


Abstract
Gene duplication is integral to evolution, providing novel opportunities for organisms to diversify in function. One fundamental pathway of functional diversification among initially redundant gene copies, or paralogs, is via alterations in their expression patterns. Although the mechanisms underlying expression divergence are not completely understood, transcription factor binding sites and nucleosome occupancy are known to play a significant role in the process. Previous attempts to detect genomic variations mediating expression divergence in orthologs have had limited success for two primary reasons. First, it is inherently challenging to compare expressions among orthologs due to variable trans-acting effects and second, previous studies have quantified expression divergence in terms of an overall similarity of expression profiles across multiple samples, thereby obscuring condition-specific expression changes. Moreover, the inherently inter-correlated expressions among homologs present statistical challenges, not adequately addressed in many previous studies. Using rigorous statistical tests, here we characterize the relationship between cis element divergence and condition-specific expression divergence among paralogous genes in Saccharomyces cerevisiae. In particular, among all combinations of gene family and TFs analyzed, we found a significant correlation between TF binding and the condition-specific expression patterns in over 20% of the cases. In addition, incorporating nucleosome occupancy reveals several additional correlations. For instance, our results suggest that GAL4 binding plays a major role in the expression divergence of the genes in the sugar transporter family. Our work presents a novel means of investigating the cis regulatory changes potentially mediating expression divergence in paralogous gene families under specific conditions.
INTRODUCTION
Gene duplication is a major driver of evolutionary innovation, allowing an organism to elaborate existing biological functions via specialization or diversification, while avoiding negative fitness effects (1–4). One of the ways in which gene duplicates diversify in function is through divergence in their expression patterns. While the mechanisms underlying this expression divergence are not completely understood, cis-regulatory elements, and in particular, transcription factor (TF)-binding sites in promoter regions are likely to play a significant role. Therefore, investigation of evolutionary changes within the TF-binding sites may yield insights into the processes mediating the expression changes among homologs.
Various studies have attempted to identify genomic variations responsible for observed expression divergence in orthologs (5). However, the investigation of expression divergence based on orthologs is handicapped not only by the difficulty in establishing analogy between differing cell types and developmental times across species, but also by the differences in trans-acting factors and regulatory programs. In contrast, gene expressions are more directly comparable in paralogs. These observations suggest a novel avenue for exploiting the relationship between expression divergence of paralogous gene families and their corresponding TF-binding sites to explore the mechanisms underlying regulatory evolution. In a prior study, Zhang et al (6) reported a weak correlation between changes in TF-binding sequences and the gene expression between pairs of yeast gene duplicates. It is worth noting that this previous study measured a global gene expression divergence based on multiple expression samples. However, regulatory mechanisms rely on a combination of diverse switches, both internal and environmental and, therefore, it is likely that the mutations in specific TF-binding sites may affect gene expression only in specific environmental conditions. Consequently, a global measure of expression divergence may obscure such condition-specific cis effects. As such, it seems most beneficial to compute expression divergence in a condition-specific manner. Previous investigations have also considered expression divergence in a pair-wise fashion. Nevertheless, a global analysis of entire gene family as opposed to individual pairs of paralogs, is likely to be more informative, and will additionally provide a global view of gene family evolution.
Here, we investigate the relationship between TF binding and condition-specific expression divergence in S. cerevisiae paralogous gene families. Our study differs from previous related studies in several important aspects. First, the analyses presented here focuses on paralogous gene families, as opposed to pairs of homologs. Second, in order to reduce the effect of confounding parameters such as trans-acting factors, expression divergence is quantified individually for specific conditions, as opposed to an overall expression similarity derived from multiple expression samples using global measures of correlations. We also employ novel, rigorous statistical technique to account for non-normal data and dependence among genes in a family. Our results demonstrate that a significant number of expression divergence patterns in yeast paralogous families strongly correlate with TF-binding site changes under specific sample conditions. We also find that incorporating nucleosome occupancy in conjunction with the TF-binding site data increases the number of strong correlations between TF binding and expression in paralogous gene families. Several interesting cases of correlated cis element and expression divergence emerge, further elucidating functional diversification in gene families. For instance, our analysis reveals GAL4-mediated expression divergence of GAL2 — a galactose transporter — from other members of the sugar transporter family. Collectively, our results suggest that in a large number of cases, functional diversification of paralogous gene families has arisen at least in part, due to condition-specific expression divergence. In turn, this expression divergence is likely to be mediated by divergence in the corresponding cis elements of the genes.
METHODS
Overview of method
Figure 1 illustrates the overall approach. For a family of N paralogous genes, we estimate the probability that a particular TF binds to the promoter (defined as the 600 bp upstream region) of each gene. Thus, for each TF x, and each family F, an N-tuple of binding scores BF(x), is obtained. Similarly, EF(s) represents the corresponding normalized expression values for the genes F in a given expression sample, s. The correlation between BF(x) and EF(s) is then compared with a background expectation for the family in order to yield a P-value representing how extreme the correlation is. Thus, a P-value is computed for each triplet of TF, gene family, and expression sample. A significant correlation is interpreted as ‘the divergence in the TF binding among the paralogs underlies the sample-specific expression divergence’. The details of the method are presented below.
Figure 1.

Overview and illustration of IPF approach. For a family of N paralogous genes (N = 10), each promoter sequence is scanned to identify putative TF-binding sites. For each TF, we obtain TF-binding probabilities in each gene promoter which are then correlated with the corresponding expression values in a specific condition. This correlation is then compared to a random distribution of correlations to obtain a P-value.
