

Abstract
Background:
Activated microglia may play a role in the pathogenesis of Alzheimer disease (AD) as they cluster around beta-amyloid (Aβ) plaques. They are, therefore, a potential therapeutic target in both AD and its prodrome amnestic mild cognitive impairment (MCI).
Objective:
To characterize in vivo with 11C-(R)-PK11195 and 11C-PIB PET the distribution of microglial activation and amyloid deposition in patients with amnestic MCI.
Methods:
Fourteen subjects with MCI had 11C-(R)-PK11195 and 11C-PIB PET with psychometric tests.
Results:
Seven out of 14 (50%) patients with MCI had increased cortical 11C-PIB retention (p < 0.001) while 5 out of 13 (38%) subjects with MCI showed increased 11C-(R)-PK11195 uptake. The MCI subgroup with increased 11C-PIB retention also showed increased cortical 11C-(R)-PK11195 binding (p < 0.036) though this increase only remained significant in frontal cortex after a correction for multiple comparisons. There was no correlation between regional levels of 11C-(R)-PK11195 and 11C-PIB binding in individual patients with MCI: only three of the five MCI cases with increased 11C-(R)-PK11195 binding had increased levels of 11C-PIB retention.
Conclusions:
Our findings indicate that, while amyloid deposition and microglial activation can be detected in vivo in around 50% of patients with mild cognitive impairment (MCI), these pathologies can occur independently. The detection of microglial activation in patients with MCI suggests that anti-inflammatory therapies may be relevant to the prevention of AD.
GLOSSARY
- Aβ
= beta-amyloid;
- AD
= Alzheimer disease;
- BP
= binding potential;
- MCI
= mild cognitive impairment;
- MMSE
= Mini-Mental State Examination;
- PBBS
= peripheral benzodiazepine binding site;
- ROI
= region of interest;
- SD
= standard deviation;
- SRTM
= simplified reference tissue model.
There has been increasing interest in the early identification of subjects who have memory impairment beyond that expected with normal aging, but who do not fulfill criteria for dementia. These cases have been labeled as having amnestic mild cognitive impairment (MCI).1
The positron emitting radiotracer 11C-PIB (Pittsburgh compound B) is a neutral derivative of thioflavin-T with nanomolar affinity for fibrillar amyloid in the AD brain.2 Using 11C-PIB (N-methyl-[11C] 2-(4′-methylaminophenyl)-6-hydroxybenzothiazole) with PET it has been possible to localize and quantify Aβ plaque load in vivo. Several PET studies have reported increased cortical 11C-PIB uptake in 90% of patients with probable AD3–8 while around 50% of patients with amnestic MCI also have significantly raised uptake.5,8–10
Although the formation of Aβ and neurofibrillary tangles are key features in AD pathogenesis, activated microglia are thought to be important and have been shown to cluster around sites of Aβ in human postmortem brain slices and in transgenic mouse models of AD.11,12 The PET radiotracer 11C-(R)-PK11195 (1-[2-chlorophenyl]-N-methyl-N-[1-methyl-propyl]-3-isoquinolinecarboxamide) is a ligand that is specific for the peripheral benzodiazepine binding site (PBBS), a receptor abnormally expressed by the mitochondria of activated microglia.13 Increased binding of 11C-(R)-PK11195 has been reported in AD,14,15 and other neurodegenerative disorders including frontotemporal dementia16 and Parkinson disease,17 indicating that microglia activation is a nonselective response to neuronal damage. The precise role of microglial activation and its relationship to Aβ remains controversial. Microglia phagocytose dead cells, remodel synapses, and remove Aβ fibrils18,19 but also have damaging effects, releasing cytokines and nitric oxide.20 Less is known about their relevance to MCI. Using the PET radiotracers 11C-PIB and 11C-(R)-PK11195 we sought to characterize and compare in vivo the patterns of amyloid deposition and microglial activation in a group of subjects with MCI.
METHODS
Subjects with MCI.
