

Abstract
T4 must be activated by its monodeiodination to T3 by type 1 or 2 iodothyronine deiodinase (D1 and D2). Recent studies show that despite an approximately 2000-fold higher Michaelis constant (Km; T4) for D1 than for D2 using dithiothreitol (DTT) as cofactor, D1 expressed in intact cells produces T3 at free T4 concentrations many orders of magnitude below its Km. To understand the factors regulating D1 and D2 catalysis in vivo, we studied a mutant D2 with a proline at position 135 of the active center of D2 replaced with a serine, as found in D1. The P135S D2 enzyme has many D1-like properties, a Km (T4) in the micromolar range, ping-pong kinetics with DTT, and sensitivity to 6n-propylthiouracil (PTU) in vitro. Unexpectedly, when the P135S D2 was expressed in HEK-293 cells and exposed to 2–200 pm free T4, the rate of T4 to T3 conversion was identical with D2 and conversion was insensitive to PTU. Using glutathione as a cofactor in vitro resulted in a marked decrease in the Km (T4) (as also occurs for D1), it showed sequential kinetics with T4 and it was sensitive to PTU but was resistant when HEK-293 cytosol was used as a cofactor. Thus, the in vivo catalytic properties of the P135S D2 mutant are more accurately predicted from in vitro studies with weak reducing agents, such as glutathione or endogenous cofactors, than by those with DTT.
Analysis of 5'-deiodinases with endogenous reducing agents improves the understanding about their physiological function.
The iodothyronine selenodeiodinases, types 1, 2, and 3 (D1, D2, and D3), constitute a family of oxidoreductases that catalyze the removal of iodine from the outer ring of T4 (D1 and D2), activating it, or from the inner ring of T3 or T4 (D3), inactivating them (1). All three deiodinases require a reducing cofactor thought to release iodide from the selenocysteine residue and regenerate the active enzyme. Early studies of D1 suggested that the endogenous cofactor for this deiodinase might be glutathione (GSH) or thioredoxin (Trx) because both of these were effective in supporting catalysis (2,3,4,5,6). Little is known as to whether the cofactors for D2 and D3 are the same or different, although GSH has been suggested as a possible cofactor for D2 (7). Identification of these cofactors is a relevant question because, like the concentration of the enzyme itself, regulation of the cofactor could have important effects on the rate of thyroid hormone activation or inactivation.
Most studies of deiodinases have used the potent synthetic thiol dithiothreitol (DTT) as a cofactor (6,7,8). This agent is very effective at maximizing deiodination by all three deiodinases and allows convenient and sensitive assays for enzyme activity (8). Using DTT, the kinetic mechanisms for the three deiodinases appear to be different, with D1 characterized by a ping-pong, double-substrate reaction mechanism (9), whereas D2 and D3 are thought to function by a sequential process in which enzyme, substrate, and cofactor form a putative catalytic complex (10,11,12). Given the uncertainties about the identity of the endogenous cofactor(s), one approach used by us and others has been to express a recombinant deiodinase in transformed cell lines and compare the reaction rates of one or the other enzymes (13,14,15). For such studies, deiodinases can be expressed at levels similar to those in normal tissues, and for studies of 5′-deiodinases, physiological concentrations of free T4 approximating hypothyroid, euthyroid, or hyperthyroid conditions can be used to reflect what may be occurring in vivo.
Such an approach was used to compare the catalytic efficiencies of D1 and D2 in HEK-293 cells at free T4 concentrations between 2 and 200 pm (15). Interestingly, despite the fact that D1 has a high Michaelis constant (Km) of about 10 μm for T4 when measured with 20 mm DTT, it was very effective at generating T3 in cell cultures when expressed at levels comparable with those in liver, despite the fact that the free T4 concentration was only 1 × 10−5 of the Km value. This efficient deiodination could be attributed to the fact that in enzymes with ping-pong reaction kinetics, the Km and maximal velocity (Vmax) are both reduced when weaker cofactors, such as GSH or Trx, are used (5,16,17). These, or similar, weaker reducing agents (relative to DTT) presumably are functioning as cofactors in these cells. When the same cells used for the cellular deiodination studies were sonicated and the Vmax for D1 was measured with 20 mm DTT and high concentrations of T4 in vitro, the reaction rate was several orders of magnitude higher than that found during the cellular deiodination, in agreement with this explanation. In contrast, the rate of T4 to T3 conversion by D2 when expressed in the same HEK-293 cells was much closer to the Vmax as measured in cell sonicates with 20 mm DTT (15). This was presumably in part due to the intrinsically lower Km of D2 for T4 (∼2 nm). These results argued that whatever cofactor is present in HEK-293 cells, it supports D2-mediated T4 to T3 conversion at rates closer to its Vmax than conversion mediated by D1. Thus, the physiological catalytic efficiency (the rate of T3 production in cells vs. that in vitro with DTT) for D2 is greater than for D1 under in vivo conditions.
Several years ago, a D1 protein that was insensitive to 6n-propylthiouracil (PTU), unlike all other known D1 enzymes, was identified in tilapia. Notably the tilapia D1, unlike other D1 enzymes (but like the PTU resistant D2 and D3), contained a proline (P) instead of a serine (S) residue at position 128, two residues carboxyterminal to the selenocysteine in the otherwise highly conserved active center (18). To determine whether the PTU insensitivity and the S to P change in tilapia D1 were related, we prepared human D2 and D3 mutants in which the proline at the position corresponding to that in D1 (position 135 in human D2) was replaced by S (19). Interestingly, this P to S change caused both D2 and D3 to become susceptible to PTU inhibition in vitro. In addition, the kinetic pattern of the P135S D2 using DTT as a cofactor became ping-pong in type, analogous to that for D1, and there was an approximately 100-fold increase in the Km of this enzyme for T4 (19).
