotinin]. The samples were resolved by sodium dodecyl sulfate (SDS)-polyacrylamide gel (BioRAD, Hercules, CA, USA) electrophoresis and transferred to polyvinylidene difluoride membrane. The membrane was probed with rabbit anti-Nrf2 (1: 500), anti-eIF4E (1: 1000), anti-phospho(S209)-eIF4E (1: 1000), anti-eIF2α(1: 1000), anti-Ser51-phospho-eIF2α (1: 1000) and anti-GAPDH (1: 5000) at 4°C overnight.After washing three times, the membrane was blotted with peroxidase-conjugated secondary antibody (1: 2500) at room temperature for 1 h. Proteins were visualized using the ECL mixture from BioRAD.
Polyribosomal profile assay
Sucrose gradient was constituted by adding 2 ml of 47, 37, 27, 17, 7% sucrose solution (10 mM NaCl, 20 mM Tris, pH 7.5, 3 mM MgCl2, 2 mM DTT, 100 µg/ml CHX) from bottom to top in a polyallomer centrifuge tube (Beckman). The layered sucrose solutions were kept at 4°C overnight to generate a continuous sucrose gradient. HEK 293 cells were cultured in 15-cm Petri dishes till achieving 75–80% confluence and treated with H2O (solvent control), 200 µM H2O2 or 50 µM SFN for 2 h. Before harvest, cells were treated with 100 µg/ml cycloheximide (CHX) for 15 min and then washed twice with cold phosphate-buffered saline (PBS). For each sample, two dishes of cells were scraped in PBS and pooled together. After centrifugation at 4000 r.p.m. for 5 min, the pellets were dissolved in 0.5 ml lysis buffer [10 mM Tris (pH 7.8), 5 mM KCl, 6 mM MgCl2, 2 mM DTT, 1X protease inhibitors (Roche), 1 mM PMSF, 1X RNasin, 100 µg/ml CHX] and physically lyzed in a Dounce homogenizer (Fishcer Scientific). The homogenized solutions were centrifuged at 12 000 r.p.m. at 4°C for 5 min. The supernatants were collected and measured at an absorption peak λ = 260 nm (A260). Twenty A260 units of samples were loaded onto the top of sucrose gradient and centrifuged at 23 000 r.p.m. at 4°C for 4 h using a Beckman L7-55 ultricentrifuge. After centrifugation, the gradient samples were monitored continuously at λ = 254 nm and recorded using a model 185 density gradient fractionator (ISCO, Inc., Lincoln, NE, USA). Fractions of 1 ml volume were collected continuously. The RNAs of each fraction was extracted using RNAeasy method (Qiagen). The RNA concentration of each fraction was measured. Same quantity of RNA of each fraction was reverse transcribed (RT) and subjected to RT-polymerase chain reaction (PCR) and quantitative PCR (qPCR) analysis.
qPCR
RT RNA samples (2 µl) were mixed with PCR primers and PCR supermix supplemented with SYBR green dye (ABI Biosystem). Each sample was prepared in duplicate. PCR reactions were performed at the condition of denaturing at 95°C for 10 min, followed by 40 cycles of thermal cycling reaction (95°C for 15 s, 55°C for 30 s and 68°C for 1 min) and concluded by a dissociation cycle (95°C 15 s, 60°C 15 s, 95°C 15 s). The qPCR data was analyzed using an SDS2.3 software (ABI Biosystem) and an absolute quantity method. The critical threshold (CT) of each sample was converted to absolute quantity according to the standard curve. The standard curve was constructed from a serial 1: 10 dilution of pcDNA3.1-Nrf2 plasmid DNA, with concentrations from 102 ng to 10−7 ng.
Toe printing
Toe printing was performed as described by Locker and Lukavsky (17) with the following modifications. Superscript II (Invitrogen Life Technologies, Carlsbad, CA, USA) was used for the RT. As an option to the [α-32P]ATP, the primer (CTCGATATGTGCATCTGTA) was labeled with IRDye™800 at the 5′ end. The cDNA products were analyzed on a standard 6% sequencing gel using model 4200 IR2 sequence analyzer (Li-cor, Lincoln, Hebraska, USA). The sequences and the toe prints were observed with e-Seq V2.0 software (Li-cor).
N-methylisatoic anhydride assay
The RNA was in vitro transcribed from the pRluc-Nrf2-Fluc which contained IRESNrf2 and 144 nt from the fluc sequence. The RNA was probed with N-methylisatoic anhydride (NMIA) following the protocol of Wilkinson et al. (18) with the modifications published by Baird et al. (19). Concentration of 65 mM NMIA gave the best result for the amount of RNA used. NMIA reactive sites were used as constraints in RNASTRUCTURE (20) to predict the secondary structure of IRESNrf2.