Yeast gene families
A list of paralogous yeast gene pairs was obtained from Ensembl’s Compara 52 homology database (http://www.ensembl.org) for S. cerevisiae (SGD 1.01). We used complete-linkage clustering of the paralogy relationship to define disjoint paralogous gene families. In addition, an alternative list of gene families was obtained using the Pfam families and their corresponding genes (pfam.sanger.ac.uk). The Ensembl-derived families are prefixed with ‘FY’ and the Pfam families are referenced by their Pfam accession numbers and prefixed with ‘PF’. We performed a number of filtering steps to improve data fidelity. For instance, we excluded families consisting primarily of hypothetical genes. In general, Ensembl-derived paralogy relationships are much more stringent than those of Pfam-derived families which are based on shared domains. To ensure that Pfam-derived families did not contain distant paralogs, we removed outlier genes from Pfam-derived families such that the minimum protein similarity of pairs of genes in Pfam families was larger than all pair-wise similarities between genes in each Ensembl-derived family. Finally, to ensure that families were mutually disjoint, if two families shared a gene, only the larger family was retained. All of our analyses were done independently on all the 16 resulting families after these filters. The family information is provided in Supplementary File 2 (worksheet Family Information).
Genome-wide expression profiles in yeast
For consistency of comparison across expression samples, we used the Affymetrix GeneChip Yeast Genome S98 Array YG-S98 platform, because it contains probes for all known 6400 yeast genes and candidate open reading frames. In addition, numerous studies have explored the use of Affymetrix data and associated noise correction (20,21). Hence, this platform served as a reasonable choice for a single, consistent platform with a large amount of data for our analysis. The data corresponds to strain S288C and GEO accession GPL90. The gene expression data is summarized in Supplementary File 2 (worksheet Gene Expression Samples). We normalized the raw CEL data using the gcRMA algorithm in Bioconductor (22).
For a given paralogous family, several expression samples have very similar expression for the genes in the family. Thus, for sample-specific analyses of this family, various expression samples cannot be considered independent. To minimize this redundancy among samples, for the gene family in question, we computed the pair-wise Kendall Tau correlation coefficient (KTC) between each pair of samples using the expression values for the genes in the family. If two expression samples have a significant (P < 0.05) KTC greater than 0.9, the samples were clustered using complete-linkage clustering into one aggregate sample in which the expression for each gene was computed as the mean value among all samples in the cluster. The sample clustering information is provided in Supplementary File 2 (worksheet Sample Expression Mapping). Note that the sample clustering is done independently for each gene family to minimize the biases caused by redundant samples while retaining the maximal amount of expression data.
Many previous studies have assumed that the expression is normally distributed, which is clearly not the case here. To account for non-normal expression data, we exploit the Box-Cox transform (23), 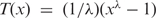 , where x ≥ 0 is the response variable and λ > 0 is the transformation parameter. Each family was transformed independently, and a family-specific λ estimated using maximum likelihood and the box.cox.powers function in the car R package. After applying the Box-Cox transformation, the distribution of the transformed expression data resembles a normal one.
, where x ≥ 0 is the response variable and λ > 0 is the transformation parameter. Each family was transformed independently, and a family-specific λ estimated using maximum likelihood and the box.cox.powers function in the car R package. After applying the Box-Cox transformation, the distribution of the transformed expression data resembles a normal one.
Since we are interested in identifying the samples in which a set of genes are either up or down-regulated, we normalize the Box-Cox transformed expressions of each gene across all samples, as follows. For each gene g in a family F, the normalized expression is given as, 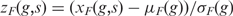 , where xF(g,s) is the Box-Cox transformed expression of g in F and sample s, and μF(g) and σF(g) are the mean and standard deviation, respectively of g in F across all expression samples.
, where xF(g,s) is the Box-Cox transformed expression of g in F and sample s, and μF(g) and σF(g) are the mean and standard deviation, respectively of g in F across all expression samples.
Promoter sequence, TF-binding probability and nucleosome occupancy
We used two sources of data to estimate the probability that a TF binds a gene promoter. The first one is based on ChIP-chip experiments reported in (8). The authors provide a P-value for each TF-gene pair, which we converted into a probability following a mixture modeling approach reported in (24). In addition to ChiP-chip, we also used the published available DNA-binding motifs for 124 TFs in (25) to estimate the TF’s-binding scores in gene promoter. We extracted the 600 bp upstream regions of all yeast genes from the UCSC database (genome.ucsc.edu), to be used as our promoters. The choice of a 600 bp upstream promoter region is consistent with many previous works (8,26,27). For each TF-binding motif (represented as a positional weight matrix or a PWM), using the previously published tool PWM_SCAN tool (9), we estimated the best overall TF-binding score (referred to as TFMax) as follows. We first measure the maximum percentile score for the PWM of a particular TF in the entire promoter and convert this score into a P-value based on the background distribution of percentile scores (all TFs, all promoters, and all positions). The minimum P-value (corresponding to the maximum percentile score) is then used as our measure of binding. We also compute another variant of motif-based binding score from the average of the three best (largest) percentile scores for the TF from non-overlapping regions in a gene promoter (referred to as TFAvg). An aggregate of these three values offers more resolution and is less prone to outliers than the using the single minimum value directly. In order to account for variations in promoter length, we also computed TF motif-binding scores for promoter lengths of 500 bp, 750 bp and 1 kb. The Pearson correlation coefficient (PCC) between both TFMax and TFAvg for a 500 bp length promoter and the default 600 bp length are R2 = 0.92 and R2 = 0.96, respectively. The PCC between both TFMax and TFAvg for a 750 bp length promoter and the default 600 bp length are R2 = 0.91 and R2 = 0.96, respectively. Finally, the PCC between both TFMax and TFAvg for a 1 kb length promoter and the default 600 bp length are R2 = 0.82 and R2 = 0.91, respectively. Therefore, we conclude that small differences in promoter length will not significantly affect our results.