Because the above-mentioned studies suggested that it was the low Km of D2 that had an important role in conferring a high efficiency to this enzyme during cellular deiodination, we designed the present studies to compare wild-type human D2 (wt D2) and the corresponding P135S mutant (P135S D2) to determine whether the enzymatic efficiency of this mutant expressed in cells with endogenous cofactors and that in vitro was more like D1 or the parent D2 and whether the P135S D2 could function as efficiently as the parent wild-type enzyme despite its much higher Km in vitro. Surprisingly, we found that the P135S D2 is as efficient as wt D2 during cellular deiodination, but its susceptibility to PTU, although retained when DTT was the cofactor in in vitro studies, was reduced when the enzyme was expressed in HEK-293 cells. This suggests that the D2-mediated deiodination supported by the endogenous cofactor functions quite differently from that with DTT. The in vitro studies with less potent physiological reducing agents such as GSH or Trx, or even crude cytosol itself, were better predictors of the characteristics of physiologic catalysis than those with DTT.
Materials and Methods
Reagents
Reagents were purchased from Sigma-Aldrich (St. Louis, MO). Outer ring-labeled [125I]rT3 and [125I]T4 (specific activity 4,400 Ci/mmol) was from PerkinElmer (Waltham, MA). Purification of [125I]rT3 and [125I]T4 was performed on LH-20 columns just before use to reduce 125I− concentration to less than 1%.
Plasmids
The wt D2 is a human wt D2 (hD2-SelP), with a FLAG peptide fused to the NH2 terminus, subcloned into a D10 vector containing a minimal selenocysteine insertion sequence element (20). In the P135S D2 construct, a Ser replaces Pro-135, as described previously (19). The rD1 plasmid was described elsewhere (21,22).
Cell culture, transfection, and deiodinase assays in intact cells
For transfection and expression studies, the HEK-293 cell line was obtained from American Type Culture Collection (Manassas, VA). Cells were grown and maintained in DMEM supplemented with 10% fetal bovine serum. For studies of the efficiency of physiological catalysis, defined as the ratio of the velocity of T4 to T3 conversion by the deiodinase expressed in cells (with a physiological cofactor) to that at a T4 concentration several times the Km using 20 mm DTT as cofactor, D2 P135S- or wt D2-expressing plasmids were transiently transfected in HEK-293 cells using Lipofectamine Plus (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The amount of plasmid transfected was adjusted to obtain comparable deiodination rates in in vivo experiments. This was achieved by transfecting 100 ng of wt D2 or 300 ng of D2 P135S into HEK-293 cells in each well of a six-well plate. Plasmid pCMVb-gal, which expresses β-galactosidase under the control of the cytomegalovirus promoter, was used in all transfections as a reporter for monitoring transfection efficiency, which was not different for the two D2 plasmids within experiments (23). At 48 h after transfection, cells in six-well plates were washed twice with sterile PBS and then cultured for 24 h in 1 ml serum-free 0.1% BSA in DMEM plus variable T4 concentrations [total (T4), 74–7,400 pm; free (T4), 2–200 pm] (15), including approximately 100,000 cpm/ml [125I]T4. The 5′-deiodinase activity in intact transfected cells was assayed as described previously (14,23). Briefly, at the end of incubation, 300 μl of medium were removed and added to 200 μl horse serum and protein precipitated by the addition of 100 μl of 50% trichloroacetic acid followed by centrifugation at 12,000 × g for 2 min. The 125I− generated was measured by a γ-counter and the appropriate corrections were made.
In vitro deiodinase assays in cell sonicates
For these assays, the remaining medium was removed and the cells washed twice with PBS, harvested, and sonicated in 0.25 m sucrose in PE buffer [0.1 m potassium phosphate and 1 mm EDTA (pH 6.9)] with 10 mm DTT. For these assays, we used 20–250 μg of cell sonicate; 10 nm T4 (for D2) or 3 μm T4 (for D2 P135S) and 20 mm DTT in a final volume of 100 μl PE. These concentrations of T4 were based on the estimated Km (T4) for each enzyme. Incubation was for 4 h at 37 C when DTT was used. The 125I− was isolated by trichloroacetic acid precipitation, as described above. For assays of PTU sensitivity, the same conditions were used, and cells were incubated with media containing 0, 10, or 100 μm of PTU. These same concentrations of PTU were used in in vitro studies.
In vitro deiodination assays using different cofactors
For experiments using GSH, Trx, and a Trx-regenerating system (15 μm of Escherichia coli Trx, 1 mm reduced nicotinamide adenine dinucleotide phosphate, and 10 μg of rat thioredoxin reductase) or cell cytosolic proteins as cofactors, rat D1- (rD1), P135S D2- or wt D2-expressing cells were harvested with 1 ml PBS and centrifuged at 3000 × g for 3 min. Cell pellets were then resuspended in 500 μl of 0.25 sucrose PE buffer and homogenized. The homogenate was centrifuged at 2000 × g for 25 min to remove large cell fragments. The supernatant fraction was then centrifuged at 100,000 × g for 1 h. The microsomes were resuspended in 0.25 m sucrose PE buffer with 10 mm DTT and stored at −80 C until use. The D2-expressing TαT1 cells (thyrotroph) (24) or untransfected HEK-293 cells were used for the preparation of cytosol, which was the 100,000 × g supernatant (S100) of homogenates after differential centrifugation. In TαT1 cells, which express D2, the D2 activity in the amount of cytosolic protein used as cofactor was negligible compared with that contained in the added microsomal preparation from HEK-293 cells transiently expressing the deiodinase of interest.
Deiodinase assays were performed as with the cell sonicates. However, a total assay volume of 50 μl Tris-EDTA buffer (pH 7.5) was used and the concentration of GSH was 20 mm. rT3 was used as substrate for rD1 and the reactions were incubated for 2.5 h. Twenty-five micrograms of HEK-293 S100 or 100 μg of TαT1 S100 were also used as cofactors. For D2 or P135S D2, assays were incubated overnight (16 h) with 1 nm T4 containing 1–2 × 105 cpm of 125I-labeled substrate. HPLC of the products of each assay showed that equimolar quantities of iodide and the appropriate product were generated during these reactions. All deiodination assays were performed in duplicate, which agreed closely in two to four different experiments.
GSH and Trx assays
GSH levels were determined by fluorescence using the DetectX detection kit for GSH (Luminos LLC, Ann Arbor, MI). Cell pellets were sonicated and then deproteinized with 5% sulfosalicylic acid. After incubation for 10 min at 4 C, samples were centrifuged at 14,000 rpm for 10 min at 4 C and the supernatants collected. ThioStar reagent (Luminos) was mixed with the samples according to the manufacturer’s instructions and incubated at room temperature for 15 min. The fluorescent product was read at a wavelength of 510 nm in a fluorescent plate reader with excitation at 390 nm. The total amount of GSH measured was then corrected for the estimated number of cells evaluated using a hemocytometer.