RESULTS
The 5′-UTR of human Nrf2 mRNA contains a functional IRES
Previous observation of oxidative stress-promoted Nrf2 translation (12) led to a hypothesis proposing that Nrf2 translation may be regulated by an unidentified IRES located in the 5′-UTR of Nrf2 mRNA (12). The 5′-UTR of human Nrf2 (hNrf2) mRNA has been previously examined using the 5′-RACE (rapid amplification of the cDNA ends) assay (21). In a total of 40 RACE clones analyzed, only 11 clones proceeded to a consensus transcription start site, whereas a majority of 27 clones (67.5%) were arrested prematurely near the AUG (+49) codon in the ORF (21) (Figure 1), suggesting the existence of a stable RNA secondary structure difficult to penetrate for the RACE assay. Indeed the mFold algorithm (22) predicted the presence of an elaborate secondary structure in the 128 nt (nt, −83 to +45, with translation start codon as +1) segment of hNrf2 mRNA, with a Gibbs free energy value (ΔG) of −65.6 kcal/mol.
Figure 1.

Schematic drawing of the structure of the 5′-UTR of human Nrf2 mRNA and plasmid constructs. Wild-type (WT) or various deleted mutants of 5′-UTR of Nrf2 mRNA were subcloned into the intercistronic region of a bicistronic vector containing a Rluc and a Fluc. 5′-UTR: 5′-untranslated region; (A)n: poly(A) tail; IE: inhibitory element; ORF: open reading frame; RBS: ribosomal binding site; TSS: transcription start site.
To analyze the function of this Nrf2 mRNA segment, we inserted it into a bicistronic vector containing a Rluc and a Fluc (13) (Figure 1). When this pRluc-Nrf2-Fluc vector is expressed in cells, the upstream Rluc cistron is translated in a cap-dependent manner, whereas the downstream Fluc cistron can only be translated if the intercistronic sequence contains a functional IRES. As a negative control, a short linker sequence was inserted into the intercistroic region, creating a pRluc-null-Fluc plasmid. The 134-nt 5′-UTR of p53 (ΔG = −65.9 kcal/mol) containing an IRES (14,15) was used as a positive control.
When the pRluc-Nrf2-Fluc vector was expressed in HeLa, HEK and HepG2 cells, ∼8-fold higher Fluc activities were observed compared with pRluc-null-Fluc (Figure 2A). The observed Fluc activities were similar in magnitude to that elicited by the IRESp53 (Figure 2A), suggesting the existence of an IRES in hNrf2 mRNA.
Figure 2.

The 5′-UTR of hNrf2 mRNA possesses a functional IRES. (A) Bicistronic analysis of the 5′-UTR of Nrf2 mRNA. When pRluc-Nrf2-Fluc vector was expressed in HeLa, HEK and HepG2 cells, robust Fluc activities were observed, comparable in magnitude to IRESp53 activities. The Fluc and Rluc activities of pRluc-null-Fluc were arbitrarily set as 1. In all figures, asterisks indicate statistical significance (t-test) of P < 0.05. (B) RT-PCR confirmation of the integrity of the bicistronic reporter mRNA (C) Examination of Nrf2 5′-UTR in the pGL3 basic vector rules out the possibility that Nrf2 5′-UTR possesses cryptic promoter activity. (D) Addition of a HP structure upstream of Nrf2 5′-UTR rules out the possibility that the observed Fluc activities result from ribosomal read through.
Previously, it was reported that observed Fluc activities may result from spurious splicing (23,24). To verify the integrity of bicistronic mRNAs, we examined the actual size of the reverse transcribed (RT)-PCR products of various constructs. When amplified by T7 and BGH primers that flank the entire ORF of Rluc-insert-Fluc, only the full-length transcripts were observed, as compared with PCR products amplified from plasmid DNA (Figure 2B). DNase I pretreatment and RT in the absence of RT proved that the RT-PCR products were indeed derived from mRNAs and not from transfected plasmid DNA (Supplementary Figure S1). Similar results were obtained in PCR reactions using internal primer 1 (P1) or 2 (P2) (Figures 1 and 2B). These data strongly suggest that the integrity of Rluc-insert-Fluc transcripts is preserved in vivo and therefore rule out the possibility of spurious splicing.
An alternative explanation for the observed Fluc activity could be that this Nrf2 segment harbors a cryptic promoter that drives transcription of Fluc thus producing a monocistronic Fluc mRNA. To rule out this possibility, we tested this Nrf2 segment in a pGL3 basic vector for promoter activity. The pGL3 basic vector contains a Fluc ORF without any upstream promoter and 3′-end enhancer. We subcloned this Nrf2 segment upstream of the Fluc ORF. When examined in HeLa cells, pGL3 basic-Nrf2 actually exhibited lower Fluc activities in comparison with pGL3 basic vector (Figure 2C), confirming that the inserted Nrf2 sequence has no cryptic promoter activity. In contrast, the presence of a SV40 promoter in the pGL3 control vector elicited robust Fluc expression (Figure 2C).