Nucleosome occupancy probability for the promoter regions was estimated using the computational model reported in (28). Once again, the Box-Cox transform was applied to the occupancy probabilities to ensure that the data distribution resembled a normal one. As in the case of expression data, a family-specific λ was computed using maximum-likelihood. Strictly speaking, after transformation the binding probabilities are no longer valid probabilities. However, we still refer to the scores as probabilities to avoid pedantry, since the transformation is only relevant to the technical details of computing the significance of correlation outlined in subsequent sections.
For a given TF and gene family with N genes, we consider this pair for analysis only if at least 1 and at most N−1 gene promoters contained a binding score above the 95th percentile. This filtering step is designed to ensure that we only analyze the gene families where there is sufficient evidence of TF-binding divergence in the gene promoters. We have tested our procedure using varying thresholds for binding scores for robustness, and found that the choice of threshold does not significantly impact the results.
To incorporate nuclesome occupancy information in estimating the TF-binding probability, we used two separate methods for computing the nucleosome occupancy probability. The first method computes the nucleosome occupancy probability by averaging the occupancy probability at each base location across the enter promoter. The second method uses the best predicted binding location based on DNA-binding motifs as the putative binding site for the TF. The nucleosome occupancy score is then computed as the average of the nucleosome occupancy predicted at each base within this specific binding site. The binding probability is then computed as the product of the nucleosome occupancy probability and TF-binding probability from ChIP. In the case of the motif-based scores (TFAvg and TFMax), we compute the nucleosome occupancy probability at the exact putative binding site (averaged across the binding site positions) and multiply this probability by the percentile score for the binding of a particular TF to the site to obtain a new score incorporating nucleosome occupancy data. We then select the largest and average of the largest three such scores to compute our new TFMax and TFAvg scores, respectively. In investigating the effect of nucleosome occupancy alone, we required a minimum nucleosome occupancy of 0.9 for at least one gene in the family.
Correlating TF-binding probabilities with expression values
Previous studies have argued that many of the so-called low affinity binding sites are likely to be functional and these should not be removed based on arbitrary thresholds (27). We therefore treat TF binding as an analog quantity as opposed to a binary variable using an arbitrary threshold. As such, we use a regression based approach to quantify the relationship between TF binding and expression. Since both the transformed TF binding probability and expressions scores are approximately normally distributed, there are a number of metrics available for computing correlation between these scores. The PCC was chosen as our measure of correlation. We measured the significance of the PCC as follows. Note that the obvious dependency of both the TF-binding scores and expression scores among paralogous genes has a profound effect on the computation of the significance of correlation. To account for this dependency, the protein sequence similarity is a reasonable proxy for evolutionary time since duplication, and thus is a practical choice for capturing the statistical dependency among paralogous genes in a family. For each pair of genes X and Y in a family, we compute the normalized protein sequence similarity as 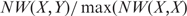 where NW(X,Y) represents the Needleman–Wunsch similarity score of proteins corresponding to genes X and Y, using the BLOSUM62 matrix. For the genes with multiple transcripts, we chose the longest transcript. This function gives an indication of the relative and overall similarity between two proteins, and produces a score in [0,1]. From these scores a correlation matrix ΣF of the genes in a family F is estimated.
where NW(X,Y) represents the Needleman–Wunsch similarity score of proteins corresponding to genes X and Y, using the BLOSUM62 matrix. For the genes with multiple transcripts, we chose the longest transcript. This function gives an indication of the relative and overall similarity between two proteins, and produces a score in [0,1]. From these scores a correlation matrix ΣF of the genes in a family F is estimated.
In order to incorporate the dependency structure of the genes in a family, we compute the significance of correlations using a form of permutation tests, called (rotation tests) (29). To perform these rotation tests, we compute a distribution of correlations based on randomly generated data, and then obtain a P-value by determining the rank of the actual correlation amongst the randomly generated correlations. To compute a random PCC, the values of the TF-binding probabilities are kept constant, but random outcomes for the expression data are generated from a multivariate normal distribution. Effectively, both the expression and TF-binding data after Box-Cox transformations can be modeled as outcomes of multivariate normal distributions of dimension N, where N is the number of genes in a family. As such, we generate random multivariate normal outcomes (for fixed TF-binding probabilities) with correlation matrix ΣF for family F, where ΣF captures the dependency structure of the genes in F. Using these random multivariate normal outcomes, a random distribution of PCC values can then be obtained. Once we have the necessary random expression outcomes, a PCC is computed between the actual binding probability scores and the random expression scores to yield a random distribution of PCC values.