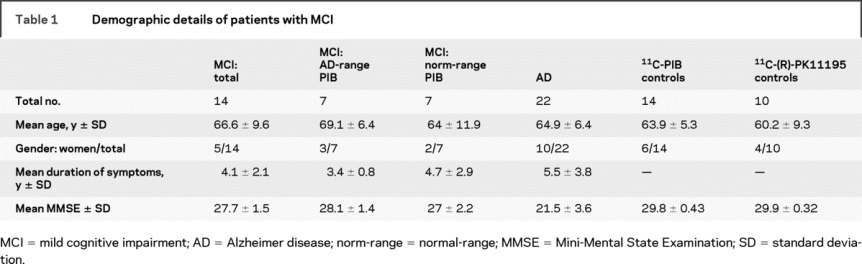
Fourteen subjects (mean age 66.6 years; SD 9.6) fulfilling Petersen's criteria for amnestic MCI1 participated in the study (table 1). All subjects underwent both 11C-PIB and 11C-(R)-PK11195 PET on separate occasions (mean interval between scans 6.4 ± 5.4 weeks). Subjects were recruited from the Hammersmith Hospitals NHS Trust (UK), the National Hospital for Neurology and Neurosurgery (UK), St. Margaret's Hospital (UK), and Victoria Hospital (UK). All patients had a comprehensive assessment including neurologic examination, neuropsychological testing, and MRI. Patients with significant white matter disease in the view of an experienced radiologist were excluded from the study. Ethical approval was granted by the Hammersmith and Queen Charlotte's Hospitals ethics committee. All patients gave informed written consent prior to their participation.
Table 1 Demographic details of patients with MCI
Subjects with AD and healthy controls.
PET data from 22 subjects (mean age 64.9 years; SD 6.4) meeting the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer's Disease and Related Disorders Association21 criteria for the diagnosis of probable AD were used for comparison with our MCI group. All 22 AD subjects had 11C-PIB (PIB) PET and, of these, 15 (mean age 63.7 years; SD 6.0) additionally had 11C-(R)-PK11195 (PK) PET (mean scan interval 3.1 ± 2.8 weeks). PIB PET data from 14 controls (mean age 63.9 years; SD 5.3) and PK PET data from 10 controls (mean age 60.2 years; SD 9.3) were also used for comparison with the MCI group. Eighteen of the 22 patients with AD and 11 of the 14 controls have had their PIB PET data previously reported.7 PK PET data for 11 of the 15 patients with AD and 8 of the 10 controls have now been accepted for publication. We have included these reference data so that our novel MCI PET data may be interpreted in context.
MRI scanning.
All subjects underwent volumetric T1-weighted MRI for the purpose of structure- function coregistration with PET and T2-weighted MRI to exclude structural lesions.
PET scanning.
Permission to administer 11C-PIB and 11C-(R)-PK11195 was obtained from the Administration of Radioactive Substances Advisory Committee of the United Kingdom. 11C-PIB and 11C-(R)-PK11195 were manufactured and supplied by GE Healthcare, Hammersmith Hospital, UK.
11C-PIB PET.
All subjects were scanned using a Siemens ECAT EXACT HR+ camera in three-dimensional acquisition mode using a scanning protocol described previously.7 Subjects received 366 ± 26.8 MBq 11C-PIB, injected IV.
Image analysis: 11C-PIB PET.
