

Abstract
The proinflammatory cytokine leukemia inhibitory factor (LIF) is induced in disease states and is known to inhibit food intake when administered centrally. However, the neural pathways underlying this effect are not well understood. We demonstrate that LIF acutely inhibits food intake by directly activating pro-opiomelanocortin (POMC) neurons in the arcuate nucleus of the hypothalamus. We show that arcuate POMC neurons express the LIF-R, and that LIF stimulates the release of the anorexigenic peptide, α-MSH from ex vivo hypothalami. Transgenic mice lacking gp130, the signal transducing subunit of the LIF-R complex, specifically in POMC neurons fail to respond to LIF. Furthermore, LIF does not stimulate the release of α-MSH from the transgenic hypothalamic explants. These findings indicate that POMC neurons mediate the acute anorectic actions of central LIF administration and provide a mechanistic link between inflammation and food intake.
Activation of POMC neurons by LIF is sufficient to inhibit food intake; a direct link between inflammatory cytokine-induced anorexia and altered central melanocortin signaling is discussed.
Cachexia is a wasting syndrome common to the late stages of many chronic diseases, including HIV, heart failure, uremia, and cancer (1). Cachexia is marked by paradoxical responses to a starved state, including 1) anorexia, 2) increased basal metabolic rate, and 3) preferential loss of lean body mass. Indeed, the severity of this synergistic catabolic state is associated with poor clinical outcomes, increased mortality, and reduced quality of life (1,2). A growing body of evidence shows that increases in central inflammatory cytokine signaling are sufficient to cause anorexia and increased basal metabolic rate (3,4,5). Administration of the bacterial endotoxin lipopolysaccharide (LPS) induces an acute cachexia syndrome associated with increases in central and peripheral proinflammatory cytokines, including IL-1β, TNF-α, and leukemia inhibitory factor (LIF) (6,7,8,9,10,11,12). The finding that central administration of each of these cytokines individually recapitulates acute cachexia strongly suggests a site of cytokine action within the central nervous system.
An identified target for central cytokine action is the arcuate nucleus of the hypothalamus (ARC). The ARC is known to be a key regulator of energy homeostasis and a major site for the integration of metabolic signals (13,14). The ARC includes two populations of neuropeptide-expressing neurons with opposing actions on energy balance. One population expresses the anorexigenic peptide, α-MSH, a cleavage product of the pro-opiomelanocortin (POMC) precursor (15). POMC is also expressed by brain stem neurons of the solitary tract nucleus (NTS) as well as pituitary corticotrophs (15). ARC POMC neurons are opposed by adjacent neurons expressing the orexigenic neuropeptides agouti-related protein (AgRP) and neuropeptide Y (NPY) (16). α-MSH derives its anorectic effect via activation of the type-4 melanocortin receptor (17), whereas AgRP acts as an endogenous antagonist and inverse agonist at the same receptor (18). It should be noted that POMC processing also results in the production of β-endorphin, which is putatively coreleased with α-MSH from axon terminals (19). The role of β-endorphin in energy homeostasis remains controversial because exogenous administration acutely increases food intake, whereas endogenous release may act to inhibit food intake (20). Energy balance is tightly regulated by the relative activity of each of these neuronal populations, which, in turn, are responsive to numerous circulating signals of energy status. Importantly, the blood brain barrier in the ARC is relatively permissive, allowing the neurons access to circulating macromolecules (14). For example, recent work in our laboratory has shown that both POMC and NPY/AgRP neurons express the receptor for IL-1β (IL-1RI) and are oppositely regulated by this cytokine to promote negative energy balance (21,22). Previous studies indicate that the ARC may act as an inflammatory amplifier within the CNS, suggesting that neurons in the ARC are likely subject to much higher concentrations of proinflammatory cytokines than found in the circulation (23,24). Furthermore, this central inflammatory response is necessary for the induction of anorexia by LPS (25).
The proinflammatory cytokine LIF is an essential component in inflammatory signaling and neuroimmune function (26,27,28). Serum LIF is elevated in chronic disease and malignancy (29,30,31), an observation that has been correlated to poor prognosis (32). Hypothalamic expression of LIF is induced in animal models of acute inflammation (33,34). The potential role of LIF in cachexia is further exemplified by the finding that, in contrast to IL-1β or LPS-induced anorexia, animals do not desensitize to chronic LIF-induced anorexia in either LIF-overexpressing tumor models (35,36) or after intracerebroventricular (i.c.v.) administration of a LIF-expressing viral vector (37). This lack of tachyphylaxis more accurately represents clinical cachexia, indicating that LIF could be an essential CNS mediator of chronic inflammation.