In order to account for multiple testing, we estimate the family-wise error rate (FWER) using Hommel’s method (7), which is more conservative than False Discovery Rate (FDR) approaches and does not assume independence among P-values. There are two contexts viewed in this paper, and in both contexts we employ the FWER. The first is in identifying significant correlations between TF binding and expression in at least one expression sample for a known TF–family regulatory relationship. In this case, for a gene family–TF pair, we obtain a P-value for the significance of correlation for each sample. We then adjust these P-values for each gene family–TF pair independently, using Hommel’s method. The second context is the case of identifying interesting biological examples of significant correlation between TF binding and expression sample divergence. Since our intent here is discovery of ‘TF-gene-sample’ triplets with significant correlations in a given family, we compute the adjusted P-values from all of the P-values obtained for that family, i.e. for all TFs and all samples.
RESULTS
Summary of data and approach
Our analysis is based on S. cerevisiae, which has been studied extensively, is well annotated, and contains a well-defined promoter (600 bp upstream region). We assembled gene families in S. cerevisiae using two resources: (i) paralogy data from Ensembl version 52 (family identifiers are prefixed with ‘FY’), and (ii) using the Pfam database (family identifiers are prefixed with ‘PF’). A summary of the family data is provided in Supplementary File 1. We restricted the analysis to families having at least five paralogous genes, and after a thorough manual filtering (see ‘Methods’ section) of candidate families, arrived at 2 Ensembl-derived and 14 Pfam-derived families. We used a compendium of 106 publicly available genome-wide S. cerevisiae expression profiles measured under a variety of experimental conditions (see ‘Methods’ section).
Figure 1 illustrates the overall approach, and the methods section provides further details. Briefly, for a given paralogous family and a given TF, we obtain the probability that the TF binds to the promoter of each gene in the family. For each gene family and for each TF regulating at least one gene in the family with high score (above the top 5th percentile of all binding scores), we tested how strongly and significantly the TF’s binding scores for the genes in the family are correlated with the genes’ expression levels in each of the expression samples. Using rigorous statistical tests, we estimated a P-value of the correlation for each gene family–TF pair and for each expression sample, separately. To correct for multiple testing, we utilized Hommel’s method for estimating the family-wise error rate (FWER) (7). However, we note that, even if we apply the very stringent Bonferroni correction in lieu of Hommel’s method, all of the following results remained unchanged, that is identical numbers and hence, percentages of significant correlations are detected. A significant P-value suggests that the divergence within the TF-binding site underlies the sample-specific expression divergence. A significant correlation is defined as a gene family–TF pair wherein the expression in at least one condition was correlated with the TF’s binding scores with P-value < 0.01 and FWER < 0.10.
TF binding in paralogous genes’ promoters are often significantly correlated with their condition-specific expressions
The global analysis reveals a few interesting results at a cursory level. Figure 2 summarizes the results, illustrating a total of 67 significant positive correlations (potential activators of expression) between TF binding and expression, as compared to 48 significant negative correlations (potential repressors of expression). Moreover, the detected significant correlations are distributed across many families and TFs, i.e., there is no bias towards specific TFs or families. Figure 3 summarizes the results for each family, showing the percentages of TFs whose binding showed significant correlations with gene expression in at least one sample. There is at least one significant correlation between TF binding and expression in all but one of the families. The families are ordered by decreasing average protein similarity of the genes within the family (see ‘Methods’ section). It is apparent that the average within-family protein similarity does not affect our ability to detect correlations. In the following sections, we elaborate on these results.
Figure 2.


Summary of results for all families. Each column in the grid corresponds to a specific TF. Rows are grouped into families, and for a family, each row represents the measure used for determining the TF-binding score. If a TF does not bind to the promoter of any gene in the family according to the corresponding binding score measure in the row, then the corresponding cell is white; these were excluded from analysis. All other cells correspond to analyzed TF–family pairs. A purple cell indicates that we did not detect a significant correlation between binding probability and expression divergence in any sample. A red or green cell indicates that we found a significant negative or positive correlation, respectively, between binding probability divergence and expression divergence in at least one sample. See text for the thresholds used to determine significance. Only families having at least one significant gene family–TF interaction are shown.
Figure 3.

Summary of significant correlations between TF-binding scores and expression in TF–family pairs. The families shown on the y-axis of the bar plot are listed in decreasing order from top to bottom of average protein similarity of the genes in the family. For instance, PF00270 has the highest average protein similarity of genes, and PF00069 has the lowest. The x-axis shows the percentage of significant correlations between TF-binding scores and expression in putative TF–family pairs, for the three different TF-binding probability metrics, both with and without nucleosome occupancy information.
Using ChIP–chip data to quantify TF–gene interaction
In vivo binding for 102 S. cerevisiae TFs in all gene promoters has been previously determined using ChIP-chip experiments (8). The authors provide a P-value for each TF–gene pair. We converted this P-value into a probability value representing the likelihood of the TF binding to the gene promoter (see ‘Methods’ section). For each of the 144 qualifying gene family and TF pairs, we tested independently for each expression sample, whether the TF’s binding scores for the genes in the family are significantly correlated with the genes’ expression levels in the expression samples.
As shown in Figure 4a, for 31 of 144 (21.5%) gene family and TF pairs analyzed, the expression in at least one condition was significantly correlated with the TF’s binding scores (P < 0.01 and FWER < 0.10). In a majority (90%) of these 31 significant cases, a significant correlation was observed in exactly one expression sample. These results suggest that TF-binding divergence correlates with expression divergence only under specific conditions. While an exhaustive analysis of all experimental conditions is not practical, it is plausible that a larger compendium of conditions will reveal a greater number of gene family and TF pairs having significantly correlated cis element and expression divergence.