Target region: cerebellar cortex ratio images of PIB retention were generated as follows: 10–90 minute (total) and 60–90 minute (late) summation images were created in Matlab. Each MRI was segmented into gray, white matter, and CSF images using Statistical Parametric Mapping software (SPM2; Wellcome Department of Imaging Neuroscience, University College London, UK). The probabilistic gray matter image was binarized by thresholding at 50% probability. Both summation PET images were then coregistered to individual subjects' MRIs which were used as a template to spatially normalize those PET images and binarized MRI probability gray matter image into Montreal Neurological Institute space. Using Analyze software (Mayo Clinic, MN), we convolved the binarized gray matter image with an in-house probabilistic brain atlas.22 This creates an individualized anatomic atlas for each subject in standard space which is used as a template to sample a priori designated regions of interest (ROIs). At this stage, 11C-PIB activity in the reference region (cerebellar cortex) is sampled. Finally, the late summation images were normalized to the mean cerebellar cortical uptake value over this 60–90-minute period, generating the late ratio image. We used a late scan reference region approach because it provides a robust measure of total: nonspecific PIB relative volumes of distribution in target brain regions without the need for arterial sampling. The cerebellum was chosen as the reference region as it is relatively free of fibrillar plaques, reflected by its lack of staining with Congo red and thioflavins.23
ROI analysis.
We quantified PIB retention in the anterior cingulate, posterior cingulate, frontal, temporal, and parietal cortex. Additionally, whole brain retention was quantified, although not considered as a region when performing statistical analysis.
11C-(R)-PK11195 PET.
All subjects were scanned using an ECAT EXACT HR ++ (CTI/Siemens, Knoxville, TN) PET Tomograph (three-dimensional mode) which has a total axial field of view of 23.4 cm. Subjects received 299 ± 16 MBq 11C-(R)-PK11195 injected IV and scanning was performed in list mode and the dynamic images rebinned over 60 minutes as 18 time frames.
Image analysis: 11C-(R)-PK11195 PET.
Parametric images of PK binding potential (BP) were generated by a simplified reference tissue model (SRTM), using RPM software written in Matlab.24 Unlike fibrillar amyloid plaques, activated microglia are distributed throughout all brain regions in AD, including the cerebellum,25 potentially making the selection of a suitable reference region for nonspecific signal difficult. We, therefore, used a supervised clustering technique to derive a nonspecific uptake tissue reference input function in all groups studied. This technique produces BP estimates that are reproducible and comparable to those derived from plasma input.26,27 In summary, the method uses six predefined kinetic classes to extract as a reference region a cluster of gray matter voxels in each individual that exhibits the kinetic behavior closest to that of gray matter in a population of healthy controls. The supervised clustering algorithm avoids regions with specific binding such as association cortex in AD and large vessels, and the resulting cluster of voxels is clear of unwanted spurious signals arising from areas of the brain known to have a rich density of PBBS receptors such as the meninges.
The resulting parametric PK BP images and 60-minute summation images were then coregistered to each subject's MRI. The 60-minute summation image contains blood flow dependent signal, providing good anatomic detail, enabling accurate coregistration. Spatial normalization and creation of gray matter object maps were performed by the same methods described for analysis of PIB images, and PK BPs quantified in the same brain regions described earlier.
Statistical analysis.
Statistical tests were performed using SPSS for Windows 14.0 statistical software. Repeated measures analysis of variance was applied to test for global differences in PIB and PK binding between MCI groups and controls, using the Greenhouse Geisser factor to correct for variance heterogeneity across regions. Univariate analysis of variance was used to detect significant differences in regional PIB and PK binding between groups, using age as a covariate and the p plot method to control for multiple comparisons.28
Because of the heterogeneity of PK binding within the two MCI subgroups (those with increased AD-range and normal control-range PIB binding), PK BPs in subjects with MCI are also described on an individual basis. We considered regional PK binding to be raised if ≥2 SD greater than the control mean BP for that region.
Pearson's rank correlation coefficient was used to interrogate the correlation between PIB and PK binding in subjects with MCI. Spearman's rank correlation coefficient was used to assess the correlation between these PET measures and Mini-Mental State Examination (MMSE) scores and duration of symptoms.
RESULTS
11C-PIB retention.