Trx assays were performed as described previously (25,26,27). Briefly, cells were lysed and then centrifuged at 13,000 × g at 4 C. The supernatant protein (25 μg) was used for measuring the Trx concentration. To this amount was added 15 μl of assay mixture, composed of 760 μm insulin and 2 mm reduced nicotinamide adenine dinucleotide phosphate in 0.2 m HEPES 20 mm EDTA solution (pH 7.6). The mixture was incubated for 37 C for 20 min, and the reaction was terminated by adding 250 μl of stop buffer (7 m guanidine-HCl, 1 mm dithionitrobenzoic acid) and absorbance was measured at 412 nm. Results were determined from a standard curve prepared at the same time.
Statistical analysis
Significant differences between two groups were determined by the two-tailed Student t test, and multiple comparisons were made using a one-way ANOVA followed by the Newman-Keuls postanalysis using GraphPad Prism 3.0 (GraphPad Inc., San Diego, CA). A probability of P < 0.05 was considered significant.
Results
T3 production in transfected cells and cell sonicates expressing D2 or P135S D2
We compared T3 production in intact HEK-293 cells expressing either wt D2 or P135S D2. We performed these experiments in the presence of free T4 at hypothyroid (2 pm), euthyroid (20 pm), and hyperthyroid (200 pm) concentrations, over a 24-h incubation period to approximate the physiological range and also to verify whether posttranslational down-regulation of D2 by T4 was preserved despite the P135S mutation (28,29,30). There was a similar T4 concentration-dependent decrease in fractional deiodination of T4 in cells expressing D2 or P135S D2, indicating a comparable saturable deiodination process for both enzymes (Fig. 1A). Despite this, T3 production steadily increased from less than 2 fmol/mg of protein · h up to about 8 fmol/mg of protein · h for wt D2 and 9 fmol/mg of protein · h for P135S D2 at euthyroid free T4 concentrations (Fig. 1B). The T3 production was comparable between the wt D2 and mutant D2 at all T4 concentrations.
Figure 1.

Cellular and in vitro T4 to T3 conversion by transiently expressed wild-type and P135S D2. A, Percent T4 to T3 conversion at variable free T4 concentrations. T4 deiodination was measured in six-well plates of HEK-293 cells transiently expressing wt D2 or D2 P135S. B, Cellular T3 production by the D2 enzymes. C, In vitro Vmax estimates for T3 production in sonicates of cells used in A at 20 mm DTT and either 10 nm T4 (wt D2) or 3 μm T4 (P135S D2). D, Physiological efficiency of cellular to in vitro T3 production by wild-type or P135S D2.
Deiodinase activity was then measured in sonicates of these cells using DTT and T4 concentrations more than 3 times the reported Km values to approximate maximal velocities. T3 production was 3.4 pmol/mg of protein · h for wt D2 (at 20 pm free T4) and about 50-fold higher for P135S D2. (Fig. 1C). This difference was 100-fold at the low and high free T4 concentrations. A decrease in the activity of both enzymes was apparent in cells incubated with the highest T4 concentration, presumably due to ubiquitination and accelerated enzyme degradation. We calculated the efficiency of physiological catalysis of the enzymes as the cellular deiodination rate divided by the Vmax with 20 mm DTT (Fig. 1D). The efficiency was low with both enzymes but was about 50- to 100-fold higher with wt D2 vs. that with the mutant enzyme because, despite the similar cellular T3 production rates of both enzymes, the in vitro deiodination of P135S D2 is 2 orders of magnitude higher than that with D2.
Enzyme kinetics
The above results in which the cellular T4 to T3 deiodination with a high Km enzyme, in this case P135S D2, is comparable to that with the low Km wt D2 despite the fact that the much higher Vmax of the mutant D2 is quite similar to that we observed with D1 vs. D2 (14,15). It argues that the Km of the mutant D2 (or D1) in vivo may be much lower than that estimated with DTT in vitro. Because there is some evidence that GSH could be an endogenous cofactor for D1 and perhaps also for D2 (7,16), we compared the in vitro reaction rates of wild-type and mutant D2 using this thiol (Fig. 2). The reaction rates with this cofactor were considerably lower than that with DTT, requiring 6–16 h of incubation to generate a fractional T4 deiodination of about 10–20%. Nonetheless, the reaction was saturated at high T4 concentrations and equimolar amounts of T3 and iodide were generated as evaluated by HPLC analysis. Also, the Lineweaver-Burk plots were linear and showed sequential reaction kinetics with GSH for both wt D2 and the mutant D2 enzyme (Fig. 2, B and D). The Km value of D2 for T4 with GSH was 2.9 nm (Fig. 2B), similar to that with 20 mm DTT (Fig. 2A) (11,31,32), but the Vmax with 20 mm GSH was about 7000-fold lower than with 20 mm DTT (Table 1). Surprisingly, for the mutant D2, the Km (T4) with GSH (1.7 nm) was about 2000-fold lower than with DTT (4.5 μm), and the kinetics were also of a sequential pattern (Fig. 2D), as opposed to the ping-pong mechanism found with DTT. A much lower apparent Km value for T4 and a shift to sequential rather than ping-pong kinetics with GSH have also been reported for D1 (5,16) and replicated in our laboratory with rat D1 (data not shown). Interestingly, the similar Vmax values for D2 and mutant D2 with GSH suggest that, for this cofactor at least, the major alteration that can explain the higher physiological catalytic efficiency than predicted from experiments with DTT is the large decrease in the Km of this enzyme (Table 1).
Figure 2.

Lineweaver-Burk plots comparing wt D2 and D2 P135S kinetics with respect to T4 using DTT or GSH as cofactor. A, Double-reciprocal plot of wt D2 activity using DTT 20 mm as cofactor and varying T4 concentrations. B, Double-reciprocal plot of wt D2 activity using GSH concentrations of 5, 10, and 20 mm. C, Double-reciprocal plots of P135S D2 activity with 20 mm and varying T4 concentrations. D, Double-reciprocal plots of T4 deiodination by P135S D2 at GSH concentrations of 7, 14, and 20 mm.