Figure 4.

(a) Stacked bar plot showing percentage of correlations in TF-family pairs between TF binding and expression divergence for putative TF binding sites identified by three different measures of binding probabilities: ChIP-chip, TFAvg and TFMax. The shaded (unshaded) area indicates the number of significant (non-significant) correlations. See text for definitions of significance. (b) Effect of using nucleosome occupancy information with respect to significant correlations between TF binding and expression divergence for three different measures of binding probability: ChIP-chip TFMax and TFAvg. The plot shows for each metric, the total number of significant correlations (red) detected if we use the TF metric with the nucleosome occupancy information as well as the number of significant family–TF pairs common (green) in the case of using the TF-binding metric without the nucleosome occupancy information.
Using motif-based methods to identify TF–gene interaction
In addition to ChIP-chip data, motif-based scanning techniques using positional weight matrices (PWMs) are commonly used for identifying potential TF-binding sites. Among these techniques, we used two different measures to quantify the strength of TF binding in a gene promoter. The first, subsequently referred to as TFMax, is the maximum percentile score using the previously published PWM_SCAN tool (9). The second hereafter referred to as TFAvg, is the average of the top three non-overlapping, maximum percentile scores for a given TF PWM in the 600 bp promoter region, and accounts for multiple putative binding sites. For a suitable comparative analysis among different TF-binding metrics, we chose thresholds for binding scores which yielded a comparable number of putative binding sites as the number obtained from the ChIP-chip binding probabilities. Based on these thresholds, the ChiP-chip binding and motif-based measures of binding yield similar numbers of significant family-TF interactions but these interactions have little intersection. Once again, Figure 2 illustrates the results from employing these two measures of TF-binding probability. Figure 3a also summarizes the results for TFAvg and TFMax binding scores, wherein 19 of 121 (15.7%) and 14 of 169 (8.3%), respectively, TF-family pairs showed significant correlation in at least one expression sample (P ≤ 0.01 and FWER ≤ 0.1). Among the significant correlations, the overlap of results between the three measures of TF binding is not significant. Thus, each binding metric reveals different yet significant correlations between TF binding and expression divergence.
Incorporation of nucleosome occupancy data yields additional significant correlations between TF binding and expression specifically for motif-based measures of TF binding
TF binding, and thus transcriptional regulation, is mediated by nucleosome occupancy by controlling accessibility of DNA to TFs. To assess the effect of nucleosome occupancy on the relationship between TF binding and expression divergence, we repeated our analysis after incorporating nucleosome occupancy in conjunction with TF-binding scores using two separate measures (see ‘Methods’ section). Since the exact location of binding in the TF promoter is unknown using ChIP-chip binding data, the first measure computes the nucleosome occupancy as the average nucleosome occupancy within the entire promoter. For the second measure, we choose the location of binding site in the promoter predicted from the best score obtained from DNA-binding motifs. The corresponding nucleosome occupancy score is then based on the predicted nucleosome occupancy of this specific location. The cases showing significant correlation (P ≤ 0.01 and FWER ≤ 0.10) based on the first measure are summarized in Figure 2, and the percentages of significant correlations are depicted in Figure 4a. For ChIP-chip derived probabilities, there is little change in the percentage of significant correlations between TF-binding scores and expression divergence, with 27 of 133 (20.3%) significant correlations. As illustrated in Figure 4b, a large majority (26) of TF-family pairs are common between results with and without nucleosome occupancy information. In terms of the second measure of nucleosome occupancy, we detected similar numbers as the first measure, 27 of 132 (20.5%) significant correlations. All 27 TF-family pairs are common between results with and without nucleosome occupancy, hence the results are consistent regardless of which measure of nucleosome occupancy is employed.
The previous trend does not hold when we incorporate nucleosome occupancy to the motif-based-binding metrics (Figure 4b). There are a total of 12 significant correlations each after incorporating nucleosome occupancy for TFAvg and TFMax. Of these significant correlations, however, the intersection of gene family-TF pairs deemed significant using nucleosome occupancy data and those deemed significant without nucleosome occupancy data is negligible, for either TFAvg or TFMax. Therefore, it seems that incorpoarting nucleosome positioning information yields additional correlations between cis element and expression divergence.
We next investigated the extent to which nucleosome occupancy exclusive of TF binding, correlates with sample-specific expression divergence. In general, the differences in nucleosome occupancy among paralogs do not significantly correlate with their condition-specific gene expression. In fact, the only significant correlation was detected in a family of histone proteins (PF00125). The biological significance of this finding is unclear. In summary, even though there is little to no correlation between nucleosome occupancy alone and sample-specific expression, we detect significant correlation if we incorporate both nucleosome occupancy and TF binding. Thus, it appears that expression divergence is mediated partially by TF-binding changes and facilitated by nucleosome positioning. Below, we discuss specific examples of paralogous expression divergence and the TFs that potentially mediate the expression divergence, as detected by our analysis. With the focus being on the discovery of the most informative results, we restrict our discussion to those cases where there is a strong evidence of in vivo binding (in the 99.5th percentile, and a more conservative FWER estimation, see ‘Methods’ section).