It is also possible that the observed IRES activity results from ribosomal read through and reinitiation at the Fluc ORF. To examine this possibility, we inserted a HP structure (16) upstream of the Nrf2 5′-UTR to block ribosomal read through. When pRluc-HP-Nrf2-Fluc vector was expressed in HeLa cells, negligible decrease of Fluc activities were observed in comparison with the pRluc-Nrf2-Fluc expression (Figure 2D), suggesting that the observed Fluc expression is unlikely to result from ribosomal read through. Unexpectedly, pRluc-HP-Nrf2-Fluc exhibited significant decrease of Rluc expression (Figure 2D). It is possible that the position of HP interferes with the termination at Rluc ORF and consequently attenuates Rluc translation. The validity of translational inhibition by the HP was further confirmed by the insertion of HP immediately upstream of the Rluc ORF (HP-Rluc-Nrf2-Fluc). In this construct, the expression of Rluc was almost completely abolished while the expression of Fluc was virtually unchanged (Figure 2D), supporting the notion that the Fluc translation does not result from Rluc read through. Furthermore, this data provide additional evidence that in the context of the bicistronic reporter system the IRESNrf2 supports internal initiation in the absence of translation of the upstream cistron. Collectively, these data show that this Nrf2 segment contains an authentic IRES.
The IRESNrf2 contains an 18S rRNA binding site and an a hairpin inhibitory element
To elucidate the functional components of the IRESNrf2, we inspected the sequence of this Nrf2 segment and observed that it contains two putative 18s rRNA binding sites (RBS). The RBS1 is located at the Nrf2 5′-UTR with 84.6% complementarity to 18s rRNA (Figures 1, 3A and 4A). The RBS2 is located within Nrf2 ORF with 87.5% complementarity to 18s rRNA (Figures 1, 3B and 4A). To investigate whether these sequences are involved in IRESNrf2 activity, we made progressive deletions. The ΔN1 construct deleted an upstream segment of the Nrf2 5′-UTR but spared the RBS1 and RBS2. The ΔN2 construct deleted the RBS1, while the ΔC construct deleted the RBS2 (Figure 1).
Figure 3.

The 5′-UTR of Nrf2 mRNA contains a putative 18S rRNA binding site (RBS). (A, B) Sequence homology of the Nrf2 5′-UTR within the RBS region among different species, and complementation between Nrf2 RBS1 and RBS2 and 18S rRNA. Open circles represent mismatches. (C–E) Expression of wild-type (WT) and deletion mutants in HeLa, HEK and HepG2 cells. Deletion of RBS1 (construct ΔN2) remarkably abrogated the Nrf2 IRES activity, while the deletion of RBS2 (construct ΔC) had only minimal effect on the activity of Nrf2 IRES.
Figure 4.

The 5′-UTR of Nrf2 contains an inhibitory element. (A) The mFold algorithm predicted the existence of a hairpin structure (boxed) upstream of the RBS1. The positions of RBS1 and RBS2 are indicated by green and yellow fonts on black box, respectively. (B) The IRESNrf2 secondary structure deduced by NMIA probing. The arrows indicate NMIA reactive sites. The RBS1 and RBS2 motifs are highlighted (C) Deletion of the IE segment (ΔN1) robustly enhanced Nrf2 IRES activity.
When the IRES activity of these deletion mutants was examined in HeLa, HEK and HepG2 cells, similar expression patterns in these three cell lines were observed. The Fluc expression was attenuated in the ΔN2 construct (Figure 3C–E), suggesting that RBS1 may participate in the recruitment of ribosome to the IRESNrf2. In contrast, deletion of the RBS2 in the ΔC construct only slightly decreased the IRESNrf2 activity (Figure 3C–E), suggesting that the RBS2 of the ORF may not be required for IRESNrf2 function.
Next, we would like to test if the IRESNrf2 is capable of direct interaction with the ribosome. The toeprinting experiment revealed that the 40S ribosomal subunits purified from HeLa cells did not bind to an in vitro transcribed IRESNrf2. Furthermore, the 48S initiation complex could not be formed on the in vitro transcribed RNA from IRESNrf2 in rabbit reticulocyte lysate in the presence of nonhydrolyzable GTP analog GMP-PNP (data not shown). These results suggest stringent requirement of oxidative stress-induced factors which likely play an active role to recruit ribosome on the IRESNrf2.