The mechanism of LIF-mediated anorexia remains unknown. One possibility is that LIF, like IL-1β, alters melanocortin signaling in the ARC. LIF is a member of the IL-6 cytokine family, along with ciliary neurotrophic factor (CNTF), which share a common gp130 signal-transducing subunit (28). LIF derives target specificity via binding to a heterodimeric receptor, consisting of LIF-R and gp130 (38). Binding stimulates gp130-mediated activation of the Janus kinase 2/signal transducer and activator of transcription (STAT3) pathway (38), the same pathway activated by the binding of leptin to its receptor (39). LIF orchestrates the pituitary response to inflammation by inducing POMC gene expression and ACTH release in corticotrophs (40,41,42), an effect dependent on Janus kinase 2/STAT3 signaling and mediated by gp130 (40,43,44,45). Although pituitary and neuronal POMC expression are differentially regulated (46), it is possible that LIF may also directly influence ARC POMC expression.
In the present work, we explore the role of POMC neurons in the acute anorectic effects of LIF. We first examine the effects of central LIF on feeding behavior and neuronal activation. We further investigate the neuroanatomic framework and the molecular mechanism to show that LIF-induced anorexia is dependent upon functional expression of gp130 in POMC neurons. Our results also demonstrate that LIF acutely induces anorexia by direct activation of ARC POMC neurons.
Materials and Methods
Animals and surgical procedures
Male Sprague Dawley rats (300–350 g; Charles River Laboratories, Wilmington, MA), wild-type C57BL/6J mice (6–9 wk of age; Jackson Laboratory, Bar Harbor, ME), transgenic C57BL/6J POMC-enhanced green fluorescent protein (EGFP) mice [6–9 wk of age; genotyping and breeding of mice were as described previously (47)], and transgenic PomcCre-gp130flox/flox mice (6–9 wk of age; genotyping and breeding described below) were maintained on a normal 12-h light, 12-h dark cycle at 22–24 C with ad libitum access to food (Purina rodent diet 5001; Purina Mills, St. Louis, MO) and water, unless otherwise noted for an experiment. Experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of Oregon Health and Science University.
Generation of PomcCre-gp130flox/flox mice
PomcCre mice (gift of Dr. Gregory Barsh, Stanford University, Palo Alto, CA) were generated and maintained on a mixed FVB/N, C57BL/6J, and 129 background as previously described (48). PomcCre heterozygotes were mated with gp130flox/flox mice (gift of Dr. Beth Habecker, Oregon Health & Science University, Portland, OR) (49) and the colony was maintained by mating PomcCre-gp130flox/flox mice with gp130flox/flox mice. The gp130flox mice were backcrossed on a C57BL/6J background for at least five generations before mating with PomcCre mice. Comparisons involving these animals were only conducted between mice of mixed background (FVB, 129, and C57BL/6). PomcCre mice were genotyped as previously reported (50), and no homozygotes were used for breeding or experiments. The gp130flox/flox mice were genotyped using previously described primers (51).
Peripheral LPS injections in mice for LIF and CNTF mRNA expression study
On the day of the experiment at 0900 h, animals received ip injections of LPS [100 μg/kg (Sigma-Aldrich Corp., St. Louis, MO) dissolved in 0.5% BSA (Sigma-Aldrich) in 0.9% saline] or 0.5% BSA in 0.9% saline alone, and were placed in clean cages without food. At 1, 4, or 8 h after treatment, animals were anesthetized with isoflurane and killed by decapitation. The brains were immediately removed, and a hypothalamic block was dissected out, preserved in RNAlater solution (Ambion, Inc., Austin, TX), and stored at 4 C overnight. RNA was extracted the next day and used for RT-PCR analysis.
Implantation of lateral ventricle cannulae
C57BL/6J, POMC-EGFP, or PomcCre-gp130flox/flox mice were anesthetized with 1–2% isoflurane and placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). A sterile guide cannula with obturator stylet was stereotaxically implanted into the lateral ventricle. The coordinates used were: 1.0-mm posterior to bregma, 0.5-mm lateral to midline, and 2.35-mm below the surface of the skull (52). The cannula was then fixed in place with dental cement. The animals were individually housed after surgery for a minimum of 1 wk, and were handled and administered 1 μl i.c.v. injections of commercial aCSF (Harvard Apparatus, Holliston, MA) daily.