GAL4 binding strongly correlates with the expression divergence of the sugar transporters
Within the sugar permease family (PF00083) shown in Figure 5, GAL4 binding is inversely correlated (TFAvg P = 1.5E–4, FWER = 0.04) with expression in a low glucose expression sample (GSM29914). As shown in Figure 6, the three genes (GAL2, HXT3 and HXT8) in this family with the greatest TF-binding score also have the lowest expression. GAL4 is known to activate GAL2 when galactose is present, but under normal conditions (including the low glucose condition) GAL4’s activity is repressed by GAL80 via interaction with GAL4.
Figure 5.

Phylogenetic tree of the sugar transporter family (PF00083) in yeast. This figure is a reproduction of Figure 1 of (30). Only the genes relevant to our analysis are shown.
Figure 6.

Correlation between GAL4 binding and expression in the sugar transport family, PF00083. The figure shows a schematic of putative GAL4 binding in the multiple alignment of the promoters of sugar transporters (locations are in scale but approximate), and the corresponding expressions of the genes in a sample cluster corresponding to GSM29914. The transcription start site (TSS) is indicated by the red arrow. The putative binding sites are categorized by percentile scores. In general, highly expressed genes (shown in green shades) have a greater number of strong binding sites.
Members of the hexose transporter family differ in their capacity to transport glucose (10). For instance, HXT6 and HXT7 are high-affinity transporters, and therefore, are active when glucose is scarce. Consistently, as shown in Figure 6, we observe that HXT6 and HXT7 have the highest expressions compared to other genes in the family. In addition, HXT3 is likely a low-affinity transporter, i.e., active only under high glucose concentrations. As expected, HXT3 is expressed at low level in the low glucose sample. The low expressions of HXT14, HXT16 and HXT8 are also consistent with previous studies which remark that these genes are either unable to transport glucose or expressed at low levels under normal conditions. Finally, GAL2 is expressed only when galactose is available. Not surprisingly, the level of GAL2 has the lowest expression in GSM29914 among the PF00083 genes, and is the sample with the third lowest expression among other samples.
Our findings are consistent with the known biology of the glucose metabolism pathway in yeast and they suggest that one specific member of the ancient sugar transport family — GAL2 — was recruited to initiate galactose transport in yeast. Sugar transport is vital to yeast, and our analysis demonstrates that the expression of the sugar transport genes strongly correlate with GAL4 binding in their promoters. In many environmental conditions the genes in this family serve redundant functions, however a closer inspection of various experimental conditions reveals that the capacity of these genes to metabolize sugar vary in subtle but important ways (10). Our results further suggest that certain aspects of these functional differences are due to differences in expression, likely to have been mediated by variable GAL4 binding in these genes’ promoters. The above findings underscore the importance of using condition-specific gene expressions, and a reliance on global or aggregated expression profiles could obscure these subtleties.
MCM1 binding strongly correlates with divergence in a family of cell wall proteins
Pfam family PF00399 includes a number of genes involved in cell wall stability and integrity. We detect a strong positive correlation (P = 1.83E–4, FWER = 0.020) between the ChIP-chip binding of MCM1 to the member genes and the corresponding expressions in a cluster of expression samples which include several stress responses induced by the addition of various chemicals and galactose (PF00399, SampleId 2, see Supplementary File 2). In particular, many of the samples included histone modifications and treatment with methyl methanesulfonate which is known to produce heat-labile DNA damage (11). This correlation remains strong in the same sample even if we incorporate nucleosome positioning data (P = 2.93E-4, FWER = 0.013). PF00399 comprises the genes PIR1, PIR2, PIR3, PIR4, TIR1 and TIR3, among which, PIR2 (HSP150) exhibits the strongest binding to MCM1. PIR2 is a heat shock protein which attaches to the cell wall to promote cell wall stability under stress conditions including heat shock, oxidative stress and nitrogen starvation (12). MCM1 is an established transcriptional activator and was recently shown to regulate PIR2 for filamentous growth (13), and PIR2 is the only gene in this family regulated by MCM1. In addition, both MCM1 and PIR2 have their highest expression levels in SampleId 2. TIR1 and TIR3 are induced by cold-shock and anaerobiosis (14) and PIR1,3,4 are primarily regulated by RLM1. Collectively, it appears that members of this family have functionally diverged and in particular PIR2 has garnered a distinct function as a MCM1-mediated stress response gene.
Divergence in a family of amino acid permeases
Ensembl-derived family, FY00077 consists of five genes—BAP2, BAP3, HIP1, AGP1 and UGA4. The first three are amino acid permeases with narrow substrate range. AGP1 is a low-affinity amino acid permease with broad substrate range and UGA4 serves as a gamma-aminobutyrate (GABA) transport protein and is involved in the utilization of GABA as a nitrogen source. Only the latter two genes—AGP1 and UGA4—are involved in nitrogen catabolism repression (15,16) while BAP2 is involved in carbon catabolism repression (17). The expression of this family is negatively and significantly (P = 9.8E-4, FWER = 0.047) correlated with the affinity to transcription fact GZF3/DEH1 based on TFAvg score in a cluster of expression samples consisting primarily of wildtype yeast in rich media conditions (FY00077, SampleId 13, see Supplementary File 1). Only UGA4 and AGP1 (involved in nitrogen catabolism repression) have high binding scores for GZF3 in their promoter. GZF3 is a key regulator of nitrogen catabolism repression and is known to directly regulate, as a negative regulator, both AGP1 (18) and UGA4 (15). For instance, under high nitrogen conditions when nitrogen catabolism is not needed, GZF3 acts as a negative regulator of UGA4 by competing for a GATA-binding site with another factor Gat1 (15). In fact, the ratio of GZF3 expression to GAT1 expression is amongst the highest in SampleId 13 versus all other samples. The observed negative correlation between the GZF3 binding and the expression of these genes is consistent with the GZF3’s role as a negative regulator of these two genes. Thus, the specific binding of GZF3 to promoters of two of the permeases is consistent with their role in nitrogen catabolism repression.