Table 1.
Effects of the P135S mutation on the kinetics of deiodination of T4 by D2
| Vmax (fmol/mg protein · min) | Km(T4) (m) | |
|---|---|---|
| DTT (20 mm) | ||
| wtD2 | 2,900 | 5.9 × 10−9 |
| P135S D2 | 22,000 | 4.5 × 10−6 |
| GSH (20 mm) | ||
| wtD2 | 0.4 | 2.9 × 10−9 |
| P135S D2 | 0.11 | 1.7 × 10−9 |
Effect of PTU on P135S D2-catalyzed deiodination
Previous analyses reported that PTU noncompetitively inhibited T4 deiodination mediated by DTT with the P135S mutant enzyme (19). We tested the effects of this inhibitor on T4 to T3 conversion at 1 nm or 3 μm of T4 for GSH and DTT as cosubstrates, respectively (Fig. 3). The results were compatible with a competitive inhibition by PTU of both DTT and GSH effects (Fig. 3, A and C). Because of the differences in the mechanism of inhibition of the DTT effect by PTU on the P135S construct from that reported earlier, we also repeated the studies of the effect of PTU when T4 concentrations were varied. As reported earlier, PTU was an uncompetitive inhibitor of T4 to T3 conversion at a fixed DTT concentration of 20 mm (Fig. 3B). However, the pattern of inhibition by PTU at a fixed GSH concentration (20 mm) was mixed, with a greater effect on Vmax than Km (Fig. 3D). To determine whether PTU was also inhibitory in HEK-293 cells transfected with the mutant enzyme, we performed the cellular conversion experiments using this agent. In intact cells and sonicates of the transfected cells, as expected, there was no significant effect of up to 100 μm PTU on T4 to T3 conversion catalyzed by wt D2 (Fig. 4, A and B). On the other hand, despite the inhibition of the conversion of T4 to T3 in sonicates of cells transfected with P135S D2 and incubated with DTT, there was no significant inhibitory effect of PTU on deiodination catalyzed by this mutant D2 in these cells (Fig. 4, A and B).
Figure 3.

Variations in the patterns of inhibition of GSH- or DTT-catalyzed T4 to T3 conversion by P135S D2 using PTU. A, Competitive inhibition of DTT by PTU at a fixed T4 concentration (3 μm). B, Uncompetitive inhibition of T4 deiodination catalyzed by fixed concentrations of DTT (20 mm). C, Competitive inhibition of GSH by PTU at a fixed T4 concentration (1 nm). D, A mixed pattern of inhibition of T4 deiodination affecting both Vmax and Km by PTU at a fixed GSH concentration (20 mm).
Figure 4.
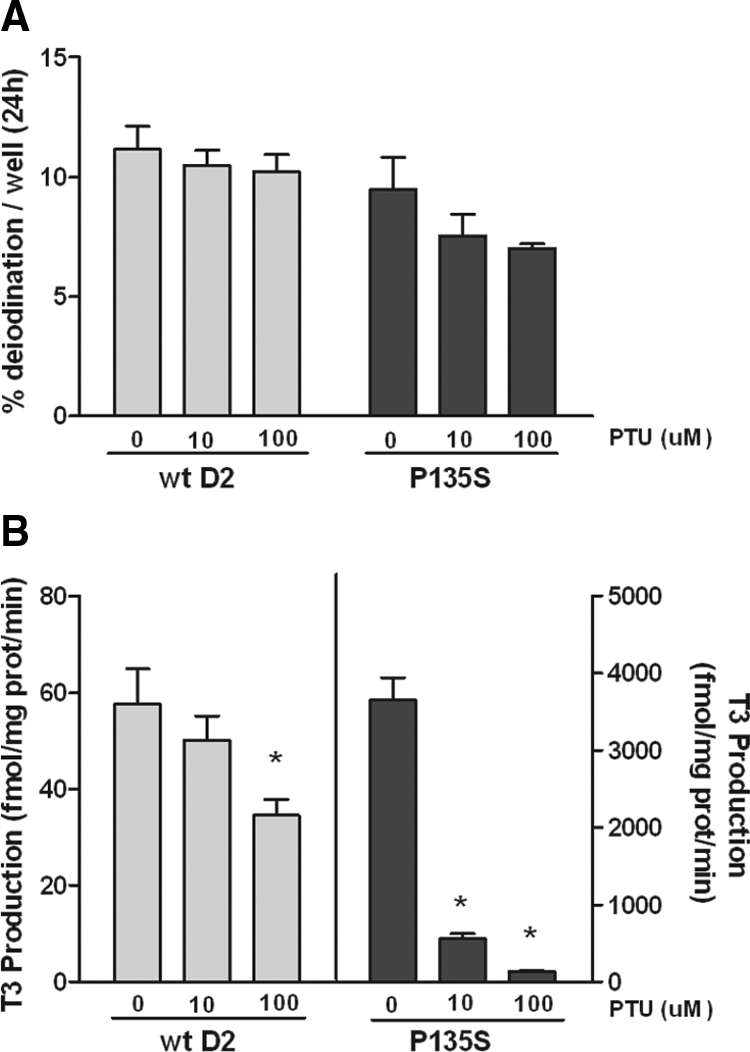
Effect of PTU on cellular and in vitro deiodination by wt D2 and P135S D2. A, Effect of PTU on T4 to T3 conversion in HEK-293 cells transiently expressing wt D2 or P135S D2 at 20 pm free T4. B, Effect of PTU on T4 to T3 conversion by the cells used in A, catalyzed by 20 mm DTT and 10 nm T4 (D2) or 3 μm T4 (P135S D2). *, Significantly different from controls (0 μm PTU) at P < 0.01.
Previous studies show that PTU is a quite effective inhibitor of D1 in HEK-293 cells, eliminating poor cellular PTU uptake as an explanation for this surprising result (15). An explanation for the disparity in the PTU effect between the cellular and in vitro conditions with P135S D2 could be the differences between the endogenous cofactor used for deiodination and DTT. In fact, when concentrations of cofactors were varied with a constant T4 concentration, the inhibitory constant for PTU inhibition with DTT (0.45 μm) for the P135S D2 was lower than that with GSH (2.6 μm) (Fig 3). Thus, higher PTU concentrations would be required for a similar degree of inhibition of deiodination with GSH or GSH-like cofactors than for DTT. Alternatively, the endogenous cofactor(s) may be even less sensitive to PTU than is the GSH-mediated reaction.