The mFold algorithm predicted that, upstream of the RBS1 there is a stable HP structure (ΔG =−11.1 kcal/mol) (Figures 1 and 4A), which was confirmed by our NMIA assay (Figure 4B, Supplementary Figure S2). Deletion of this HP structure (the ΔN1 construct) elicited a robust augmentation of Fluc expression (Figure 4C). Therefore this stem–loop structure appears to function as an IE (25) for the IRESNrf2 activity, although the precise mechanism of this inhibition is not clear. Deletion of this IE segment may have affected the secondary structure of IRESNrf2 resulting in the exposure of the RBS1 and/or its proximal region in an open conformation. Alternatively, this HP segment may contain an unidentified translation repressor.
IRESNrf2 mediates redox-sensitive translation of Nrf2
Nrf2 plays the key role in antioxidative defense. Oxidative conditions promote Nrf2 translation (12). We therefore examined whether IRESNrf2-driven translation is redox sensitive. Rluc-Nrf2-Fluc expressing HEK and HeLa cells were treated with H2O2 (200 µM) or phyto-oxidant sulforaphane (SFN, 50 µM), two potent activators of Nrf2 and antioxidant/phase II genes inducers (26), as well as reducing agents glutathione (GSH, 1 mM) and N-acetyl-cysteine (NAC, 1 mM), for 2 h. Importantly, the treatment of cells with H2O2 and SFN enhanced the IRESNrf2 activity 1.5- and 2.5-fold, respectively (Figure 5A). H2O2 and SFN treatments also increased Nrf2 immunoreactivities substantially (Figure 5B), whereas the mRNA level of Nrf2, as measured by quantitative real-time PCR, appeared mainly unchanged (Figure 5C). Since a variety of stresses induce phosphorylation of the Ser51 residue of eIF2α (4,5), we also examined eIF2α phosphorylation. Both H2O2 and SFN treatments resulted in the phosphorylation of eIF2α (Figure 5B). In contrast, treatments with GSH and NAC had virtually no impact on IRESNrf2 activity (Figure 5A). Thus, IRESNrf2-driven translation initiation appears to be upregulated by oxidative signals.
Figure 5.

IRESNrf2-dependent translation initiation is redox sensitive. (A) Two hours treatment of H2O2 (200 µM) and sulforaphane (SFN, 50 µM) significantly enhanced Nrf2 IRES activity. Treatment with reducing compounds glutathione (GSH, 1 mM) and N-acetyl-cysteine (NAC, 1 mM) virtually had no effect. (B) H2O2 and SFN treatment enhanced Nrf2 immunoreactivities and eIF-2α phosphorylation. In addition, H2O2 and SFN had opposite effect on eIF-4E phosphorylation. (C) Real-time PCR showed that H2O2 and SFN treatments failed to change the mRNA level of hNrf2. (D–F) Redox-sensitivity remained in the ΔN1 (D) and ΔC mutants (F) but completely diminished in the ΔN1 mutant (E).
It is very interesting to observe that whereas both H2O2 and SFN could enhance IRESNrf2-dependent translation, yet they exhibited opposite effect on cap-dependent translation. H2O2 could enhance both IRES- and cap-dependent translation (as measured by Fluc and Rluc expression), SFN significantly enhanced IRES-dependent translation but suppressed cap-dependent translation (Figure 5A). This differential induction effect may be correlated with the observation that H2O2 treatment enhanced phosphorylation of eIF4E at the Ser209 residue (Figure 5B), whereas SFN treatment significantly attenuated eIF4E phosphorylation (Figure 5B).
To further identify which functional component of the IRESNrf2 may account for redox sensitivity, we investigated the redox-reactivity of ΔN1, ΔN2 and ΔC deletion mutants. While the redox-sensitivities of both ΔN1 and ΔC mutants were still preserved (Figure 5D and F), the redox-sensitivity of ΔN2 mutant was abolished (Figure 5E), suggesting that the redox sensitivity is likely dependent on the presence of the RBS1 and/or its surrounding sequences.
