

Abstract
Estrogens play a critical role in brain development by acting on areas that express estrogen receptors. In the rodent cortex, estrogen receptor α (ERα) mRNA expression is high early in postnatal development but declines starting at postnatal day (PND) 10 and is virtually absent in the adult cortex. The mechanisms controlling this regulation are largely unknown. Methylation is important for gene silencing during development in many tissues, including the brain. In the present study, we examined the methylation status of ERα 5′ untranslated exons during early postnatal development in male and female mice using methylation-specific PCR and pyrosequencing. Several regions of ERα promoter displayed a significant increase in methylation at PND 18 and 25 compared with PND 4. DNA methyltransferases (DNMT) are important for the initiation and maintenance of methylation. Real-time PCR showed that DNMT3A, the de novo DNMT peaked at PND 10 and was decreased by PND 25. DNMT1, which is important for maintenance of methylation, increased across development and stayed high in adult cortex. The methyl-CpG-binding protein 2 (MeCP2) is also important for stabilization of methylation. A chromatin immunoprecipitation assay showed a correlation between association of MeCP2 with ERα promoter and the increase in methylation and decrease in ERα expression after PND 10. In mice containing a mutant MeCP2 protein, ERα mRNA expression and promoter methylation patterns across development were different compared with wild-type mice. These data suggest that methylation of ERα promoters regulates ERα mRNA expression in the cortex during postnatal development in a MeCP2-dependent fashion.
Estrogen receptor-α gene expression in the cerebral cortex is suppressed after the first week of life by DNA methylation and the subsequent binding of MeCP2 to the promoter.
Estrogens are a group of steroid hormones that play a crucial role in sexual development and reproduction. Estrogens are also involved in numerous other physiological processes including modulating fine motor control (1), influencing pain sensitivity (2,3) and memory (4,5,6), and functioning as a protective factor (7,8). The primary biologically active estrogen, 17β-estradiol (E2), plays a large role in early nervous system development by influencing mechanisms such as apoptosis (9,10), neurite outgrowth (11,12), synaptogenesis (13), and cell differentiation (14,15). E2 exerts its effects at target tissues through interactions with nuclear steroid receptors as well as membrane receptors. The intracellular receptors act as transcription factors to stimulate or repress gene expression (16,17). Many of the physiological effects of E2 on target tissues are mediated by specific estrogen receptors α (ERα) and β (ERβ). The effects of E2 on the brain during early postnatal development are largely related to regional and temporal expression of ERα and ERβ.
Receptor binding studies have demonstrated that E2 binds in reproductive areas of the brain such as the hypothalamus in addition to areas of the brain not associated with reproduction, such as the cortex (18,19). Interestingly, binding in the cortex is high in early postnatal development but diminished in the adult (19). This developmental decrease in binding in the cortex has recently been shown to be a result of a decline in ERα and not ERβ mRNA expression levels (20,21). Changes in ERα mRNA expression in the cortex not only occur during development but have also been observed after brain injury. For example, ERα mRNA expression is increased in the cortex after a model of stroke, and this rise is thought to confer estradiol’s neuroprotective activity (22,23,24). Additionally, changes in ERα mRNA expression have also been observed in breast cancer cells. In breast cancer, regulation of ERα gene expression is mediated by methylation of DNA and acetylation of histone proteins (25,26,27). The ERα promoter of ER-negative breast cancer cells is methylated, whereas in ER-positive cells, the ERα promoter is unmethylated (27,28). In the same breast cancer model system, treatment with 5-aza-deoxycytidine, a DNA methyltransferase inhibitor, leads to the release of methyl-CpG-binding protein complexes, and ERα mRNA is reexpressed (25).
Not only is methylation of DNA implicated in pathological processes such as breast cancer, but it is also critical for proper development in all organisms (29). Recently, methylation of the rat ERα promoter was shown to be important for regulating ERα gene expression in the brain (30). ERα promoter was differently methylated depending on maternal care in early life (30). Methylation of DNA involves recruitment of several families of proteins including the DNA methyltransferases (DNMTs) and the methyl-CpG-binding domain proteins (MBDs). The DNMTs add a methyl group to cytosines of CpG residues, and the MBDs are recruited to the methylated DNA to maintain methylation (31,32). Recruitment of these proteins to the 5′ untranslated region of a gene results in gene silencing. MBDs such as methyl-CpG-binding protein 2 (MeCP2) have been shown to play a crucial role in neural development (32). For example, a mutation in MeCP2 is believed to be associated with the neurological disorder Retts syndrome (32). Here we investigated the role of MeCP2 in normal, nonpathological regulation of ERα gene expression across development.
The importance of methylation in neural development along with recent studies showing epigenetic regulation of ERα in peripheral mammalian tissues suggest that methylation of the ERα promoter may be a potential mechanism for the regulation of ERα mRNA expression in the developing mouse cortex.
Materials and Methods
Animals
The animal care and use committee of the University of Kentucky approved all experimental procedures. C57BL/6 mice were bred at the University of Kentucky and maintained in our own colony. For some studies, B6.129S-Mecp2tm1Hzo/J MeCP2 mutant mice were purchased from The Jackson Laboratory (Bar Harbor, ME) (005439). All animals were maintained in constant temperature conditions on a 12-h light, 12-h dark cycle and were provided food and water ad libitum.