To examine this issue, we designed an in vitro assay system in which we used the S100 fraction of HEK-293 or the D2-expressing TαT1 cells as cofactors to support D1- or D2-mediated deiodination (Fig. 5). The endogenous GSH concentration in HEK-293 cytosol was 0.37 μg GSH per 106 cells, whereas that in TαT1 cells was undetectable (<10 ng/106 cells). The Trx concentrations in both cell lines were 1.9 ng/μg of cell protein. We first used the in vitro cofactor system with microsomes prepared from cells expressing rD1, which were supplemented with GSH, Trx, 100 μg of TαT1 S100, or 25 μg of S100 from nontransfected HEK-293 cells. These amounts of S100 proteins were those found to give maximal stimulation. With respect to D1, GSH, Trx, and the TαT1 and HEK-293 S100 fractions, all stimulated the deiodination of rT3. Catalysis by all four cofactors was readily inhibited by 10 μm PTU (Fig. 5A). There was no significant activity when either of the S100 fractions was assayed in the absence of the deiodinase-containing microsomes and equal quantities of labeled I− and 3,3′ T2 were generated during the incubation as evaluated by HPLC analyses. We also tested D2-mediated T4 deiodination, which was significantly stimulated by both GSH and Trx but much less effectively by the S100 fractions (Fig. 5B). There was a small, but statistically significant, inhibition by PTU of GSH stimulated D2 activity, which was not concentration dependent. The D2-catalyzed deiodination mediated by the other cofactors was not affected by PTU (Fig. 5B). Monodeiodination of T4 by P135S D2 was also stimulated by both thiol cofactors and the S100 fractions tested (Fig. 5C). PTU inhibited the GSH, Trx, and TαT1 S100 stimulated activity of P135S D2 but not that by HEK-293 S100 protein. This is consistent with the reduced sensitivity to PTU of this enzyme when expressed in HEK-293 cells.
Figure 5.
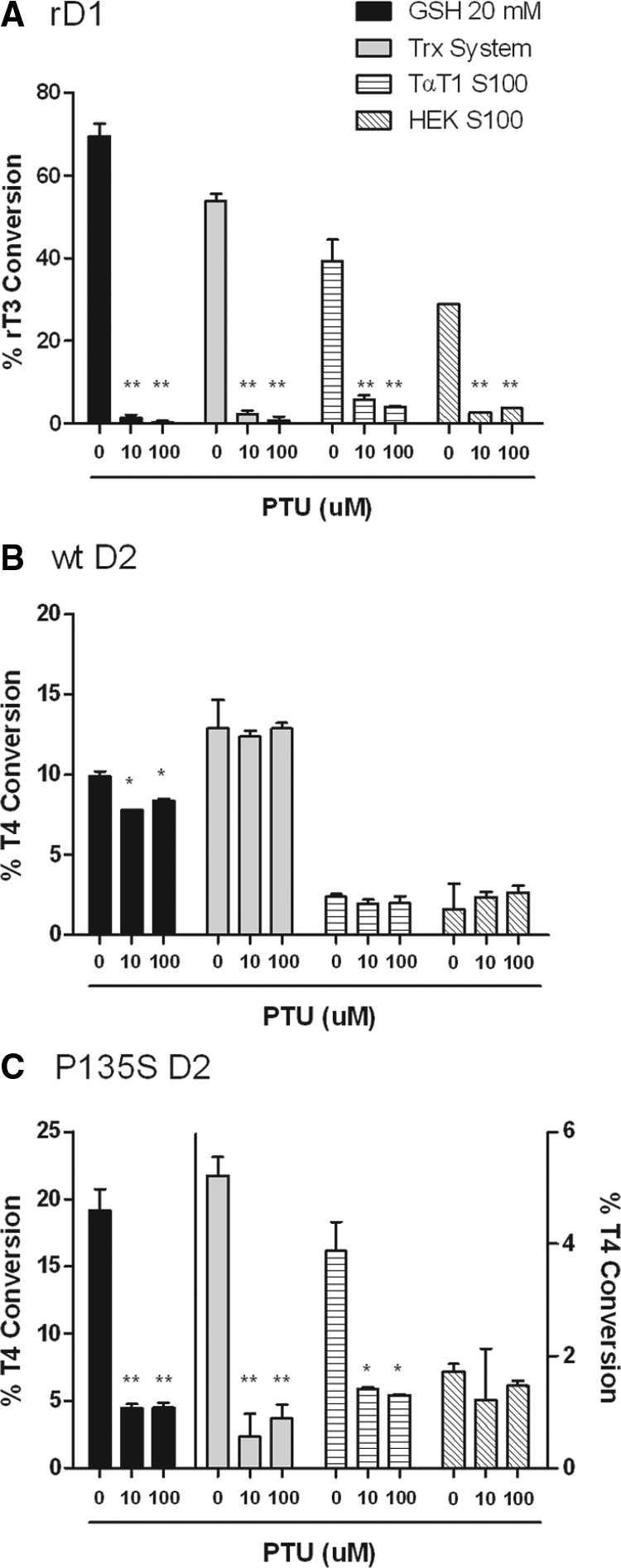
Effect of PTU on in vitro deiodination by rD1, wt D2, or P135S in the presence of different cofactor systems. A, Fractional deiodination of 1 nm rT3 by rD1 and 20 mm GSH, a Trx system, TαT1 S100, or HEK-293 S100, in the absence or presence of PTU (10 or 100 μm). B, Deiodination rate under conditions in A using T4 (1 nm) by wt hD2 as in A. C, Deiodination rate of 1 nm T4 by P135S D2 under the conditions in A. *, Significantly different from controls (0 μm PTU) at P < 0.05; **, P < 0.01.