Central IL-1β injection in mice for LIF and CNTF mRNA expression study
On the day of the experiment at 0900 h, mice received icv injections of 10 ng murine IL-1β (R&D Systems, Minneapolis, MN) dissolved in 1 μl aCSF, or aCSF alone, and were placed in clean cages without food. At 8 h after treatment, mice were anesthetized with isoflurane and killed by decapitation. The brains were immediately removed, and a hypothalamic block was dissected out, preserved in RNAlater solution, and stored at −80 C until RNA extraction and RT-PCR analysis.
RNA preparation and RT-PCR
Hypothalamic blocks with median eminence attached were isolated and RNA was extracted for quantitative RT-PCR analysis as described previously (22). Raw Ct values from 18S endogenous controls were compared between groups to validate observed changes in target genes.
Central LIF injection in mice for feeding studies
Animals were transferred to clean cages and food was removed at lights off, the evening before the experiment. On the day of the experiment at 0830 h, mice received ip injections of ketorolac [40 mg/kg (Sigma-Aldrich) dissolved in 0.9% saline]. At 0900 h, mice received i.c.v. injections of 50 ng murine LIF (molecular mass = 47 kDa; Santa Cruz Biotechnology, Santa Cruz, CA) dissolved in 1 μl 0.5% BSA (carrier) in 0.9% saline, or 0.5% BSA alone, and given a weighed quantity of food. At 1, 2, 4, 8, and 12 h after treatment, food was weighed and returned to the cage. For the PomcCre-gp130flox/flox feeding study, animals were given 1 wk to recover, and the experiment was repeated with all animals switching i.c.v. treatments. All animals were randomly assigned to treatment group.
Central LIF injection in mice for immunohistochemistry
On the day of the experiment at 0830 h, mice received ip injections of ketorolac [40 mg/kg (Sigma-Aldrich) dissolved in 0.9% saline]. At 0900 h, mice received i.c.v. injections of 10 ng or 100 ng murine LIF (Santa Cruz Biotechnology) dissolved in 1 μl 0.5% BSA in 0.9% saline, or 0.5% BSA alone, and placed in clean cages with food removed. For the cFos and EGFP IHC, 90 min after i.c.v. injection, POMC-EGFP mice were anesthetized with a ketamine cocktail and transcardially perfused with 0.01 m PBS followed by 4% paraformaldehyde in PBS for fixation. For the pSTAT3 and ACTH IHC, PomcCre-gp130flox/flox mice and WT littermates were killed and fixed as described above 30 min after i.c.v. treatment. Brains were removed and postfixed 2–4 h, cryopreserved in 20% sucrose, and stored at −80 C until sectioning and staining. All animals were randomly assigned to treatment group.
Immunohistochemistry and cell counting
Dual immunofluorescence histochemistry was performed as previously described (21). Detailed methods can be found in the supplemental Materials and Methods published on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org. The number of cFos- or pSTAT3-immunoreactive cells and double-labeled cells was counted by eye in sections representing the ARC by investigators blinded to the treatments as previously reported (21).
Hypothalamic peptide secretion
Male C57BL/6J mice or PomcCre-gp130flox/flox mice and WT littermates were anesthetized with isoflurane and killed quickly by decapitation. The brain was removed (with care taken to ensure that there was no contamination of the hypothalamic portion with residual pituitary), and a 2-mm slice including the median eminence was prepared using a vibrating microtome (Leica VS 1000) that included the paraventricular and arcuate nuclei. Individual hypothalami received a 1-h equilibration period with aCSF [126 mm NaCl, 0.09 mm Na2HPO, 6 mm KCl, 4 mm CaCl2, 0.09 mm MgSO4, 20 mm NaHCO3, 8 mm glucose, 0.18 mg/ml ascorbic acid, and 0.6 trypsin inhibitory unit aprotinin/ml] at 37 C. Hypothalami were then incubated for 45 min at 37 C in 700 μl aCSF (basal period) before being challenged with a single concentration of murine LIF (Santa Cruz Biotechnology) (0.1–50 nm) in 700 μl aCSF for 45 min at 37 C. Tissue viability was verified by a 45-min exposure to 700 μl aCSF containing 56 mm KCl. At the end of each treatment period, supernatants were removed and frozen at −80 C until assayed by RIA. Explants that failed to show peptide release above basal release in response to aCSF containing 56 mm KCl were excluded from data analysis.