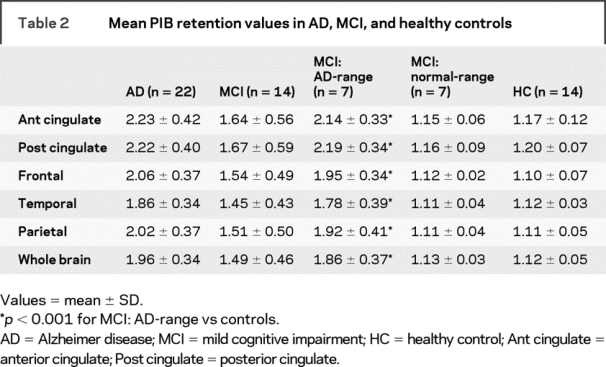
Visual inspection of the late PIB ratio images showed increased uptake in 7 of 14 (50%) subjects with MCI. Quantitative analysis revealed PIB uptake ratio values comparable to our AD group, with twofold increased uptake in cingulate and frontal regions. These seven patients with MCI were therefore considered as a subgroup, having mean AD-range PIB binding which was greater than control mean binding in all cortical ROIs (p < 0.001). The remaining seven subjects with MCI had normal levels and patterns of cortical PIB uptake, with no mean binding difference in this normal-range subgroup compared to that of controls (p = 0.69) (table 2). Two of our 22 subjects with AD had regional 11C-PIB uptake within control-range.7 One control (64-year-old man) had increased PIB retention (>2 SD greater than control mean) in the anterior cingulate, frontal, and temporal cortex (see discussion).
Table 2 Mean PIB retention values in AD, MCI, and healthy controls
11C-(R)-PK11195 binding.
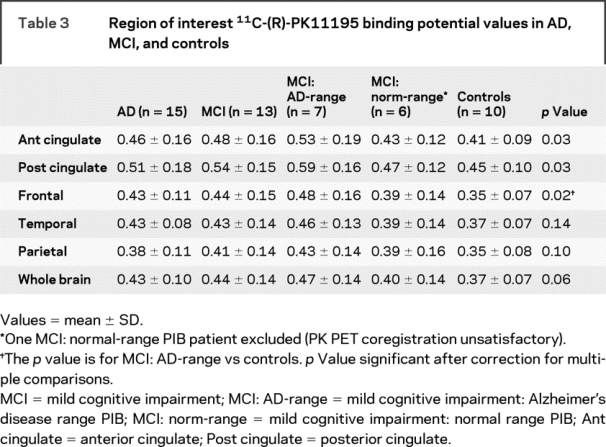
Coregistration of the PK BP image with MRI in one subject with MCI who had normal PIB uptake (55-year-old man) was unsatisfactory. He was, therefore, excluded from further PK PET analysis. Repeated measures analysis of variance showed a significant difference (p = 0.036) between mean PK BPs in AD-PIB range patients with MCI and controls. AD-PIB range patients with MCI had significantly increased mean PK binding in the anterior cingulate, posterior cingulate, and frontal cortex. However, after correction for multiple comparisons, only frontal cortex PK uptake remained significantly raised. There was no difference between mean regional PK binding in the AD-PIB range MCI and AD groups (p = 0.31), or between normal-PIB range MCI and controls (p = 0.67). Grouped mean PK BPs in the subject groups are shown in table 3.
Table 3 Region of interest 11C-(R)-PK11195 binding potential values in AD, MCI, and controls
Individually, PK binding was increased in 5 of 13 (38%) subjects with MCI, three with AD-range and two with normal-range PIB binding (table e-1 on the Neurology® Web site at www.neurology.org). PK BP values in the MCI subgroups compared to that of controls in the frontal cortex are displayed in figure 1. PET images showing PK binding in patients with MCI are displayed in figure 2.