Discussion
In the present study, we explored the effects of the substitution of a serine for a proline in position 135 of human D2 on its function in vitro using various cofactors and in HEK-293 cells. The mutation produces important changes in D2-mediated deiodination as assessed in in vitro studies, making its characteristics more D1-like as would be expected. Using DTT as cofactor, there is an increase in the Vmax and Km for T4 (Table 1) (19) as well as an enhanced susceptibility to PTU inhibition. Surprisingly although, minimal changes are observed in its capacity to convert T4 to T3 when it is transiently expressed in HEK-293 cells (Fig 1, A and B). A similar discrepancy occurs between cellular deiodination rates and in vitro assays with D1 (15) and has been described for other mutant D1 enzymes (13).
We used the P135S D2 with these D1 characteristics to address the potential causes for this discrepancy between physiological T4 deiodination in cells and the in vitro results with DTT. We hypothesized that this marked difference could be attributed to discrepancies in the effects of endogenous cofactors vs. that of the highly potent dithiol, DTT. Whereas the cofactor is not known for D2 or D3, there is evidence that GSH and Trx may serve this role with D1. In fact, we found both of these agents support D2-mediated T4 to T3 conversion in vitro (Table 1 and Figs. 2, 3, and 5), although the rates are much slower than with D1 (Fig. 5). Stimulation of catalysis by GSH and Trx has not been previously shown with recombinant D2. Interestingly, the P135S mutant was significantly more responsive to GSH stimulation than D2, and the Km (T4) of the GSH-catalyzed reaction was reduced more than 2000-fold. Surprisingly, the Km (T4) of D2 remained unchanged with GSH, and both D2 and the mutant had kinetics suggesting a sequential pattern (Fig. 2, B and D). For the D2 mutant, this corresponded to a change from the ping-pong kinetics observed when P135S D2 is stimulated by DTT (Fig. 3) (19). The marked dependence of the Km (T4) on the cofactor has previously been reported for D1 and has been demonstrated using GSH, Trx, and low DTT concentrations (4,5,16). The Km (T4) of D2 and the mutant with GSH are virtually identical, which may well explain the similarity of the cellular T3 production rates. A comparison of the T3 production rates by cells transiently expressing these enzymes with that found with DTT or GSH in vitro is illustrative (Table 2). T3 production by wt D2 was about 4000-fold higher in in vitro assays when using 20 mm DTT compared with the same conditions with 20 mm GSH as cofactor. For the P135S D2, this ratio was 23,000-fold. Interestingly, when 20 mm GSH was used, cellular T3 production for wt D2 (and P135S D2) was similar to the amount produced in vivo by the same enzyme. Thus, use of GSH as a cofactor gives results that more closely mimic the physiological rate of T4 to T3 conversion by these enzymes than does DTT, even though it may, or may not, be the endogenous cofactor. The same arguments could be made with respect to the much higher efficiency of D1 for cellular T4 to T3 conversion than would be predicted in light of its high Km (T4) with DTT (10 μm) (15). The very low level of GSH in the TαT1 cytosol argues against this as a cofactor for D1 in the TαT1 cell S100 fraction.
Table 2.
Comparison of T3 production in cells with that under different cofactor conditions for wtD2 and P135S D2
| Cellular T3 production (fmol/mg protein · min)a |
In vitro T3 production (fmol/mg protein · min)b
|
Cellular to in vitro T3 production ratio
|
|||
|---|---|---|---|---|---|
| DTT (20 mm) | GSH (20 mm) | DTT (20 mm) | GSH (20 mm) | ||
| wtD2 | 0.25 | 230 | 0.06 | 1.1 × 10−3 | 4.2 |
| P135S D2 | 0.23 | 6275 | 0.27 | 3.7 × 10−5 | 0.85 |
Cellular T3 production of HEK-293 cells transiently expressing either wt or P135S D2, incubated with 20 pm free T4 in six-well plates for 24 h.
In vitro T3 production by wtD2- and P135S D2-expressing microsomes from each well. T4 concentrations for in vitro assays with 20 mm DTT were 10 nm T4 (for D2) or 3 μm T4 (for D2 P135S). For in vitro assays with 20 mm GSH, 1 nm T4 was used for both enzymes. The T4 concentrations in the in vitro assays were based on the Km (T4) for each enzyme and designed to provide an estimate of the total potential activity of the enzyme in each well with each cofactor. This permits the determination of the efficiency of the cellular deiodination rate relative to that in vitroin the presence of an excess of either cofactor as well as substrate.
We were also surprised that the in vivo sensitivity to PTU of the P135S D2 was much lower during cell-based deiodination than during its in vitro studies with DTT from the same cell sonicates (Fig. 4). Kinetic studies showed that both DTT- and GSH-mediated T4 to T3 conversion were competitively inhibited by PTU similar to what has been reported for D1. The results of the PTU inhibition studies in the presence of fixed GSH (20 mm) and variable T4 are markedly different from the uncompetitive inhibition seen here with 20 mm DTT as cofactor (Fig. 3, B vs. D).
The mechanism thought to explain PTU inhibition of D1 is the formation of an enzyme-Se-S-PTU complex (9). In case of D1, the putative SeI intermediate that forms in the ping-pong mechanism is the target for inhibition by PTU (22,33,34,35), which is thought to compete with the cofactor to form a relatively stable Se-S-PTU intermediate, blocking enzyme regeneration. However, because this putative intermediate should not exist for D2-mediated T4 monodeiodination due to its sequential kinetics in which the oxidized enzyme is immediately reduced by the cofactor, it should be PTU resistant. Therefore, the susceptibility of P135S D2 to PTU with DTT as cofactor is probably due to the ping-pong kinetic mechanism observed in vitro with this cofactor. The higher apparent inhibitory constant for PTU with GSH when compared with DTT (2.6 vs. 0.45 μm) for the P135S D2 may reflect a more complex mixture of ping-pong and sequential mechanisms for the mutant D2. This would contribute to the reduced PTU sensitivity of the cellular deiodination by P135S D2.