α-MSH RIA
α-MSH immunoreactivity was measured as described previously (53). The lowest detectable level that could be distinguished from the zero standard was 0.22 fmol/tube. The intraassay coefficient of variation was determined by replicate analysis (n = 6) of two samples at α-MSH concentrations of 2 and 10 fmol/tube, and the results were 6.7 and 9.7%, respectively. The interassay coefficients of variation were 22.7 and 12.5% for the range of values measured.
Double-label in situ hybridization histochemistry
Simultaneous visualization of POMC and LIF-R mRNA in the rat brain (n = 3) was performed as previously reported (21), with slight modifications. Determination of cells expressing both LIF-R and POMC mRNA was performed using criteria previously described (21). This technique is detailed further in supplemental Materials and Methods.
Results
Induction of hypothalamic LIF mRNA expression after ip LPS and i.c.v. IL-1β administration
To examine the induction of LIF mRNA by LPS, male mice were randomly assigned to receive ip LPS (100 μg/kg) or saline. We found that hypothalamic LIF mRNA was increased 10.67 ± 2.8-fold in LPS-treated mice 1 h after treatment, but that this effect was transient, as LIF was induced only 4.34 ± 2.01-fold at 4 h, and no increase was observed by 8 h (Fig. 1A).
Figure 1.
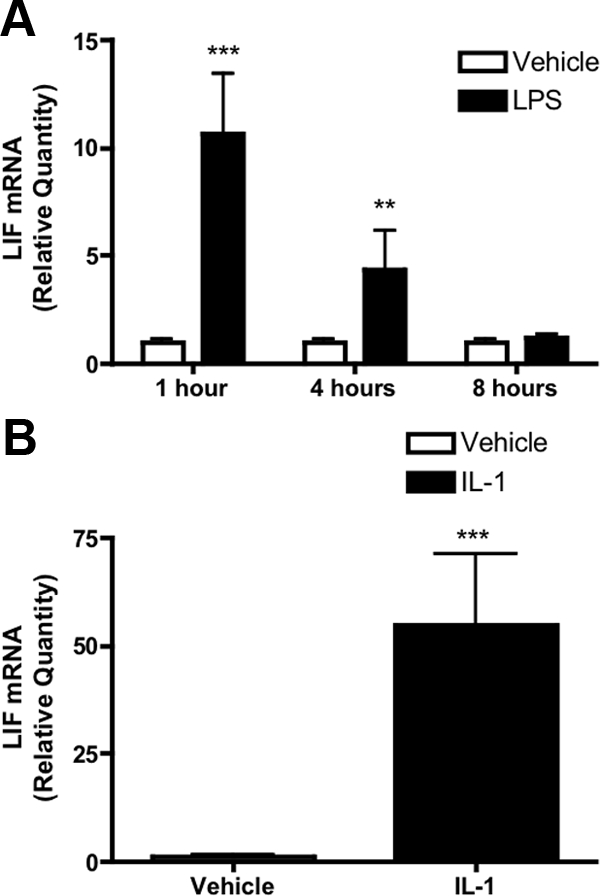
Hypothalamic LIF expression in response to LPS and IL-1β. A, Relative quantity of hypothalamic LIF mRNA after ip LPS (100 μg/kg) compared with vehicle in mice. LPS transiently induces hypothalamic LIF expression at 1 and 4 h after injection, although this effect is abolished at 8 h. B, Central (i.c.v.) IL-1β (10 ng) potently induces hypothalamic LIF expression 8 h after administration. Data normalized to vehicle at each time point. Results expressed as mean ± sem. Statistics calculated by two-way ANOVA followed by post hoc analysis using a Bonferroni corrected t test (A) or two-tailed Student’s t test (B) (**, P < 0.01 vs. vehicle; ***, P < 0.001 vs. vehicle).
To evaluate the LIF-inducing capacity of central IL-1β, mice received i.c.v. injection of either IL-1β (10 ng) or vehicle. IL-1β treatment increased hypothalamic LIF mRNA by 54.89 ± 15.2-fold compared with vehicle (Fig. 1B). Hypothalamic IL-6 mRNA expression was also transiently induced after both LPS and IL-1β treatment, although no elevation in CNTF mRNA was observed (supplemental Fig. S1).