Quantitative real-time PCR
To examine gene expression, we isolated and collected the isocortex from male and female mice killed at several developmental time points from postnatal day (PND) 4 to PND 25 with PND 0 considered the day of birth (n = 6–8 per time point). The brain region collected included tissue from approximately Bregma 1.2 to −0.38 mm and was dissected from the nucleus accumbens, dorsal to the corpus callosum and lateral to the beginning of the hippocampal formation. Fifty to 100 mg tissue from this region was isolated and homogenized in 1 ml Trizol reagent (Invitrogen, Carlsbad, CA). The resulting RNA pellet was briefly air dried and suspended in 50 μl ribonuclease (RNase)-free water. The suspended RNA was incubated at 56 C for 10 min and stored at −80 C until reverse transcription. Diethylpyrocarbonate-treated H2O was added to bring 1 μg total RNA for each sample up to a final volume of 10 μl. One microliter of random primers (Invitrogen) and 1 μl 10 mm dNTPs were added to each reaction. The samples were incubated at 65 C for 5 min and then chilled on ice. Four microliters of 5× first-strand buffer, 2 μl 0.1 m dithiothreitol, 1 μl RNasin, and 1 μl Superscript reverse transcriptase (Invitrogen) was then added to each sample. Samples were then incubated at room temperature for 10 min, 42 C for 50 min, and 70 C for 15 min. For real-time PCR, each reaction contained 21.25 μl diethylpyrocarbonate-treated H2O, 25 μl 2× SYBR Green Brilliant Master Mix (Stratagene, La Jolla, CA), 1 μl upstream primer at a concentration of 250 nm, 1 μl downstream primer at a concentration of 50 nm, 0.75 μl reference dye (Stratagene; diluted 1:500), and 1 μl appropriate cDNA. Primer concentrations were previously optimized for each gene and result in a single DNA PCR product with no primer-dimer formation (22). Each 96-well plate contained a nontemplate control, and each sample was run in triplicate. Cycling parameters were as follows: one cycle at 95 C for 10 min, 40 cycles of 95 C for 30 sec, annealing temperature for 1 min, 72 C for 30 sec, and one cycle of 95 C for 1 min and 55 C for 30 sec. Real-time fluorescent measurements were taken at every cycle, and change in threshold cycle (ΔCt) was calculated. The ΔCt for each sample was normalized to the earliest developmental time point. All data were normalized to the housekeeping gene histone 3.1. The previously prescribed primers used for ERα and histone 3.1 were used (20). Primers for DNMT primers DNMT1 and DNMT3A have also been previously described (33).
Western blot
To determine levels of protein expression, total cell lysates were obtained from 50–100 mg isolated tissue from each side of the cortex as isolated for gene expression analysis (n = 6 per time point). Tissue was combined and homogenized in lysis buffer containing PBS, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 50 mm sodium fluoride, and 0.5% deoxycholate with Complete protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Cellular debris was separated by centrifugation at 13,000 × g for 30 min at 4 C. Total protein (20 μg) was separated by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane. Membrane was washed in PBS and then blocked with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) for 1 h at room temperature. After blocking, primary antibody was added, and the membrane was incubated overnight at 4 C. Membranes were washed in PBS plus Tween 20 and then incubated in Odyssey secondary antibody (LI-COR; 700dx, 1:1000) for 1 h at room temperature. Membranes were then dried overnight and developed using Odessey software.
Methylation-specific PCR
For methylation-specific PCR, genomic DNA was extracted from isocortex tissue from male and female mice on PND 4, 10, 18, and 25 (n = 6–8 per time point) using previously described methods (34). Briefly, 20–50 mg tissue was homogenized in 250 μl lysis solution [10 mm Tris-HCl (pH 8.0), 150 mm EDTA, 1% SDS, and 100 g/ml proteinase K] and incubated for 20 min at 55 C; 100 μl RNase solution [10 mm Tris-HCl (pH 8.0), 1 mm EDTA (TE), and 30 U RNase] was added; and the mixture was incubated at 37 C for 1 h. Two rounds of phenol-chloroform extraction were performed and 150 μl 1.0 m sodium acetate (pH 7.0) and 2 vol 100% ethanol was added to separated supernatant, and ethanol precipitation was performed. Sodium bisulfite modification was conducted on 500 ng genomic DNA using the EZ Methylation Gold kit (Zymo Research, Orange, CA).
For methylation-specific PCR, the sodium bisulfite-modified DNA was amplified in a 25-μl reaction. Each PCR contained 1× PCR Buffer (Invitrogen), 1.2 mm MgCl2 (Invitrogen), 0.1 mm dNTPs, 2 pm forward primer, 2 pm reverse primer, and 2.5 U Platinum Taq polymerase (Invitrogen). The cycling conditions were 95 C for 2 min and 35 cycles of 95 C for 30 sec, appropriate annealing temperature for 30 sec, and 72 C for 30 sec, followed by 72 C for 10 min. The primers used for each promoter were designed using Methprimer (http://www-genome.wi.mit.edu/genomesoftware/other/primer3.html) and are shown in supplemental Table 1 (published as supplemental data on The Endocrine Society’s Journals Online web site at http://endo. endojournals.org).