Another possibility for the PTU resistance of the in vivo reaction is that the cellular cofactor that supports D2-mediated deiodination in HEK-293 cells is not GSH and is not sensitive to PTU. To address that question, we compared deiodinase activity with various exogenous or endogenous cell type-specific microenvironments, the latter using the S100 cytosol fraction from HEK-293 and TαT1 cells. This is an approximation to physiological conditions, although the deiodination is less efficient than in transfected cells. This could be for a number of reasons including the absence of a mechanism that could maintain the physiological cofactor concentrations constant over the in vitro incubation period or because of the potential oxidation or other damage to the cofactor during the preparation of the S100 fraction. Nonetheless, to our knowledge, this is the first evidence that crude cytosolic proteins can support D2-catalyzed deiodination. Using this system, we found robust deiodination by D1 in the presence of GSH, the Trx system, HEK-293 S100, and even with TαT1 S100 (Fig. 5), which does not contain detectable GSH, although it does contain Trx. Moreover, TαT1 cytosol supports D1 activity even better than the HEK-293 cytosol, when both are used in amounts giving maximal stimulation, suggesting there may be cell type-specific quantitative differences in the cofactors expressed. D1 activity is quite susceptible to PTU inhibition with all cofactors (Fig. 5A). In addition, as is the case with DTT, GSH, Trx, and the S100 fractions from TαT1 and HEK-293 cells all support D1 activity better than that of D2.
D2 activity is stimulated by GSH and especially the Trx system but has significantly lower but still measurable activity when S100 fractions of different cells are used to mediate deiodination. Only minimal inhibition of the GSH-stimulated reaction by D2 occurs with PTU. The P135S D2 enzyme is more efficient than wt D2 with all potential cofactors except Trx. However, the low activity of this enzyme found using HEK-293 S100 is not sensitive to PTU, although that with TαT1 is (Fig. 5C). This could be due to different mechanisms of deiodination supported by these two cytosols or perhaps merely to the low rate of deiodination. If it is the cofactor per se, this would contribute to the PTU insensitivity we observed in HEK-293 cells.
It is known that D2 activity is down-regulated by T4 via a posttranscriptional mechanism that increases its rate of degradation/inactivation via its ubiquitination (28,30). This mechanism depends on deiodination of a substrate that requires either Sec or Cys in the catalytic center (29). Because there is a decrease in the in vivo T3 production during catalysis at high T4 concentrations for both wild-type and mutant D2, the Pro in position 135 is not essential for recognition by ubiquitination machinery responsible for this posttranscriptional regulation (Fig. 1A).
Taken together, the present results show that the use of DTT as a cofactor does not allow accurate predictions of the behavior of the deiodinases in vivo. As assessed by DTT-mediated deiodination, the P135S mutation in D2 results in important modifications of the in vitro biochemical properties of D2, but those have minimal effects on its function in vivo. Studies with putative endogenous cofactors, although giving much lower deiodination rates, provide more accurate information about how the mutant D2 as well as the wild-type D1 will function in vivo. An in vitro method for assessing the role of various cellular cofactors, illustrated here, should be helpful in allowing a better analysis of how these enzymes function under physiological circumstances.
Acknowledgments
We thank Dr. Stephen Huang and Ms. Michelle A. Mulcahey for HPLC analyses, Dr. Ann Marie Zavacki and Arne Holmgren for review of the manuscript and helpful discussions, and Dr. Holmgren for reagents and advice regarding the thioredoxin assay.
Footnotes
This work was supported by the National Institutes of Health Grants ROI DK36256 and FIC TW007559.
Disclosure Summary: A.L.M. and P.R.L. are supported by National Institutes of Health grants. The other authors have nothing to disclose.
First Published Online December 4, 2009
Abbreviations: D1, Deiodinase type 1; D2, selenodeiodinase type 2; D3, selenodeiodinase type 3; DTT, dithiothreitol; GSH, glutathione; Km, Michaelis constant; P, proline; PE, buffer of potassium phosphate and EDTA; PTU, 6n-propylthiouracil; S, serine; Trx, thioredoxin; Vmax, maximal velocity.
References
- Bianco AC, Larsen PR 2005 Cellular and structural biology of the deiodinases. Thyroid 15:777–786 [DOI] [PubMed] [Google Scholar]
- Visser TJ, Mol JA, Holmgren A 1986 The possible role of glutathione and glutaredoxin in the enzymatic deiodination of the thyroid hormone. In: Holmgren A, Branden C, Jornvall H, Sjoberg B, eds. Thioredoxin and glutaredoxin systems: structure and function. New York: Raven Press; 369–376 [Google Scholar]
- Balsam A, Ingbar SH 1979 Observations on the factors that control the generation of triiodothyronine from thyroxine in rat liver and the nature of the defect induced by fasting. J Clin Invest 63:1145–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami A, Rosenberg IN 1987 Thioredoxin stimulates enzymatic outer ring monodeiodination of reverse triiodothronine. Endocrinology 121:1937–1945 [DOI] [PubMed] [Google Scholar]
- Sharifi J, St Germain DL 1992 The cDNA for the type I iodothyronine 5′-deiodinase encodes an enzyme manifesting both high Km and low Km activity. Evidence that rat liver and kidney contain a single enzyme which converts thyroxine to 3,5,3′-triiodothyronine. J Biol Chem 267:12539–12544 [PubMed] [Google Scholar]
- Bhat GB, Iwase K, Hummel BC, Walfish PG 1989 Kinetic characteristics of a thioredoxin-activated rat hepatic and renal low-Km iodothyronine 5′-deiodinase. Biochem J 258:785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami A, Rosenberg IN 1990 Regulation of iodothyronine 5′-deiodinases: effects of thiol blockers and altered substrate levels in vivo and in vitro. Endocrinology 126:2597–2606 [DOI] [PubMed] [Google Scholar]