Intracerebroventricular LIF induces anorexia associated with increased ARC POMC neuron activity
Male C57BL/6J mice were injected i.c.v. with 100 ng LIF (n = 13) or vehicle (n = 12) to test whether i.c.v. LIF acutely inhibits food intake in mice. LIF-treated mice showed reduced food intake at all time points and ate significantly less at 2 and 4 h after treatment than vehicle-treated mice, although by 8 h no statistical differences in food intake were found (Fig. 2, A and B). For this and all subsequent in vivo LIF physiology studies, mice were pretreated with ketorolac (40 mg/kg) to block LIF-induced local prostaglandin production in the CNS.
Figure 2.
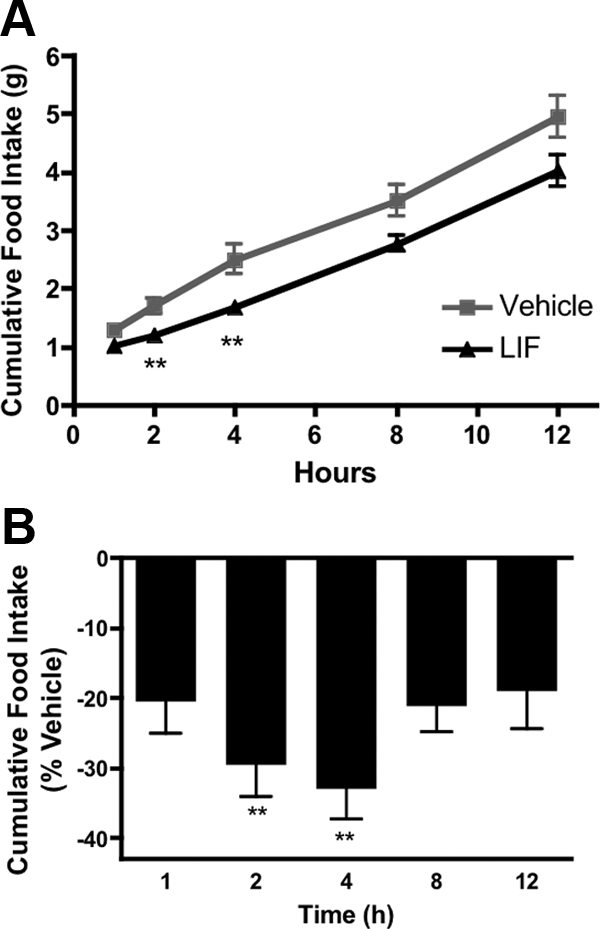
Effects of central LIF on food intake in WT mice. A, Cumulative food intake after i.c.v. bolus injection of LIF (100 ng) vs. vehicle in wild-type mice. Mice fasted overnight and pretreated with ipketorolac (40 mg/kg). B, Cumulative food intake in LIF-treated mice expressed as percent reduction compared with vehicle. Inhibition of food intake was significant at 2 and 4 h posttreatment. Data are expressed as mean ± sem. Statistics calculated two-way ANOVA followed by post hoc analysis using a Bonferroni corrected t test (**, P < 0.01 vs. vehicle).
To evaluate the role of ARC POMC neurons in mediating LIF-induced anorexia, we repeated this experiment in POMC-EGFP mice, and POMC neuron involvement was assessed using cFos-like immunoreactivity (CFLIR) as a marker for neuronal activation (54) (Fig. 3, A–I). LIF administration (n = 4) elicited significant increases in the number of cells displaying CFLIR per section in the ARC compared with vehicle (n = 3) (10 ng LIF, 49.51 ± 2.7 cells per section; vehicle, 9.69 ± 0.8 cells per section; P < 0.0001) (Fig. 3, A, D, G, and J). LIF also elicited significant increases in CFLIR in POMC-EGFP neurons (10 ng LIF, 30.48 ± 1.3 cells per section; vehicle, 5.97 ± 0.9 cells per section; P < 0.0001) (Fig. 3, C, F, I, and K). No additional increase in CFLIR was seen in POMC neurons when higher doses of LIF (100 ng) were used (Fig. 3, J and K). CFLIR was also examined in NTS POMC neurons. Although an increase in total CFLIR per section was observed, there was no change in the percentage of NTS POMC neurons showing CFLIR (supplemental Fig. S2).
Figure 3.

LIF activates POMC-EGFP neurons in the hypothalamus. A, D, and G, POMC-EGFP expression in the arcuate nucleus. B, Low cFos IR (red) in vehicle-treated mice (n = 3). C, Few POMC neurons show nuclear cFos IR after vehicle treatment. E and H, cFos IR is increased in LIF-treated (10 ng, n = 4) mice. F and I, LIF increases cFos IR in POMC neurons. G–I, Enlargement of D–F shown for clarity (white boxes indicate regions of enlargement). J, LIF increases cFos IR per section. K, Approximately 30% of POMC neurons contain cFos immunoreactivity after either 10 or 100 ng LIF treatment. Data are expressed as mean ± sem, and statistics calculated by one-way ANOVA followed by a post hoc analysis using a Bonferroni corrected t test (***, P < 0.001 vs. vehicle). Scale bars, 100 mm (A–F) and 50 mm (G–I). 3V, Third ventricle.