Analysis of DNA methylation of ERα
We examined the methylation status of specific CpG dinucleotides within the promoter region of mouse ERα. Genomic DNA was extracted from the isocortex of female mice at PND 4, 18, and 25 (n = 6–8), as described above. Pyrosequencing was performed by EpigenDx. Briefly, sodium bisulfite modification was conducted on the genomic DNA (Zymo Research EZ Methylation Gold kit), and 1 μg DNA was used for bisulfite modification followed by the PCR. PCR was performed using HotStar Taq polymerase (QIAGEN, Valencia, CA; catalog no. 203205). Each reaction contained the following: 1× PCR buffer, 3 mm MgCl2, 200 μm of each dNTP, 6 pmol forward primer, 6 pmol reverse primer, 0.75 U HotStar Taq polymerase, 1 μl bisulfite-treated DNA, and water to adjust to proper volume. PCR conditions were as follows: 95 C for 15 min; 30 cycles of 95 C for 30 sec, annealing temperature for 30 sec, and 72 C for 30 sec; and 72 C for 5 min. The annealing temperature for exon A is 60 C and for exon C is 57 C. The primers used for exons A and C were forward 5′-TGGGTTATTTGTGTTTTGTAGGATAG-3′ and reverse 5′-CTTAAATCTAATACAACAAAACCATTC-3′. The primers used to amplify exon C were forward 5′-TGTTAAGTGTTTTGTTTATTGGTTG-3′ and reverse 5′-CCTCTTTCCAAAAATATTCCATAAATT-3′. Universally methylated and unmethylated DNA samples (Millipore Bioscience Research Reagents, Billerica, MA) were used as controls in these experiments.
Chromatin immunoprecipitation (ChIP)
For ChIP assays, we used the Upstate Biotechnologies (Temecula, CA) ChIP assay kit (catalog item 17-295). Briefly, cortex was dissected and minced with a clean razor blade. Proteins were cross-linked in DMEM plus 1% formaldehyde at 37 C. Cells were then lysed in cold lysis buffer containing 1% SDS, 10 mm EDTA, 50 mm Tris (pH 8.1). This lysis buffer also included Roche Complete Mini protease inhibitor cocktail, which contains several protease inhibitors with broad specificity. These inhibitors work to inhibit serine, cysteine, and metalloproteases (Roche; catalog item 11836153001). One tablet was added for every 10 ml buffer. Cross-linked DNA was sheared to approximately 400–500 bp with three sets of 5-sec pulses using a Fisher Scientific (Pittsburgh, PA) Ultrasonic Dismembrator (model D100) sonicator with 5-mm tip and set to 30% maximum power. Sonicated cell supernatant was diluted 10-fold in ChIP dilution buffer [0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl (pH 8.1), 167 mm NaCl] with Roche Complete Mini protease inhibitor cocktail. A sample was removed after dilution and called the input sample. The remaining diluted cell supernatant was precleared with salmon sperm DNA/protein A agarose 50% slurry. At this point, another sample was removed and used for the no-antibody control. Primary MeCP2 antibody (Upstate Biotechnology, Lake Placid, NY; 05−764, 1:1000) was added to the remaining precleared supernatant and incubated overnight. Agarose was pelleted by centrifugation to separate the beads with bound chromatin from the supernatant containing unbound chromatin. The protein A/antibody/chromatin complex was washed with low-salt immune complex wash buffer [0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.1), 150 mm NaCl] and high-salt immune complex wash buffer [0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.1), 500 mm NaCl] followed by LiCl complex wash buffer (0.25 m LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid), 1 mm EDTA, 10 mm Tris (pH 8.1), and TE buffer (10 mm Tris-HCl, 1 mm EDTA). The protein A agarose/antibody/chromatin complex was eluted in elution buffer (1% SDS, 0.1 m NaHCO3). Cross-links were reversed by adding 20 μl 5 m NaCl to eluates and heating at 65 C for 4 h. DNA was recovered by phenol/chloroform extraction and ethanol precipitation. Standard PCR with 30 cycles was performed on the resulting sample as well as on the input incubated samples that did not undergo any preclearing or immunoprecipitation. The resulting sample was also immunoprecipitated with a nonimmune antibody. The cycling conditions were 95 C for 2 min and 30 cycles of 95 C for 30 sec, 60 C for 30 sec, 72 C for 30 sec, followed by 72 C for 10 min. The primers for ERα promoters A and C are shown in supplemental Table 1.
Statistical analysis
All data were analyzed using a two-way ANOVA comparing postnatal day and position of CG pair for each exon and for each gender. Significant differences within a group were determined by Fisher post hoc tests. An additional three-way repeated-measures ANOVA was also used to compare genotype, postnatal age, and base pair position for each promoter. All differences were considered significant at P < 0.05.
Results
Methylation of ERα exons A and C increases across early postnatal development in both the male and female mouse cerebral cortex
This study was designed to test the hypothesis that changes in ERα mRNA expression across development correspond with changes in the methylation status of ERα 5′ untranslated exons (Fig. 1A). Male and female mice were killed at PND 4, 10, 18, and 25. The cortex was dissected away from the whole brain, and genomic DNA was isolated and treated with sodium bisulfite. The resulting DNA was used for methylation-specific PCR using primers designed to detect the methylation status of ERα 5′ untranslated exons A and C (supplemental Table 1). Methylation of exons A and C was detectable at PND 10 in both males and females, but it appeared to be greater for exon C (Fig. 1B). This time point corresponds to the decline in ERα mRNA previously described (20) and to the decrease in ERα protein shown here in supplemental Fig. 1. Primers designed to recognize unmethylated regions amplified DNA only at PND 4 for both exons A and C in male and female mice. The methylation status of the other promoters was not examined because they either are not expressed in the brain or did not contain predicted CpG islands. These changes in methylation of the ERα promoters across development were further investigated using pyrosequencing.
Figure 1.