DISCUSSION
Gene duplication events provide the necessary ‘spare parts’ for evolutionary innovation by facilitating elaboration of existing biological functions (1–4). Diversification of gene functions can involve at least two distinct pathways: (i) alteration of gene expression pattern, and (ii) alteration of protein’s sequence, structure and eventually its interactions and biochemical activity. Alteration of expression patterns is likely to involve changes in the cis regulatory elements. Extensive work has been carried out to identify, model, and analyze regulatory evolution and evolution of novel gene function. Typically, many computational analyses have exploited highly conserved non-coding regions as a proxy for putative functional elements. Such analyses fail to capture divergent aspects of sequence that might underlie functional diversification of gene families. Other approaches have attempted to correlate sequence changes in cis regulatory regions with expression divergence in pairs of orthologs. However, not only is it inherently difficult to compare expressions across multiple species, but expression profiles across multiple samples obscure the effects of regulation in specific, individual conditions.
In view of the above remarks, we have investigated the correlated changes between TF-binding sites and the condition-specific expressions of paralogous genes in the model organism S. cerevisiae, using rigorous statistical analysis. Moreover, we have attempted to characterize the impact of regulatory sequence evolution on expression divergence in paralogous gene families, as opposed to analyzing paralogous pairs of genes, as was done previously. Our genome-wide analysis in yeast has revealed several significant correlations between changes in TF-binding scores in the promoters of paralogous genes and their expression values in specific experimental conditions. We have also observed that diverse measures of TF binding appear to capture different aspects of TF-binding site variation and evolution, which underscores the value of incorporating TF-binding data from a variety of sources. In general, incorporating nucleosome occupancy probabilities in the promoters yields additional significant correlations. It is worth emphasizing that since ChIP-chip captures in vivo binding and thus implicitly incorporates nucleosome occupancy, we observed negligible additional significant correlations after incorporating nucleosome occupancy data to ChIP-chip-binding probabilities. The additional significant correlations retrieved are most likely due to the fact that ChIP-chip experiments are executed for a specific and limited set of conditions, and may not capture the effect of nucleosome occupancy in other experimental conditions. This fact reinforces the value of our analysis, as we are able to gain further insights from utilizing several different experimental samples. Finally, we have highlighted a few specific examples of significant correlations between TF-binding site divergence with expression divergence in specific conditions. Collectively, our findings suggest that during evolution, alterations in TF-binding sites contribute to condition-specific expression changes among paralogous genes. Our results further suggest that evolution of nucleosome occupancy within paralogous families potentially underlie the expression divergence among the paralogs, as noted in a recent review (19).
Although we have identified a number of significant correlations between TF-binding site and expression, there are obviously a large number of cases in which we could not detect any significant correlations in any expression sample. Expression divergence is potentially mediated by a multitude of factors including genomic, epigenomic and transcriptional changes that are not captured solely by mutations in the proximal promoters of genes. There are also other mitigating factors including the lack of known TF-binding sites and the relatively small gene families, thereby reducing the power of the statistical tests. In addition, our compendium of expression samples is far from being comprehensive, and so many correlations would not be detected due to the unavailability of relevant expression samples. Despite these limitations, we have found significant correlations between cis regulatory elements and sample-specific expression, and even capture the effect of nucleosome positioning in transcriptional evolution.
The statistical challenges involved in performing the analysis presented here should not be understated. Expressions and TF-binding scores in paralogous genes cannot be assumed to be either normally distributed or statistically independent. While many studies neglect or discount such details, we have adopted conservative, but rigorous statistical technique. It is also worth mentioning that due to stringent multiple testing corrections, a genome-wide application of our technique potentially diminishes the percentage of significant correlations. In view of this observation, we believe that a careful application of our method, perhaps even in additional organisms, to specific gene families utilizing a smaller but more relevant subset of putative TFs and expression samples will be more fruitful.
The methods presented here will uniquely reveal novel functional cis elements that may underlie the expression divergence among paralogs, as illustrated by several known cases. For instance, we found that GAL4 binding may underlie the expression divergence within the sugar permease family in yeast. We were able to capture relevant correlations between TF binding and expression in this family, precisely because we investigated sequence-expression relationships at the level of families in a condition-specific manner. Sugar transporter genes have evolved to perform related, but subtly distinct functions. Our analysis suggests that this functional diversification is facilitated, at least in part, by expression divergence, which in turn is mediated by divergence in TF binding. Moreover, by analyzing the entire family, as opposed to one gene at a time, our approach affords a greater statistical power. From a broader perspective, genome-wide applications of our method may provide a means of generating testable hypotheses as well as insights into the evolutionary process by which members of gene families diversify their functions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: National Institute of Health (NIH) grants T32-HG-000046 (LNS) and R01GM085226 (SH). Funding for open access charges is provided by NIH grant, R01GM085226.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Prof. Andreas Buja for helpful discussions regarding rotation tests. They also thank two anonymous reviewers for helpful and constructive comments for improving the manuscript.