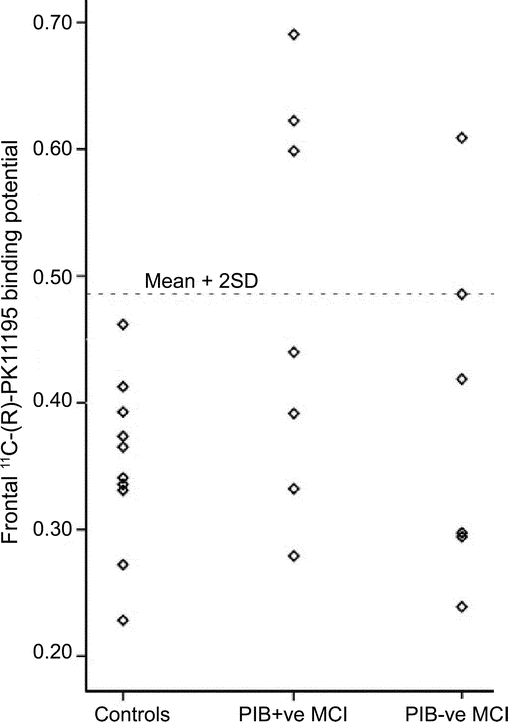
Figure 1 Scatterplot showing 11C-(R)-PK11195 binding potentials in frontal cortex of controls, and mild cognitive impairment (MCI) subgroups; PIB-positive (+ve) and PIB-negative (−ve) MCI
The dotted horizontal line shows controls' mean + 2 SD.

Figure 2 11C-(R)-PK11195 binding potentials and corresponding PIB ratio images in two patients with PIB-positive mild cognitive impairment (MCI)
MCI-I has normal PK binding and MCI-II has increased PK binding.
Correlations.
We found no correlation between individual regional PK binding and PIB retention in our MCI groups. Additionally, no correlation was found between duration of symptoms or MMSE scores with either PIB retention or PK binding in our AD-PIB range subgroup. However, in the PIB-negative subgroup, lower MMSE scores correlated with higher 11C-(R)-PK11195 binding in the anterior cingulate (Spearman rho, ρ = −0.812, p < 0.05) and temporal cortex (Spearman rho, ρ = −0.812, p < 0.05).
Follow-up.
Five PIB-positive subjects with MCI have been followed for 24–36 months after their baseline PIB PET scan and 3 (60%) of these have clinically converted to AD. Three PIB-negative MCI subjects have also been followed up for 2–3 years and none have converted to AD, although they continue to fulfill the criteria for a diagnosis of MCI. These three include the two MCI subjects with regionally increased PK binding but normal PIB uptake.
DISCUSSION
In this combined 11C-PIB and 11C-(R)-PK11195 PET study, we found that 7 (50%) of our 14 subjects with amnestic MCI had evidence of increased amyloid deposition. This is consistent with neuropathologic studies which report Aβ deposition in a significant proportion of subjects with MCI,29,30 and with the concept that amyloid deposition occurs early in the clinical evolution to AD. During a 2–3-year follow-up, three out of five of our PIB-positive but no PIB negative patients with MCI have converted to clinically probable AD. A longitudinal follow-up of the MCI cohort is ongoing.
Only one of 14 healthy controls showed increased PIB retention; this targeted the anterior cingulate and frontal and temporal cortices. Increased amyloid deposition has been reported in 10% of elderly healthy adults31 and its prevalence increases with age. Our relatively young control group (mean age 63.9 years; SD 5.3) might account for the low prevalence of significant PIB retention in our controls.
Compared to controls, our patients with MCI with increased PIB retention had significantly higher mean levels of cortical PK binding, although, after correction for multiple comparisons, only frontal PK uptake remained significantly raised, probably reflecting the low power of our small series. We detected regional increases in PK binding in our patients with MCI less frequently than raised PIB binding and there are a number of possible explanations for this. First, the specific signal associated with 11C-(R)-PK11195 uptake is low compared with 11C-PIB uptake, in part due to the lower density of activated microglia compared with amyloid plaques, as seen in AD,32 and also the higher background nonspecific signal seen with PK compared to that seen with 11C-PIB. This may have resulted in false negative findings in some patients. When delineating ROIs, we used a probabilistic atlas that takes into account variations in individual brain structures and atrophy. However, a limitation of this study is that we did not formally perform partial volume correction. This also may have resulted in an underestimation of cortical PK binding and false negative findings. Against this, we detected increased binding in the anterior and posterior cingulate, relatively small structures compared to our other interrogated ROIs. Finally, evidence exists for multiple factors apart from Aβ that may influence reactivity of microglia within brain, for example ApoE4 status33 and region-specific differences in the reactivity of microglial populations.34 We did not perform apolipoprotein E genotyping in our subjects. Different ApoE alleles influence amyloid load and so future PET studies may allow the interaction between ApoE genotype, amyloid deposition, and microglial activation to be explored in vivo.