Methylation status of ERα upstream-untranslated exons changes during development. A, Schematic of known ERα upstream-untranslated exons (A). All exons are spliced to a common splice site just before the translational start site and yield the same ERα gene product. B, There was an increase in methylation of mouse ERα exons A and C across early postnatal development. Genomic DNA was extracted from female and male mouse cortex at PND 4, 10, 18, and 25. DNA was treated with sodium bisulfite and amplified using primers specific for methylated or unmethylated regions of promoter A and promoter C. There was differential methylation of both promoters across early postnatal development, but no gender difference across postnatal days. Methylation was increased starting at PND 10. Experiments were repeated at least three times, and samples were analyzed using control unmethylated β-actin primers.
Analysis of DNA methylation of ERα gene across development
Our previous studies have shown that changes in ERα gene expression are associated with an increase in DNA methylation. To further define potential sites of methylation and regulation, we performed pyrosequencing (Fig. 2). All pyrosequencing data were analyzed using a two-way ANOVA comparing postnatal day and position of CG pair. In females, cytosine methylation analysis within promoter A of the ERα gene revealed a main effect of postnatal time point [F(2,154) = 45.844; P < 0.0001] (Fig. 2A). Fisher’s post hoc tests revealed that cortex from both PND 18 (P < 0.0009) and PND 25 (P = 0.0001) had significantly more methylation than PND 4 and that PND 25 had significantly more methylation than PND 18 (P < 0.0001) (Fig. 2A). In males, analysis within promoter A revealed a main effect of postnatal time point [F(2,99) = 267.316; P < 0.0001] (Fig. 2B). Fisher’s post hoc tests revealed significantly more methylation at PND 18 and 25 than PND 4 (P < 0.0001) (Fig. 2B). There was also a main effect of CG position [F(10,99) = 2.711; P < 0.005] and a significant interaction [F(20,99) = 89.677; P < 0.0001] (Fig. 2B).
Figure 2.

Pyrosequencing analysis of genomic DNA from female and male mouse cortex collected at PND 4, 18, and 25 (n = 6–8 per time point). A and B, Percent methylation of CG sites found in exon A in female (A) and male (B) cortex; C and D, percent methylation of CG sites found in exon C of the ERα gene in female (C) and male (D) cortex. Asterisks indicate significant differences from PND 4 determined by Tukey post hoc tests after a two-way ANOVA (P < 0.05).
A similar pattern was observed with promoter C. Analysis within promoter C of the ERα gene in females revealed a main effect of postnatal time point [F(2,60) = 15.414; P < 0.0001] (Fig. 2C). Fisher’s post hoc tests revealed that cortex from both PND 18 (P < 0.0001) and PND 25 (P = 0.0001) had significantly more methylation than PND 4 (Fig. 2C). In males, cytosine methylation analysis within promoter C revealed a main effect of postnatal time point [F(2,45) = 56.394; P < 0.0001] (Fig. 2D). Fisher’s post hoc tests revealed that PND 4 was significantly less than PND 18 and 25 (P < 0.0001) (Fig. 2D). There was also a main effect of CG position [F(4,45) = 5.172; P = 5.172] and an interaction between postnatal day CG position [F (8,45) = 4.578; P < 0.0004] (Fig. 2D). These data indicate an overall increase in methylation of CG regions of both promoters that occurs with increasing age in both males and females.
Expression of DNMT3A and DNMT1 mRNA changes across early postnatal development in the male and female mouse cortex
DNMTs are involved in the methylation of cytosine residues. DNMT3A is the de novo methyltransferase responsible for initial methylation, whereas DNMT1 is responsible for maintenance of methylation status (35,36,37,38). We investigated both DNMT3A and DNMT1 mRNA expression in the cortex using real-time PCR. RNA was isolated from male and female mice at PND 1, 4, 10, 18, and 25. DNMT3A mRNA levels were significantly increased at PND 10 in male mice [F(4,14) = 32.95; P < 0.001] (Fig. 3B). Females also demonstrated a 4-fold increase in DNMT3A mRNA [F(4,14) = 11.19; P = 0.002] (Fig. 3A). There was no statistically significant effect of gender (P = 0.084). DNMT1 mRNA levels were significantly increased beginning at PND 10 and continued to increase at all time points that we investigated in both females [F(4,14) = 34.49; P < 0.001] (Fig. 3C) and males [F(4,14) =62.00; P < 0.001] (Fig. 3D), and there was no significant effect of gender (P = 0.250). These data suggest that changes in DNMT mRNA expression occur at the same time that ERα mRNA expression decreases and promoter methylation increases across early postnatal development.
Figure 3.

DNMT mRNA expression in female and male mouse cortex across early postnatal development. RNA was extracted from cortical tissue taken from female and male mice at PND 1, 4, 10, 18, and 25. RNA was reverse transcribed, and the resulting cDNA was used for real-time PCR using DNMT3A- or DNMT1-specific primers. A, DNMT3A mRNA expression was significantly increased in female cortex at PND 10 and 18. B, DNMT3A mRNA was also significantly increased in male cortex at PND 10 and 18. This increase in DNMT3A was only transient, and expression was no longer significantly increased at PND 25 in either female or male cortex. This increase is correlated with the increase in ERα mRNA expression in the cortex at PND 10. C, In female cortex, DNMT1 mRNA was significantly increased at PND 10, 18, and 25. D, In male cortex, DNMT1 mRNA was significantly increased at PND 18 and 25. Asterisks on the graphs indicate significant differences from PND1 (P < 0.05).