Nrf2 mRNAs are recruited into polysomes upon oxidant exposure
In agreement with a previous report (12), H2O2 and SFN treatments could remarkably increase Nrf2 protein levels (Figure 5B) in the absence of concomitant increase of endogenous Nrf2 mRNA levels (Figure 5C), indicating that Nrf2 regulation occurs at the translational or posttranslational level, and not at the transcriptional level. To examine whether endogeneous Nrf2 mRNAs are actively translated in a redox-sensitive manner, we performed a polyribosomal profile assay in HEK 293 cells (Figure 6). The ribosomes were fractionated by an ultracentrifugation through a sucrose gradient. In comparison with the control sample treated with solvent (H2O, Figure 6A), 2-h treatment with 200 µM H2O2 increased the size of 60S/80S peaks and attenuated the magnitude of polysomal fractions (Figure 6B), indicating polysomal disassembly and global translational repression under oxidative stress. Likewise, 2-h treatment with 50 µM SFN also enlarged 60S/80S peaks and induced a severe rundown of polysomal fractions (Figure 6C). RT-PCR analyses showed that, in control cells, the hNrf2 mRNAs mainly distributed in the fractions 7–9, equivalent to 80S monosome and low polysomal fractions (Figure 6D). Importantly, both H2O2 and SFN treatments shifted hNrf2 mRNAs to high polysomal fractions (Figure 6D), indicating active translation of hNrf2 mRNAs. In contrast, H2O2 and SFN treatments shifted β-actin mRNAs in a reverse direction (Figure 6E), indicating a translational suppression of this housekeeping gene. Our RT-PCR results were confirmed by qPCR assays. Indeed, H2O2 and SFN treatments shifted Nrf2 mRNAs to high polysomal fractions (Figure 6F). In contrast, both H2O2 and SFN treatments considerably decreased the amount of β-actin mRNA associated with high polysomal fractions, indicative of reduced translation (Figure 6G). These data confirmed the redox-sensitivity of endogenous hNrf2 translation in vivo in the cells.
Figure 6.

Polyribosomal profile assay of HEK cells treated with H2O (control), 200 µM H2O2 or 50 µM SFN for 2 h. (A–C) Typical polyribome profiles after sucrose gradient ultracentrifugation monitored at 254 nm from the top (7% sucrose, left) to the bottom (47% sucrose, right) are shown. Each sucrose gradient was separated into 11 fractions as indicated. (D, E) RT-PCR data shows that Nrf2 mRNAs were recruited into polysomal fractions in response to H2O2 and SFN treatment as verified by real time PCR measurement (F, G). Results are representative of three independent experiments.
DISCUSSION
Early studies have suggested that Nrf2 activation may be partially attributed to augmented Nrf2 biosynthesis (12). In the present study, we identified within the 5′-UTR of hNrf2 mRNA an IRES. Previously, an IRES has also been identified in the leading sequence of yeast AP-1 (YAP1) (27), a yeast homolog of Nrf2.
The IRESNrf2 has two putative RBSs (Figure 3A and B). RBS1 is highly conserved among human, mouse (NM_010902/U20532), rat (NM_031789) and bovine (NM_001011678) Nrf2 mRNAs (Figure 3A). RBS2 is conserved among human, mouse (U20532) and rat (NM_031789) Nrf2 (Figure 3B). Deletion analyses showed that RBS1 but not RBS2 is required for the IRESNrf2 function both under normal and oxidative conditions (Figures 3 and 5). The RBS1 is complementary to the 749–761 nt of human 18S rRNA (Figure 3A). It was shown previously that the 746–784 nt of 18S rRNA in 40S ribosomal subunit was accessible to a complementary probe and thus is likely accessible to mRNAs (28). We attempted to demonstrate that the IRESNrf2 can directly interact with the ribosome in vitro, but were unable to do so. Furthermore, we were unable to form the 48S initiation complex on the in vitro transcribed RNA from IRESNrf2 in rabbit reticulocyte lysate (RBL) in the presence of GMP-PNP (data not shown). These data does not exclude the possibility that the RBS1 is indeed participating in the IRESnrf2 function. Rather, it suggests that modifications of factors such as IRES trans-acting factors (ITAFs) or initiation factors by oxidative stress, a condition that cannot be recapitulated in RRL in vitro, would be required for proper functioning of the IRESNrf2.
The IRESNrf2 possesses a hairpin structured IE (Figure 4A and B). High sequence homology was observed in this stem-loop segment among the 5′-UTR of human, mouse (NM_010902) and bovine (NM_001011678) Nrf2 mRNA. Surprisingly, deletion of this stem–loop structure robustly enhanced translation efficiency of the Nrf2 IRES (Figure 4C). The identification of an IE within the IRESNrf2 may provide some clue to answer a long-time puzzle why cells would waste energy to constantly synthesize proteins, such as Nrf2, that are merely destined for degradation under normal cellular conditions. Our present data suggest that the antioxidant machinery operates in an efficient and economical fashion. Under unstressed conditions, the Nrf2 translation is sustained at low level of activity (Figure 6D and F). In an equilibrium with Keap1-mediated Nrf2 degradation, this low-activity of constitutive translation of Nrf2 is likely sufficient for cells to keep surveillance of the slight redox fluctuation and maintain basal antioxidant activity. Upon being challenged by oxidative stress, the IE-mediated suppression may be ‘overruled’ resulting in enhanced Nrf2 translation, as illustrated by the recruitment of Nrf2 mRNAs into translationally active polysomal fractions (Figure 6D and F). Magnified Nrf2 translation coupled with inhibited Keap1-mediated Nrf2 degradation could instantaneously elicit an effective antioxidant response. Further studies are needed to examine the nature and regulation of this IE.