An early 11C-PK11195 PET study in patients with AD with a lower sensitivity PET camera failed to detect increased binding compared to controls.35 However, there are a number of important methodologic differences between that and the present study. First, they used a cerebellar reference region where specific binding can be found. This may have led to apparent absent binding in other brain areas. Second, their control group consisted predominantly of patients with small cerebral gliomas which can bind PK. Third, they used racemic PK11195 whereas we used the active (R)-enantiomer of PK11195 which has been shown to bind PK binding sites with nanomolar affinity.36 More recently, SPECT15 and PET14 studies in patients with AD demonstrated significant increases in cortical PK binding, confirming this tracer is capable of detecting microglial activation in vivo. The PET study14 included one patient with isolated memory impairment who demonstrated increased binding in subregions of the temporal lobe, although 18F-FDG PET was normal at this time, suggesting that microglial activation can occur before metabolic deficits become evident, a viewpoint that is backed up by a histopathologic study in patients with AD.37 Here, microglial activation along with Aβ and congophilic deposits were found in the middle frontal gyrus in the absence of neurofibrillary tangles. These observations and our finding of comparable levels of PK binding in our PIB-positive MCI and AD group provide a rationale for assessing microglial activation in patients with MCI, a significant proportion of whom already have amyloid deposition at the time of clinical presentation.
We found no correlation between individual levels of amyloid deposition, as measured by 11C-PIB PET, and microglial activation, as assessed by 11C-(R)-PK11195 PET. This is in contrast to immunohistochemistry studies which have found activated microglial cells clustered at sites of neuritic plaques. However, it has also been demonstrated that not all amyloid plaques are associated with surrounding microglia,38 again implying that multiple factors may be involved in microglial activation as a response to amyloid pathology. Additionally, we found no correlation between MMSE scores or disease duration with PIB retention and PK binding in our PIB-positive subgroup. It is likely, however, that MMSE and other neuropsychometry measures in these patients are influenced by additional pathology such as the presence or absence of neurofibrillary tangles. The recent development of higher affinity PET radioligands which bind to the PBBS, such as 11C-DAA110639 and 11C-vinpocetine,40 may provide greater sensitivity for assessing and quantifying microglial activation in cognitive impairment and allow one to further explore the relationship between microglial activation, fibrillar amyloid, and cognition. The follow-up of PIB-positive patients with MCI with in vivo PBBS markers will enable a better understanding of the time course of microglial activation and its significance as a predictor of clinical progression to AD, and provide the opportunity to monitor the pathophysiologic effects of anti-inflammatory agents in vivo.
Finally, two subjects with MCI with increased PK binding had normal PIB uptake. The first patient (MCI-5) was a 70-year-old woman who had small focal increases in PK binding in the frontal and parietal cortices. Since her symptoms began 5 years ago she has shown no progression in her symptoms, supported by annual clinical, neuropsychological, and neuroimaging assessments. While these do not suggest a neurodegenerative cause for her memory impairment, her diagnosis, and the cause for the PK findings, remains unclear. Continued follow-up will be important in assessing its significance. The second patient (MCI-3), who had increased PK binding in all cortical regions of interest, was a 74-year-old man with a history of memory impairment dating back at least 5 years. This was associated with ventricular enlargement on magnetic resonance neuroimaging but annual clinical and neuropsychological assessments showed no convincing progression over this time. Subsequently he suffered a subacute deterioration in his cognition associated with increasing ventricular enlargement. A ventriculoperitoneal shunt was inserted which led to an improvement in his symptoms. An underlying neurodegenerative condition, again, seems unlikely given the long period of stability in his symptoms. A more definitive diagnosis has not yet been made and it remains to be seen whether shunting remains beneficial over the long term.