MeCP2 is associated with the ERα promoter in the cortex after PND 10
Methyl binding proteins also play an important role in regulation of gene expression by stabilizing methylation of CpG sites (33). Because MeCP2 has been shown to be involved in the regulation of ERα mRNA expression in other tissues (25), we investigated the direct interaction of MeCP2 with ERα promoters in the cortex. Cortical tissue was taken from female mice at PND 4, 10, and 18 and used for a ChIP assay with an anti-MeCP2 antibody. MeCP2 was associated the ERα promoters A and C at PND 10 and 18. MeCP2 was not associated at the earlier time point PND 4 when ERα mRNA expression was elevated (Fig. 4). For each condition, we performed PCR with ERα 5′ untranslated exon A and C primers, the input, and the specific ChIP product. The experiments were repeated three times. The input sample was compared to demonstrate equal starting sample quantities. The correlation between MeCP2 associations with the ERα upstream-untranslated exon A and C after PND 10 suggests that methylation and recruitment of MeCP2 could account for the changes in the ERα gene expression after early postnatal development.
Figure 4.
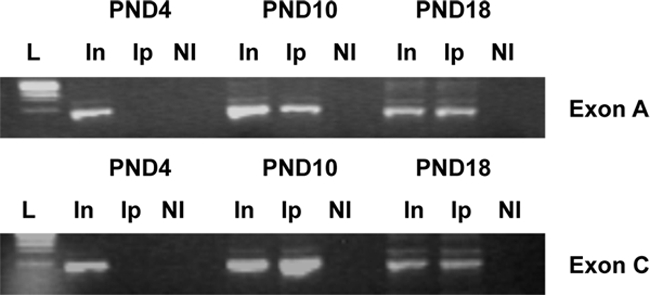
To determine whether MeCP2 was associated with ERα promoters, we conducted a ChIP assay. Cortical tissue was taken from female mouse pups at PND 4, 10, and 18. Nuclear proteins were then cross-linked, and sheared chromatin was immunoprecipitated with MeCP2 antibody (Ip). Recovered chromatin was amplified by standard PCR using primers spanning regions of exon A or exon C. Nonimmune IgG was used in all ChIP reactions as a control. An ethidium bromide-stained gel of PCR products showed a representative of ChIP analysis. In, Input; NI, nonimmune IgG antibody.
ERα mRNA expression in the cortex is altered in MeCP2 mutant mice across development
To investigate the influence of MeCP2 on ERα mRNA expression across development, we collected tissue at several postnatal time points for real-time PCR from the cortex of MeCP2 mice with defective MeCP2 protein (Fig. 5A). There was a significant overall effect of genotype [F(1,16) = 24.426; P = 0.0001] and of postnatal day [F(3,16) = 6.787; P = 0.0037]. There was also a significant interaction [F(3,16) = 9.688; P = 0.0007] such that the differences in ERα mRNA expression between wild type and MeCP2 mutant were dependent on the postnatal day. The wild-type mice showed the expected pattern where ERα mRNA expression was significantly greater at PND 4 than PND 18 (P = 0.0002) and PND 25 (P = 0.0018). ERα mRNA expression in PND 10 mice was also significantly greater than PND 18 (P = 0.002) and PND 25 (P = 0.0017). The MeCP2 mutant mice had a significant increase in ERα mRNA expression at PND 18 (P = 0.0033) and PND 25 (P = 0.0026) compared with PND 4. This increase was also seen at the protein level (Fig. 5B). These data are consistent with the idea that MeCP2 and methylation regulate the decrease in ERα mRNA expression that occurs as part of normal postnatal development. These data also confirm the hypothesis that disruption of MeCP2 produces a different pattern of ERα expression across development.
Figure 5.

Mutations in MeCP2 change the developmental pattern of ERα mRNA and protein expression in the cortex. A, ERα mRNA expression in MeCP2 mutant females was significantly increased across early postnatal development. In contrast, in wild-type females, there was a significant decrease in ERα mRNA expression starting at PND 10. B, Western blot showing ERα protein expression in wild-type and MeCP2 mutant mice. C, Pyrosequencing analysis of genomic DNA from MeCP2 mutant female mice collected at PND 4, 18, and 25. C, Percent methylation of CG sites found in exon A. D, Percent methylation of CG sites found in exon C of the ERα gene. Asterisks indicate significant differences from PND 4 determined by Tukey post hoc tests after a two-way ANOVA (P < 0.05).
We also investigated the degree of cytosine methylation in MeCP2 mutant mice to determine the pattern of methylation across development in the absence of functional MeCP2. In MeCP2 mutant mice, cytosine methylation analysis within exon A of the ERα gene revealed no main effect of postnatal time point (Fig. 5C). A similar analysis of cytosine methylation within exon C of the ERα gene revealed a main effect of postnatal time point [F(2,60) = 7.143; P = 0.0016] and a main effect of CG position [F(8,60) = 3.210; P = 0.0187] but no interaction (Fig. 5C). Fisher’s post hoc tests revealed that cortex from PND 25 had significantly more methylation than PND 4 (P = 0.0228) and PND 18 (P < 0.0001) (Fig. 5D). These data indicate a different methylation pattern in both promoters than the pattern seen in wild-type cortex.