All cellular IRES described to date require ITAFs to regulate IRES activity (2). Our data suggest that the IRESNrf2-mediated translation suppression and initiation may be regulated by ITAFs in a redox-sensitive manner. Further studies on the interaction of IRESNrf2-ITAFs will deepen our mechanistic understanding of Nrf2 translational regulation under normal physiological versus oxidative stress conditions.
The IRESNrf2-driven translation appears to be redox-sensitive. IRESNrf2-mediated internal initiation can be significantly upregulated by the treatment of oxidants H2O2 and SFN. In contrast, treatments of reducing compounds GSH and NAC have no effect (Figure 5A). Redox-sensitivity of IRES-driven internal initiation has been reported before. The IRES activity of Apaf-1 was significantly down-regulated when exposed to anoxia for 24 and 48 h (29). Transient exposure (30 min) to 200 µM H2O2 enhanced translation mediated by the IRES of Hepatitis C virus (IRESHCV) in HepG2 and A549 cells, coupled with eIF2α phosphorylation (30).
In the present study, treatments with H2O2 and SFN enhance phosphorylation of eIF2α (Figure 5B). Phosphorylation of eIF2α at the Ser51 residue is a signature event of translational response to stress. A variety of stresses induce phosphorylation of eIF2α, a composite element of ternary eIF2-GTPase complex. Phosphorylation of eIF2α disables eIF2-GTPase, which mediates Met-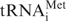 binding to 40S ribosome, and consequently suppresses global translation. Paradoxically, eIF2α phosphorylation enhances translation of some stress factors (4,5). The precise role of eIF2α phosphorylation in stress-induced translational regulation is unclear, since eIF2α phosphorylation alone appears not sufficient to elicit internal initiation (12). Under hypoxia stress, oxidative stress, viral infection, amino acid starvation and heat shock, the eIF2α can be phosphorylated by heme-regulated inhibitor kinase, PKR-like endoplasmic reticulum (ER) kinase (PERK), protein kinase RNA (PKR) and general control non-derepressible-2 (GCN2) (4,5). PERK is activated in response to ER stress and the unfolded protein response (4,5). Previously, it was reported that PERK could activate Nrf2 signaling (31,32). Further studies are needed to test whether PERK may facilitate Nrf2 translation under oxidative and ER stress via eIF2α phosphorylation.
binding to 40S ribosome, and consequently suppresses global translation. Paradoxically, eIF2α phosphorylation enhances translation of some stress factors (4,5). The precise role of eIF2α phosphorylation in stress-induced translational regulation is unclear, since eIF2α phosphorylation alone appears not sufficient to elicit internal initiation (12). Under hypoxia stress, oxidative stress, viral infection, amino acid starvation and heat shock, the eIF2α can be phosphorylated by heme-regulated inhibitor kinase, PKR-like endoplasmic reticulum (ER) kinase (PERK), protein kinase RNA (PKR) and general control non-derepressible-2 (GCN2) (4,5). PERK is activated in response to ER stress and the unfolded protein response (4,5). Previously, it was reported that PERK could activate Nrf2 signaling (31,32). Further studies are needed to test whether PERK may facilitate Nrf2 translation under oxidative and ER stress via eIF2α phosphorylation.
It is very interesting to observe differential inductive effect of two phase II inducers (H2O2 and SFN) with distinct chemical structure (26,33). H2O2 can enhance both cap-dependent and cap-independent translation (Figure 5A). Similar H2O2 effect was also observed in the IRESHCV (30). In contrast, SFN can selectively augment cap-independent translation but suppress cap-dependent translation (Figure 5A). It warrants further examination whether this differential effect results from distinct regulation of eIF4E phosphorylation by H2O2 and SFN (Figure 5B). Phosphorylation of eIF4E enhances cap binding affinity and favors the assembly of eIF4F complex (34,35). The eIF4E can be directly phosphorylated by Mnk1/2 (MAP kinase-interacting kinases) at the Ser209 residue (36,37). In addition, eIF4E can also be phosphorylated in a PI3K (phosphtidylinositol 3 kinase)-dependent manner (34). Further study is needed to elucidate the signaling events underlying H2O2− and SFN-induced effect. The differential induction elicited by H2O2 and SFN also raised the question whether other structurally distinct phase II inducers (26,33) also have differential effect on Nrf2 translation. It is interesting to observe that curcumin, a β-diketone with Michael acceptor functionality that is known to induce Nrf2 expression (38), can suppress phosphorylation of eIF4E binding protein (39,40) and may eventually regulate eIF4E availability.