ACKNOWLEDGMENT
The authors thank Hope McDevitt, Andreanna Williams, James Anscombe, and Andrew Blyth for help with scanning.
Supplementary Material
Address correspondence and reprint requests to Dr. Aren Okello, Cyclotron Building, Hammersmith Hospital, Du Cane Road, London, W12 0NN, UK aren.okello@imperial.ac.uk
Supplemental data at www.neurology.org
Disclosure: This work was conducted in collaboration with Imanet, GE Healthcare. The Dementia Research Centre is an Alzheimer's Research Trust Coordinating Centre. UCLH/UCL received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. A. Okello is an Alzheimer's Research Trust research associate; P. Edison is a Medical Research Council clinical research fellow; H. Archer received funding from the Alzheimer's Research Trust; J. Kennedy is supported by funding from the Alzheimer's Research Trust; Z. Walker has received consultancy fees from GE Healthcare; N. Fox is a Medical Research Council senior clinical fellow; David Brooks is Head of Neurology, Medical Diagnostics, GE Healthcare.
Received July 1, 2008. Accepted in final form September 25, 2008.
REFERENCES
- 1.Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol 2001;58:1985–1992. [DOI] [PubMed] [Google Scholar]
- 2.Klunk WE, Wang Y, Huang GF, et al. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci 2003;23:2086–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 2004;55:306–319. [DOI] [PubMed] [Google Scholar]
- 4.Lopresti BJ, Klunk WE, Mathis CA, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med 2005;46:1959–1972. [PubMed] [Google Scholar]
- 5.Price JC, Klunk WE, Lopresti BJ, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab 2005;25:1528–1547. [DOI] [PubMed] [Google Scholar]
- 6.Archer HA, Edison P, Brooks DJ, et al. Amyloid load and cerebral atrophy in Alzheimer's disease: an 11C-PIB positron emission tomography study. Ann Neurol 2006;60:145–147. [DOI] [PubMed] [Google Scholar]
- 7.Edison P, Archer HA, Hinz R, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology 2007;68:501–508. [DOI] [PubMed] [Google Scholar]
- 8.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 2007;68:1718–1725. [DOI] [PubMed] [Google Scholar]
- 9.Forsberg A, Engler H, Almkvist O, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging 2008;29:1456–1465. [DOI] [PubMed] [Google Scholar]
- 10.Kemppainen NM, Aalto S, Wilson IA, et al. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology 2007;68:1603–1606. [DOI] [PubMed] [Google Scholar]
- 11.Haga S, Akai K, Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol (Berl) 1989;77:569–575. [DOI] [PubMed] [Google Scholar]
- 12.Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature 2008;451:720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banati RB, Newcombe J, Gunn RN, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain 2000;123:2321–2337. [DOI] [PubMed] [Google Scholar]
- 14.Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. Lancet 2001;358:461–467. [DOI] [PubMed] [Google Scholar]
- 15.Versijpt JJ, Dumont F, Van Laere KJ, et al. Assessment of neuroinflammation and microglial activation in Alzheimer's disease with radiolabelled PK11195 and single photon emission computed tomography: a pilot study. Eur Neurol 2003;50:39–47. [DOI] [PubMed] [Google Scholar]
- 16.Cagnin A, Rossor M, Sampson EL, Mackinnon T, Banati RB. In vivo detection of microglial activation in frontotemporal dementia. Ann Neurol 2004;56:894–897. [DOI] [PubMed] [Google Scholar]
- 17.Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis 2006;21:404–412. [DOI] [PubMed] [Google Scholar]
- 18.Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD. Scavenging of Alzheimer's amyloid beta-protein by microglia in culture. J Neurosci Res 1996;43:190–202. [DOI] [PubMed] [Google Scholar]