To investigate the difference between methylation in wild-type and MeCP2 mutant mice, we conducted an additional three-way ANOVA comparing genotype, postnatal age, and base pair position for each promoter. For exon A, there was an overall main effect of postnatal day [F(2,120) = 13.348; P < 0.0001] and genotype [F(1,120) = 28.739; P < 0.0001]. There was also an interaction between postnatal day and genotype [F(2,120) = 14.014; P < 0.0001]. This interaction is important because it suggests that the significant differences between postnatal day are dependent on the genotype of the animal. For exon C, there was an overall main effect of postnatal day [F(2,120) = 13.348; P < 0.0001] and genotype [F(1,120) = 28.739; P < 0.0001]. There was also an interaction between postnatal day and genotype [F(2,120) = 14.014; P < 0.0001]. These data indicate a clear role for MeCP2 in regulating ERα expression in the cortex during early postnatal development.
Discussion
ERα expression in the rodent cortex decreases across early postnatal development (20) and increases after ischemic injury (23). Our previous studies indicate that the increase in ERα mRNA expression after injury is due to a decrease in methylation of an ERα upstream exon (23). In the present study, we found that ERα 5′ exons A and C were methylated at PND 10, the developmental time point when there is a significant decrease in ERα mRNA expression (20). We found corresponding changes in methylation-specific enzymes such as the DNMTs during this time period. Our final set of experiments demonstrates a direct role for the methyl-CpG binding protein MeCP2 in the regulation of ERα gene expression. To our knowledge, these data are the first to demonstrate a developmental pattern of ERα methylation and DNMT expression in the cerebral cortex.
Epigenetic regulation of nuclear receptors has been thoroughly studied in cancer biology (27), but the potential role of epigenetic modulation of the ERα 5′ untranslated exons during brain development has not been studied. Other labs have demonstrated a difference in methylation of ERα promoter in another brain region, the medial preoptic area, in adult rats depending on maternal care early in life (30). Here, we found that in the cortex, ERα 5′ exons A and C were methylated at developmental time points corresponding with a significant decrease in ERα mRNA expression in both male and female mice (20). Further analysis of methylation by pyrosequencing also revealed an increase in methylation of most CG loci across development. These data suggest that broad changes in methylation of ERα exons are responsible for the silencing of ERα gene expression in the cortex after PND 10. Changes in methylation are associated with increased DNMT mRNA expression (39,40).
Here we measured DNMT mRNA expression in both male and female mouse cortex across development. These DNMTs are important enzymes that add a methyl group (CH3) to cytosine residues in the CpG dinucleotides sequence (41,42). This reaction is accomplished by DNMTs, such as the developmentally regulated DNMT3A (43,44,45). As expected, DNMT3A, the de novo or initiation enzyme, peaked at PND 10 and returned to a lower level by PND 25. PND 10 is also the developmental time point in which methylation of ERα upstream-untranslated exons was detected and when ERα mRNA expression drastically decreases (20). DNMT1 is responsible for maintaining methylation status of methylated CpG loci (37,38). Interestingly, DNMT1 mRNA expression increased across development and stayed high even at the latest time point that we measured. These data demonstrate that the methylation machinery required for gene silencing is expressed in the appropriate spatial and temporal pattern in the cortex across postnatal development.
The most direct mechanism by which DNA methylation can obstruct transcription is to prevent the binding of the basal transcriptional machinery (46). Methylation of CpG residues impedes or abolishes binding by these factors. A complex of methyl-CpG-binding proteins assembles on the DNA blocking transcriptional activation of the promoter. The methyl-CpG-binding protein family consists of five members, including MBD1, -2, -3, and -4 and MeCP2. MBD2 and MeCP2 have the ability to bind the promoter of the ERα gene (25,47). Because of MeCP2’s role in neural development, its ability to bind to the ERα untranslated exons was examined. We determined by using ChIP that MeCP2 is associated with ERα 5′ untranslated exon C at PND 10 and beyond, which correspond with the time that ERα mRNA is drastically decreased and methylation of the untranslated exons is first detected. Furthermore, in MeCP2 mutant mice, developmental ERα regulation and promoter methylation is disrupted. It remains possible, however, that other methyl-CpG-binding proteins are involved in the regulation of ERα in addition to MeCP2 and that the loss of MeCP2 alters DNA methylation and the activities of other methyl-CpG-binding proteins. These data do suggest that MeCP2 plays a crucial role in regulation of ERα mRNA expression in the developing mouse cortex.
Although E2 is responsible for generating gender differences in a variety of brain functions, it has been demonstrated that there are no sex differences in ERα mRNA expression in the isocortex across development (20). Similarly, we found no gender differences between DNMT expression or methylation status of ERα 5′ untranslated exons in the current study. Other labs have shown developmental sex differences in MeCP2 mRNA expression in parts of the brain that exhibit sex differences (48). Although we did not measure MeCP2 mRNA specifically across development, we did look at association of MeCP2 with ERα promoter in both male and female mice (supplemental Fig. 2). There were no gender differences in the association of MeCP2 with ERα promoters (supplemental Fig. 2). These observations suggest that it is the availability of the ligand and not differences in receptor expression that is responsible for gender differences seen in cognitive-like behaviors and cortical morphology (49,50).
The current study establishes that methylation of ERα promoters in conjunction with methyl-CpG-island-binding proteins play a crucial role in regulating the developmental decrease in ERα mRNA. Understanding methylation of the ERα promoters will allow for the elucidation of the mechanisms that regulate ERα mRNA expression in the cortex. This not only is important to the understanding of basic developmental neurobiology but also has potential future applications in understanding the role of ERα in receptor-mediated neuroprotective actions of estrogens. This series of studies appear to support the general hypothesis that neurons revert back to an early developmental stage of gene expression after a neuronal insult or injury.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health National Center for Research Resources Grant P20 RR 15592 and National Science Foundation IOS-0919944.
Disclosure Summary: The authors have nothing to disclose.