In conclusion, we characterized a novel redox-sensitive IRES in the 5′-UTR of Nrf2 mRNA. In combination with previous studies, our data suggest that Nrf2 expression is not only regulated at the transcriptional (41) and post-translational (42–45) levels, but also at a level of protein synthesis. Importantly, the redox-sensitive translation and degradation of Nrf2 enables the cells to respond and adjust to a rapidly changing redox environment.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Institutes of Health (R01 CA94828 and NIEHS P30ES005022 to A.-N.K., partial); Canadian Institutes of Health Research (MOP 89737 to M.H.). M.H. is the CHEO Volunteer Association endowed scholar. Funding for open access charge: The National Institutes of Health (R01 CA94828 and NIEHS P30ES005022 to A.N.T.); Canadian Institutes of Health Research (MOP 89737 to M.H.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Xinfu Jiao and Mike Kiledjian for providing the bicistronic vector; Drs Terri Kinzy, Anthony Esposito, Wonjun Oh and Hua Wei of UMDNJ, Yi-Ching Hsieh of CINJ for their kind assistance in polysomal profile assay; Dr Rita Hahn and Shaojun Yang in EOHSI for their assistance in ultracentrifugation; and Irina Aranovich for her assistance with real time PCR.
REFERENCES
- 1.Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 2.Stoneley M, Willis AE. Cellular internal ribosome entry segments: structures, trans-acting factors and regulation of gene expression. Oncogene. 2004;23:3200–3207. doi: 10.1038/sj.onc.1207551. [DOI] [PubMed] [Google Scholar]
- 3.Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell. Biol. 2004;5:827–835. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell. Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 5.Komar AA, Hatzoglou M. Internal ribosome entry sites in cellular mRNAs: mystery of their existence. J. Biol. Chem. 2005;280:23425–23428. doi: 10.1074/jbc.R400041200. [DOI] [PubMed] [Google Scholar]
- 6.Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl Acad. Sci. USA. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Yu SW, Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J. Biol. Chem. 2006;281:27251–27263. doi: 10.1074/jbc.M602746200. [DOI] [PubMed] [Google Scholar]
- 11.Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol. Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 12.Purdom-Dickinson SE, Sheveleva EV, Sun H, Chen QM. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol. Pharmacol. 2007;72:1074–1081. doi: 10.1124/mol.107.035360. [DOI] [PubMed] [Google Scholar]
- 13.Stoneley M, Paulin FE, Le Quesne JP, Chappell SA, Willis AE. C-Myc 5′ untranslated region contains an internal ribosome entry segment. Oncogene. 1998;16:423–428. doi: 10.1038/sj.onc.1201763. [DOI] [PubMed] [Google Scholar]
- 14.Ray PS, Grover R, Das S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006;7:404–410. doi: 10.1038/sj.embor.7400623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang DQ, Halaby MJ, Zhang Y. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene. 2006;25:4613–4619. doi: 10.1038/sj.onc.1209483. [DOI] [PubMed] [Google Scholar]
- 16.Coldwell MJ, deSchoolmeester ML, Fraser GA, Pickering BM, Packham G, Willis AE. The p36 isoform of BAG-1 is translated by internal ribosome entry following heat shock. Oncogene. 2001;20:4095–4100. doi: 10.1038/sj.onc.1204547. [DOI] [PubMed] [Google Scholar]
- 17.Locker N, Lukavsky PJ. A practical approach to isolate 48S complexes: affinity purification and analyses. Methods Enzymol. 2007;429:83–104. doi: 10.1016/S0076-6879(07)29005-6. [DOI] [PubMed] [Google Scholar]
- 18.Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- 19.Baird SD, Lewis SM, Turcotte M, Holcik M. A search for structurally similar cellular internal ribosome entry sites. Nucleic Acids Res. 2007;35:4664–4677. doi: 10.1093/nar/gkm483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl Acad. Sci. USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto T, Yoh K, Kobayashi A, Ishii Y, Kure S, Koyama A, Sakamoto T, Sekizawa K, Motohashi H, Yamamoto M. Identification of polymorphisms in the promoter region of the human NRF2 gene. Biochem. Biophys. Res. Commun. 2004;321:72–79. doi: 10.1016/j.bbrc.2004.06.112. [DOI] [PubMed] [Google Scholar]