- 19.Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer's disease amyloid beta-protein by microglial cells. J Biol Chem 1997;272:29390–29397. [DOI] [PubMed] [Google Scholar]
- 20.McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging 2001;22:799–809. [DOI] [PubMed] [Google Scholar]
- 21.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 22.Hammers A, Allom R, Koepp MJ, et al. Three-dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp 2003;19:224–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joachim CL, Morris JH, Selkoe DJ. Diffuse senile plaques occur commonly in the cerebellum in Alzheimer's disease. Am J Pathol 1989;135:309–319. [PMC free article] [PubMed] [Google Scholar]
- 24.Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ. Parametric imaging of ligand-receptor binding in PET using a simplified reference region model. Neuroimage 1997;6:279–287. [DOI] [PubMed] [Google Scholar]
- 25.Larner AJ. The cerebellum in Alzheimer's disease. Dement Geriatr Cogn Disord 1997;8:203–209. [DOI] [PubMed] [Google Scholar]
- 26.Anderson AN, Pavese N, Edison P, et al. A systematic comparison of kinetic modelling methods generating parametric maps for [(11)C]-(R)-PK11195. Neuroimage 2007;36:28–37. [DOI] [PubMed] [Google Scholar]
- 27.Turkheimer FE, Edison P, Pavese N, et al. Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J Nucl Med 2007;48:158–167. [PubMed] [Google Scholar]
- 28.Turkheimer FE, Smith CB, Schmidt K. Estimation of the number of “true” null hypotheses in multivariate analysis of neuroimaging data. Neuroimage 2001;13:920–930. [DOI] [PubMed] [Google Scholar]
- 29.Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol 2006;63:38–46. [DOI] [PubMed] [Google Scholar]
- 30.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 2006;63:665–672. [DOI] [PubMed] [Google Scholar]
- 31.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 2006;67:446–452. [DOI] [PubMed] [Google Scholar]
- 32.Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC, Wegiel J. Contribution of glial cells to the development of amyloid plaques in Alzheimer's disease. Neurobiol Aging 2004;25:663–674. [DOI] [PubMed] [Google Scholar]
- 33.Lombardi VR, Garcia M, Cacabelos R. Microglial activation induced by factor(s) contained in sera from Alzheimer-related ApoE genotypes. J Neurosci Res 1998;54:539–553. [DOI] [PubMed] [Google Scholar]
- 34.Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol 2001;101:249–255. [DOI] [PubMed] [Google Scholar]
- 35.Groom GN, Junck L, Foster NL, Frey KA, Kuhl DE. PET of peripheral benzodiazepine binding sites in the microgliosis of Alzheimer's disease. J Nucl Med 1995;36:2207–2210. [PubMed] [Google Scholar]
- 36.Shah F, Hume SP, Pike VW, Ashworth S, McDermott J. Synthesis of the enantiomers of [N-methyl-11C]PK 11195 and comparison of their behaviours as radioligands for PK binding sites in rats. Nucl Med Biol 1994;21:573–581. [DOI] [PubMed] [Google Scholar]
- 37.Arends YM, Duyckaerts C, Rozemuller JM, Eikelenboom P, Hauw JJ. Microglia, amyloid and dementia in Alzheimer disease. A correlative study. Neurobiol Aging 2000;21:39–47. [DOI] [PubMed] [Google Scholar]
- 38.Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev 2005;48:234–239. [DOI] [PubMed] [Google Scholar]
- 39.Venneti S, Wagner AK, Wang G, et al. The high affinity peripheral benzodiazepine receptor ligand DAA1106 binds specifically to microglia in a rat model of traumatic brain injury: implications for PET imaging. Exp Neurol 2007;207:118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vas A, Shchukin Y, Karrenbauer VD, et al. Functional neuroimaging in multiple sclerosis with radiolabelled glia markers: Preliminary comparative PET studies with [(11)C]vinpocetine and [(11)C]PK11195 in patients. J Neurol Sci 2008;264:9–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.