First Published Online December 4, 2009
Abbreviations: ChIP, Chromatin immunoprecipitation; DNMT, DNA methyltransferase; E2, 17β-estradiol; ER, estrogen receptor; MBD, methyl-CpG-binding domain protein; MeCP2, methyl-CpG-binding protein 2; PND, postnatal day; RNase, ribonuclease; SDS, sodium dodecyl sulfate.
References
- Boulware MI, Mermelstein PG 2005 The influence of estradiol on nervous system function. Drug News Perspect 18:631–637 [DOI] [PubMed] [Google Scholar]
- Li L, Fan X, Warner M, Xu XJ, Gustafsson JK, Wiesenfeld-Hallin Z 2009 Ablation of estrogen receptor α or β eliminates sex differences in mechanical pain threshold in normal and inflamed mice. Pain 143:37–40 [DOI] [PubMed] [Google Scholar]
- Sanoja R, Cervero F 22 July 2009 Estrogen-dependent changes in visceral afferent sensitivity. Auton Neurosci 10.1016/j.autneu.2009.07.001 [DOI] [PubMed] [Google Scholar]
- Hill RA, Boon WC 2009 Estrogens, brain, and behavior: lessons from knockout mouse models. Semin Reprod Med 27:218–228 [DOI] [PubMed] [Google Scholar]
- Söderström I, Strand M, Ingridsson AC, Nasic S, Olsson T 2009 17β-Estradiol and enriched environment accelerate cognitive recovery after focal brain ischemia. Eur J Neurosci 29:1215–1224 [DOI] [PubMed] [Google Scholar]
- Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH, Simpkins JW 2008 Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci USA 105:15148–15153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM 1998 Estradiol protects against ischemic injury. J Cereb Blood Flow Metab 18:1253–1258 [DOI] [PubMed] [Google Scholar]
- Rusa R, Alkayed NJ, Crain BJ, Traystman RJ, Kimes AS, London ED, Klaus JA, Hurn PD 1999 17β-Estradiol reduces stroke injury in estrogen-deficient female animals. Stroke 30:1665–1670 [DOI] [PubMed] [Google Scholar]
- Chung WC, Swaab DF, De Vries GJ 2000 Apoptosis during sexual differentiation of the bed nucleus of the stria terminalis in the rat brain. J Neurobiol 43:234–243 [PubMed] [Google Scholar]
- Forger NG 2006 Cell death and sexual differentiation of the nervous system. Neuroscience 138:929–938 [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD 1976 Golgi-Cox modifications for the impregnation of whole mount preparations of organotypic cultures of the CNS. Brain Res 118:293–298 [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD 1976 Sex steroids and the development of the newborn mouse hypothalamus and preoptic area in vitro: implications for sexual differentiation. Brain Res 106:407–412 [DOI] [PubMed] [Google Scholar]
- Mong JA, Roberts RC, Kelly JJ, McCarthy MM 2001 Gonadal steroids reduce the density of axospinous synapses in the developing rat arcuate nucleus: an electron microscopy analysis. J Comp Neurol 432:259–267 [DOI] [PubMed] [Google Scholar]
- De Vries GJ, Rissman EF, Simerly RB, Yang LY, Scordalakes EM, Auger CJ, Swain A, Lovell-Badge R, Burgoyne PS, Arnold AP 2002 A model system for study of sex chromosome effects on sexually dimorphic neural and behavioral traits. J Neurosci 22:9005–9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simerly RB 2002 Wired for reproduction: organization and development of sexually dimorphic circuits in the mammalian forebrain. Annu Rev Neurosci 25:507–536 [DOI] [PubMed] [Google Scholar]
- Welboren WJ, Sweep FC, Span P, Stunnenberg H 23 July 2009 Genomic actions of estrogen receptor α: what are the targets and how are they regulated? Endocr Relat Cancer 10.1677/ERC-09-0086 [DOI] [PubMed] [Google Scholar]
- Tsai MJ, O'Malley BW 1994 Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 63:451–486 [DOI] [PubMed] [Google Scholar]
- Stumpf WE, Sar M 1976 Steroid hormone target sites in the brain: the differential distribution of estrogin, progestin, androgen and glucocorticosteroid. J Steroid Biochem 7:1163–1170 [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Stumpf WE, MacLusky NJ, Zielinski JE, Hochberg RB 1990 Developmental changes in estrogen receptors in mouse cerebral cortex between birth and postweaning: studied by autoradiography with 11β-methoxy-16α-[125I]iodoestradiol. Endocrinology 126:1112–1124 [DOI] [PubMed] [Google Scholar]
- Prewitt AK, Wilson ME 2007 Changes in estrogen receptor-α mRNA in the mouse cortex during development. Brain Res 1134:62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda RC, Toran-Allerand CD 1992 Developmental expression of estrogen receptor mRNA in the rat cerebral cortex: a nonisotopic in situ hybridization histochemistry study. Cereb Cortex 2:1–15 [DOI] [PubMed] [Google Scholar]
- Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM 1999 Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci 19:6385–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westberry JM, Prewitt AK, Wilson ME 2008 Epigenetic regulation of the estrogen receptor α promoter in the cerebral cortex following ischemia in male and female rats. Neuroscience 152:982–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Rau SW, Shughrue PJ, Zhu H, Yu J, Cashion AB, Suzuki S, Gerhold LM, Bottner MB, Dubal SB, Merchanthaler I, Kindy MS, Wise PM 2006 Differential modulation of estrogen receptors (ERs) in ischemic brain injury: a role for ERα in estradiol-mediated protection against delayed cell death. Endocrinology 147:3076–3084 [DOI] [PubMed] [Google Scholar]
- Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE 2005 Release of methyl CpG binding proteins and histone deacetylase 1 from the Estrogen receptor α (ER) promoter upon reactivation in ER-negative human breast cancer cells. Mol Endocrinol 19:1740–1751 [DOI] [PubMed] [Google Scholar]
- Hayashi SI, Eguchi H, Tanimoto K, Yoshida T, Omoto Y, Inoue A, Yoshida N, Yamaguchi Y 2003 The expression and function of estrogen receptor α and β in human breast cancer and its clinical application. Endocr Relat Cancer 10:193–202 [DOI] [PubMed] [Google Scholar]
- Lapidus RG, Nass SJ, Butash KA, Parl FF, Weitzman SA, Graff JG, Herman JG, Davidson NE 1998 Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer Res 58:2515–2519 [PubMed] [Google Scholar]
- Alvarez S, Diaz-Uriarte R, Osorio A, Barroso A, Melchor L, Paz MF, Honrado E, Rodríguez R, Urioste M, Valle L, Díez O, Cigudosa JC, Dopazo J, Esteller M, Benitez J 2005 A predictor based on the somatic genomic changes of the BRCA1/BRCA2 breast cancer tumors identifies the non-BRCA1/BRCA2 tumors with BRCA1 promoter hypermethylation. Clin Cancer Res 11:1146–1153 [PubMed] [Google Scholar]
- Jaenisch R 1997 DNA methylation and imprinting: why bother? Trends Genet 13:323–329 [DOI] [PubMed] [Google Scholar]
- Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ 2006 Maternal care associated with methylation of the estrogen receptor-α1b promoter and estrogen receptor-α expression in the medial preoptic area of female offspring. Endocrinology 147:2909–2915 [DOI] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H 2002 Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35:243–254 [DOI] [PubMed] [Google Scholar]
- Nan X, Bird A 2001 The biological functions of the methyl-CpG-binding protein MeCP2 and its implication in Rett syndrome. Brain Dev 23(Suppl 1):S32–S37 [DOI] [PubMed] [Google Scholar]
- Park IY, Sohn BH, Choo JH, Joe CO, Seong JK, Lee YI, Chung JH 2005 Deregulation of DNA methyltransferases and loss of parental methylation at the insulin-like growth factor II (Igf2)/H19 loci in p53 knockout mice prior to tumor development. J Cell Biochem 94:585–596 [DOI] [PubMed] [Google Scholar]
- Nishino K, Hattori N, Tanaka S, Shiota K 2004 DNA methylation-mediated control of Sry gene expression in mouse gonadal development. J Biol Chem 279:22306–22313 [DOI] [PubMed] [Google Scholar]
- Yokochi T, Robertson KD 2002 Preferential methylation of unmethylated DNA by mammalian de novo DNA methyltransferase Dnmt3a. J Biol Chem 277:11735–11745 [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E 1999 DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257 [DOI] [PubMed] [Google Scholar]
- Ting AH, Jair KW, Suzuki H, Yen RW, Baylin SB, Schuebel KE 2004 Mammalian DNA methyltransferase 1: inspiration for new directions. Cell Cycle 3:1024–1026 [PubMed] [Google Scholar]
- Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, MacLeod AR 2003 DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet 33:61–65 [DOI] [PubMed] [Google Scholar]
- Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, Sasaki H 2001 Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 97:1172–1179 [DOI] [PubMed] [Google Scholar]
- Kanai Y, Ushijima S, Kondo Y, Nakanishi Y, Hirohashi S 2001 DNA methyltransferase expression and DNA methylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int J Cancer 91:205–212 [DOI] [PubMed] [Google Scholar]
- Dunn BK 2003 Hypomethylation: one side of a larger picture. Ann NY Acad Sci 983:28–42 [DOI] [PubMed] [Google Scholar]
- Bird A 2002 DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21 [DOI] [PubMed] [Google Scholar]
- Xie S, Wang Z, Okano M, Nogami M, Li Y, He WW, Okumura K, Li E 1999 Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene 236:87–95 [DOI] [PubMed] [Google Scholar]
- Feng J, Chang H, Li E, Fan G 2005 Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J Neurosci Res 79:734–746 [DOI] [PubMed] [Google Scholar]
- Semeralul MO, Boutros PC, Likhodi O, Okey AB, Van Tol HH, Wong AH 2006 Microarray analysis of the developing cortex. J Neurobiol 66:1646–1658 [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP 1999 Methylation-induced repression: belts, braces, and chromatin. Cell 99:451–454 [DOI] [PubMed] [Google Scholar]
- Müller HM, Fiegl H, Goebel G, Hubalek MM, Widschwendter A, Müller-Holzner E, Marth C, Widschwendter M 2003 MeCP2 and MBD2 expression in human neoplastic and non-neoplastic breast tissue and its association with oestrogen receptor status. Br J Cancer 89:1934–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian JR, Forbes-Lorman RM, Auger AP 2007 Sex difference in mecp2 expression during a critical period of rat brain development. Epigenetics 2:173–178 [DOI] [PubMed] [Google Scholar]
- Juraska JM 1991 Sex differences in cognitive regions of the rat brain. Psychoneuroendocrinology 16:105–109 [DOI] [PubMed] [Google Scholar]
- Reid SN, Juraska JM 1992 Sex differences in the gross size of the rat neocortex. J Comp Neurol 321:442–447 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.