- 22.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holcik M, Graber T, Lewis SM, Lefebvre CA, Lacasse E, Baird S. Spurious splicing within the XIAP 5′-UTR occurs in the Rluc/Fluc but not the betagal/CAT bicistronic reporter system. RNA. 2005;11:16051609. doi: 10.1261/rna.2158605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Eden ME, Byrd MP, Sherrill KW, Lloyd RE. Demonstrating internal ribosome entry sites in eukaryotic mRNAs using stringent RNA test procedures. RNA. 2004;10:720–730. doi: 10.1261/rna.5225204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornelis S, Tinton SA, Schepens B, Bruynooghe Y, Beyaert R. UNR translation can be driven by an IRES element that is negatively regulated by polypyrimidine tract binding protein. Nucleic Acids Res. 2005;33:3095–3108. doi: 10.1093/nar/gki611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc. Natl Acad. Sci. USA. 1993;90:2965–2969. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou W, Edelman GM, Mauro VP. Transcript leader regions of two Saccharomyces cerevisiae mRNAs contain internal ribosome entry sites that function in living cells. Proc. Natl Acad. Sci. USA. 2001;98:1531–1536. doi: 10.1073/pnas.98.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu MC, Tranque P, Edelman GM, Mauro VP. rRNA-complementarity in the 5′ untranslated region of mRNA specifying the Gtx homeodomain protein: evidence that base- pairing to 18S rRNA affects translational efficiency. Proc. Natl Acad. Sci. USA. 1999;96:1339–1344. doi: 10.1073/pnas.96.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nevins TA, Harder ZM, Korneluk RG, Holcik M. Distinct regulation of internal ribosome entry site-mediated translation following cellular stress is mediated by apoptotic fragments of eIF4G translation initiation factor family members eIF4GI and p97/DAP5/NAT1. J. Biol. Chem. 2003;278:3572–3579. doi: 10.1074/jbc.M206781200. [DOI] [PubMed] [Google Scholar]
- 30.MacCallum PR, Jack SC, Egan PA, McDermott BT, Elliott RM, Chan SW. Cap-dependent and hepatitis C virus internal ribosome entry site-mediated translation are modulated by phosphorylation of eIF2alpha under oxidative stress. J. Gen. Virol. 2006;87:3251–3262. doi: 10.1099/vir.0.82051-0. [DOI] [PubMed] [Google Scholar]
- 31.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 33.Talalay P, Fahey JW, Holtzclaw WD, Prestera T, Zhang Y. Chemoprotection against cancer by phase 2 enzyme induction. Toxicol. Lett. 1995;82–83:173–179. doi: 10.1016/0378-4274(95)03553-2. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Fu H, Khuri FR, Sun SY. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol. Cell. Biol. 2007;27:7405–7413. doi: 10.1128/MCB.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Li Y, Yang DQ. Phosphorylation of eIF-4E positively regulates formation of the eIF-4F translation initiation complex following DNA damage. Biochem. Biophys. Res. Commun. 2008;367:54–59. doi: 10.1016/j.bbrc.2007.12.118. [DOI] [PubMed] [Google Scholar]
- 36.Mahalingam M, Cooper JA. Phosphorylation of mammalian eIF4E by Mnk1 and Mnk2: tantalizing prospects for a role in translation. Prog. Mol. Subcell. Biol. 2001;27:132–142. [PubMed] [Google Scholar]
- 37.Pyronnet S. Phosphorylation of the cap-binding protein eIF4E by the MAPK-activated protein kinase Mnk1. Biochem. Pharmacol. 2000;60:1237–1243. doi: 10.1016/s0006-2952(00)00429-9. [DOI] [PubMed] [Google Scholar]
- 38.Dinkova-Kostova AT, Talalay P. Relation of structure of curcumin analogs to their potencies as inducers of Phase 2 detoxification enzymes. Carcinogenesis. 1999;20:911–914. doi: 10.1093/carcin/20.5.911. [DOI] [PubMed] [Google Scholar]
- 39.Beevers CS, Li F, Liu L, Huang S. Curcumin inhibits the mammalian target of rapamycin-mediated signaling pathways in cancer cells. Int. J. Cancer. 2006;119:757–764. doi: 10.1002/ijc.21932. [DOI] [PubMed] [Google Scholar]
- 40.Yu S, Shen G, Khor TO, Kim JH, Kong AN. Curcumin inhibits Akt/mammalian target of rapamycin signaling through protein phosphatase-dependent mechanism. Mol. Cancer Therap. 2008;7:2609–2620. doi: 10.1158/1535-7163.MCT-07-2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002;22:2883–2892. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 43.Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- 44.Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during antioxidant response. Mol. Cell. Biol. 2009;34:663–673. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li W, Yu S, Liu T, Kim JH, Blank V, Li H, Kong AN. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim. Biophys. Acta. 2008;1783:1847–1856. doi: 10.1016/j.bbamcr.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.