REFERENCES
- 1.Ohno S. Evolution by Gene Duplication. London: Allen and Unwin; 1970. [Google Scholar]
- 2.Wagner A. Selection and gene duplication: a view from the genome. Genome Biol. 2002;3:1012. doi: 10.1186/gb-2002-3-5-reviews1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 4.Vavouri T, McEwen GK, Woolfe A, Gilks WR, Elgar G. Defining a genomic radius for long-range enhancer action: duplicated conserved non-coding elements hold the key. Trends Genet. 2006;22:5–10. doi: 10.1016/j.tig.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Tirosh I, Weinberger A, Bezalel D, Kaganovich M, Barkai N. On the relation between promoter divergence and gene expression evolution. Mol. Syst. Biol. 2008;4:159. doi: 10.1038/msb4100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z, Gu J, Gu X. How much expression divergence after yeast gene duplication could be explained by regulatory motif evolution? Trends Genet. 2004;20:403–407. doi: 10.1016/j.tig.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Hommel G. A stagewise rejective multiple test procedure based on a modified Bonferroni test. Biometrika. 1988;75:383–386. [Google Scholar]
- 8.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy S, Hannenhalli S. Identification of transcription factor binding sites in the human genome sequence. Mamm. Genome. 2002;13:510–4. doi: 10.1007/s00335-002-2175-6. [DOI] [PubMed] [Google Scholar]
- 10.Ozcan S, Johnston M. Function and regulation of yeast hexose transporters. Microbiol. Mol. Biol. Rev. 1999;63:554–569. doi: 10.1128/mmbr.63.3.554-569.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, Helleday T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005;33:3799–3811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapteyn JC, Van Egmond P, Sievi E, Van Den Ende H, Makarow M, Klis FM. The contribution of the O-glycosylated protein Pir2p/Hsp150 to the construction of the yeast cell wall in wild-type cells and beta 1,6-glucan-deficient mutants. Mol. Microbiol. 1999;31:1835–1844. doi: 10.1046/j.1365-2958.1999.01320.x. [DOI] [PubMed] [Google Scholar]
- 13.Birkaya B, Maddi A, Joshi J, Free SJ, Cullen PJ. Role of the cell wall integrity and filamentous growth mitogen-activated protein kinase pathways in cell wall remodeling during filamentous growth. Eukaryot. Cell. 2009;8:1118–1133. doi: 10.1128/EC.00006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abramova N, Sertil O, Mehta S, Lowry CV. Reciprocal regulation of anaerobic and aerobic cell wall mannoprotein gene expression in Saccharomyces cerevisiae. J. Bacteriol. 2001;183:2881–2887. doi: 10.1128/JB.183.9.2881-2887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luzzani C, Cardillo SB, Bermudez Moretti M, Correa Garcia S. New insights into the regulation of the Saccharomyces cerevisiae UGA4 gene: two parallel pathways participate in carbon-regulated transcription. Microbiology. 2007;153(Pt 11):3677–3684. doi: 10.1099/mic.0.2007/010231-0. [DOI] [PubMed] [Google Scholar]
- 16.Schreve JL, Sin JK, Garrett JM. The Saccharomyces cerevisiae YCC5 (YCL025c) gene encodes an amino acid permease, Agp1, which transports asparagine and glutamine. J. Bacteriol. 1998;180:2556–2559. doi: 10.1128/jb.180.9.2556-2559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peter GJ, During L, Ahmed A. Carbon catabolite repression regulates amino acid permeases in Saccharomyces cerevisiae via the TOR signaling pathway. J. Biol. Chem. 2006;281:5546–5552. doi: 10.1074/jbc.M513842200. [DOI] [PubMed] [Google Scholar]
- 18.Abdel-Sater F, Iraqui I, Urrestarazu A, Andre B. The external amino acid signaling pathway promotes activation of Stp1 and Uga35/Dal81 transcription factors for induction of the AGP1 gene in Saccharomyces cerevisiae. Genetics. 2004;166:1727–1739. doi: 10.1534/genetics.166.4.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segal E, Widom J. From DNA sequence to transcriptional behaviour: a quantitative approach. Nat. Rev. Genet. 2009;10:443–456. doi: 10.1038/nrg2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irizarry RA, Wu Z, Jaffee HA. Comparison of Affymetrix GeneChip expression measures. Bioinformatics. 2006;22:789–794. doi: 10.1093/bioinformatics/btk046. [DOI] [PubMed] [Google Scholar]
- 21.Harrison AP, Johnston CE, Orengo CA. Establishing a major cause of discrepancy in the calibration of Affymetrix GeneChips. BMC Bioinformatics. 2007;8:195. doi: 10.1186/1471-2105-8-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Box GEP, Cox DR. An analysis of transformations. J. Roy. Stat. Soc. 1964;26:211–252. [Google Scholar]
- 24.Chen G, Jensen ST, Stoeckert CJ., Jr Clustering of genes into regulons using integrated modeling-COGRIM. Genome Biol. 2007;8:R4. doi: 10.1186/gb-2007-8-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat. Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- 27.Tanay A. Extensive low-affinity transcriptional interactions in the yeast genome. Genome Res. 2006;16:962–972. doi: 10.1101/gr.5113606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langrund O. Rotation tests. Stat. Comput. 2005;15:53–60. [Google Scholar]
- 30.Wieczorke R, Krampe S, Weierstall T, Freidel K, Hollenberg CP, Boles E. Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett. 1999;464:123–128. doi: 10.1016/s0014-5793(99)01698-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.