- Visser TJ, Does-Tobé I, Docter R, Hennemann G 1976 Subcellular localization of a rat liver enzyme converting thyroxine into tri-iodothyronine and possible involvement of essential thiol groups. Biochem J 157:479–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser TJ 1979 Mechanism of action of iodothyronine-5′-deiodinase. Biochim Biophys Acta 569:302–308 [DOI] [PubMed] [Google Scholar]
- Visser TJ, Leonard JL, Kaplan MM, Larsen PR 1982 Kinetic evidence suggesting two mechanisms for iodothyronine 5′-deiodination in rat cerebral cortex. Proc Natl Acad Sci USA 79:5080–5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser TJ, Kaplan MM, Leonard JL, Larsen PR 1983 Evidence for two pathways of iodothyronine 5′-deiodination in rat pituitary that differ in kinetics, propylthiouracil sensitivity, and response to hypothyroidism. J Clin Invest 71:992–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Klootwijk W, Visser TJ 2003 Substitution of cysteine for selenocysteine in the catalytic center of type III iodothyronine deiodinase reduces catalytic efficiency and alters substrate preference. Endocrinology 144:2505–2513 [DOI] [PubMed] [Google Scholar]
- Croteau W, Bodwell JE, Richardson JM, St Germain DL 1998 Conserved cysteines in the type 1 deiodinase selenoprotein are not essential for catalytic activity. J Biol Chem 273:25230–25236 [DOI] [PubMed] [Google Scholar]
- Buettner C, Harney JW, Larsen PR 2000 The role of selenocysteine 133 in catalysis by the human type 2 iodothyronine deiodinase. Endocrinology 141:4606–4612 [DOI] [PubMed] [Google Scholar]
- Maia AL, Kim BW, Huang SA, Harney JW, Larsen PR 2005 Type 2 iodothyronine deiodinase is the major source of plasma T3 in euthyroid humans. J Clin Invest 115:2524–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami A, Rosenberg IN 1988 Effects of glutathione on iodothyronine 5′-deiodinase activity. Endocrinology 123:192–202 [DOI] [PubMed] [Google Scholar]
- Leonard JL, Visser TJ 1986 Biochemistry of deiodination. In: Hennemann G, ed. Thyroid hormone metabolism. New York: Marcel Dekker; 193–196 [Google Scholar]
- Sanders JP, Van der Geyten S, Kaptein E, Darras VM, Kühn ER, Leonard JL, Visser TJ 1997 Characterization of a propylthiouracil-insensitive type I iodothyronine deiodinase. Endocrinology 138:5153– 5160 [DOI] [PubMed] [Google Scholar]
- Callebaut I, Curcio-Morelli C, Mornon JP, Gereben B, Buettner C, Huang S, Castro B, Fonseca TL, Harney JW, Larsen PR, Bianco AC 2003 The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J Biol Chem 278:36887–36896 [DOI] [PubMed] [Google Scholar]
- Curcio-Morelli C, Gereben B, Zavacki AM, Kim BW, Huang S, Harney JW, Larsen PR, Bianco AC 2003 In vivo dimerization of types 1, 2, and 3 iodothyronine selenodeiodinases. Endocrinology 144:937–946 [DOI] [PubMed] [Google Scholar]
- Berry MJ, Banu L, Larsen PR 1991 Type I iodothyronine deiodinase is a selenocysteine-containing enzyme. Nature 349:438–440 [DOI] [PubMed] [Google Scholar]
- Berry MJ, Kieffer JD, Harney JW, Larsen PR 1991 Selenocysteine confers the biochemical properties characteristic of the type I iodothyronine deiodinase. J Biol Chem 266:14155–14158 [PubMed] [Google Scholar]
- Canettieri G, Celi FS, Baccheschi G, Salvatori L, Andreoli M, Centanni M 2000 Isolation of human type 2 deiodinase gene promoter and characterization of a functional cyclic adenosine monophosphate response element. Endocrinology 141:1804–1813 [DOI] [PubMed] [Google Scholar]
- Christoffolete MA, Ribeiro R, Singru P, Fekete C, da Silva WS, Gordon DF, Huang SA, Crescenzi A, Harney JW, Ridgway EC, Larsen PR, Lechan RM, Bianco AC 2006 Atypical expression of type 2 iodothyronine deiodinase in thyrotrophs explains the thyroxine-mediated pituitary thyrotropin feedback mechanism. Endocrinology 147:1735–1743 [DOI] [PubMed] [Google Scholar]
- Arner E, Zhong L, Holmgren A 1999 Preparation and assay of mammalian thioredoxin and thioredoxin reductase. In: Packer L, ed. Methods in enzymology. San Diego: Academic Press; 226–239 [DOI] [PubMed] [Google Scholar]
- Holmgren A 1979 Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J Biol Chem 254:9627–9632 [PubMed] [Google Scholar]
- Holmgren A 1979 Reduction of disulfides by thioredoxin. Exceptional reactivity of insulin and suggested functions of thioredoxin in mechanism of hormone action. J Biol Chem 254:9113–9119 [PubMed] [Google Scholar]
- Steinsapir J, Harney J, Larsen PR 1998 Type 2 iodothyronine deiodinase in rat pituitary tumor cells is inactivated in proteasomes. J Clin Invest 102:1895–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinsapir J, Bianco AC, Buettner C, Harney J, Larsen PR 2000 Substrate-induced down-regulation of human type 2 deiodinase (hD2) is mediated through proteasomal degradation and requires interaction with the enzyme’s active center. Endocrinology 141:1127–1135 [DOI] [PubMed] [Google Scholar]
- Gereben B, Goncalves C, Harney JW, Larsen PR, Bianco AC 2000 Selective proteolysis of human type 2 deiodinase: a novel ubiquitin-proteasomal mediated mechanism for regulation of hormone activation. Mol Endocrinol 14:1697–1708 [DOI] [PubMed] [Google Scholar]
- Salvatore D, Bartha T, Harney JW, Larsen PR 1996 Molecular biological and biochemical characterization of the human type 2 selenodeiodinase. Endocrinology 137:3308–3315 [DOI] [PubMed] [Google Scholar]
- St Germain DL 1988 The effects and interactions of substrates, inhibitors, and the cellular thiol-disulfide balance on the regulation of type II iodothyronine 5′-deiodinase. Endocrinology 122:1860–1868 [DOI] [PubMed] [Google Scholar]
- Visser TJ, van Overmeeren-Kaptein E 1981 Substrate requirement for inactivation of iodothyronine-5′-deiodinase activity by thiouracil. Biochim Biophys Acta 658:202–208 [DOI] [PubMed] [Google Scholar]
- Leonard JL, Rosenberg IN 1978 Thyroxine 5′-deiodinase activity of rat kidney: observations on activation by thiols and inhibition by propylthiouracil. Endocrinology 103:2137–2144 [DOI] [PubMed] [Google Scholar]
- Leonard JL, Rosenberg IN 1980 Iodothyronine 5′-deiodinase from rat kidney: substrate specificity and the 5′-deiodination of reverse triiodothyronine. Endocrinology 107:1376–1383 [DOI] [PubMed] [Google Scholar]