LIF-R is expressed by POMC neurons in the ARC
To determine whether a neuroanatomical framework exists for direct LIF activation of POMC neurons, we examined the expression of LIF-R in the rat brain using in situ hybridization. Within the hypothalamus, LIF-R expression was densest in the ARC (Fig. 4A). Double label in situ hybridization showed clusters of silver grains representing LIF-R mRNA overlying POMC neurons (red precipitate) (Fig. 4B). Semiquantitative analysis using a signal to background ratio cutoff of 2.5 indicated that radiolabeled LIF-R was expressed by 20.37 ± 1.3% of digoxigenin-labeled POMC neurons. We also performed double label in situ hybridization for gp130 expression by POMC neurons (data not shown). gp130 labeling ARC was too dense to resolve individual neuron clusters, as the glycoprotein is likely expressed by numerous glia and neurons.
Figure 4.

Expression of LIF-R in the hypothalamus. A, Representative dark-field photomicrograph showing expression of LIF-R mRNA (silver grain clusters) in the ARC of WT rats. B, Double-label in situ hybridization showing expression of LIF-R (silver grain clusters) by cells expressing POMC mRNA (red precipitate). Arrows point to POMC neurons that coexpress LIF-R mRNA. Open arrowheads signify POMC neurons that do not express LIF-R mRNA. Filled arrowheads denote cells that express LIF-R mRNA but not POMC mRNA. Scale bars, 100 mm (A) and 25 mm (B). 3V, Third ventricle.
LIF increases α-MSH release from ARC POMC neurons
We used a murine hypothalamic explant model to assess whether central LIF stimulates the release of the anorexigenic peptide, α-MSH, from ARC POMC neurons. Hypothalami were incubated ex vivo in the presence of LIF (50 nm, n = 10) or aCSF alone (n = 9). LIF significantly increased α-MSH release from the explants by 240.77 ± 25.4% compared with aCSF (Fig. 5). This result demonstrates that increases in local LIF concentration, as seen in inflammatory states, are able to acutely induce α-MSH release from ARC POMC neurons. No increase in α-MSH release in response to lower concentrations of LIF (0.1 nm, 1.0 nm, 5.0 nm, n = 5–6 per concentration) was observed (data not shown).
Figure 5.
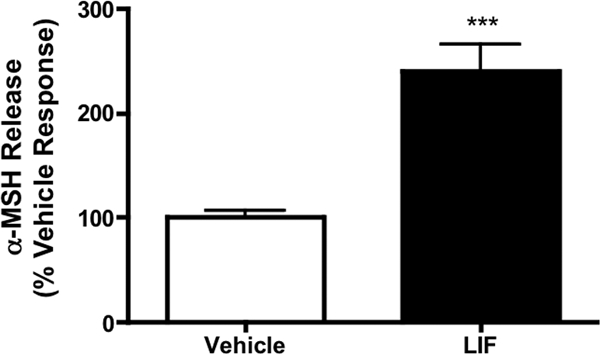
LIF stimulates α-MSH release from murine hypothalamic explants. LIF (50 nm, n = 10) increased α-MSH release in vitro by approximately 2.5-fold over vehicle (n = 9). Results are expressed as mean ± sem. Statistics calculated by two-tailed Student’s t test (***, P < 0.001 vs. vehicle).
Generation of mice lacking gp130 in POMC-expressing cells
To evaluate the role of intact LIF-R/gp130 signaling in POMC neurons, we generated a transgenic mouse in which gp130 was functionally deleted specifically in POMC neurons (PomcCre-gp130flox/flox). We verified that the transgenic animals show no differences in feeding behavior, body weight, refeeding after fast, wheel running activity, or metabolic rate as measured by oxygen consumption (supplemental Fig. S4). These animals also displayed a normal anorectic response to LPS-induced global inflammation (supplemental Fig. S5). To verify loss of LIF signaling in POMC neurons, PomcCre-gp130flox/flox mice and wild-type (WT) littermates received i.c.v. LIF (50 ng, WT n = 6; PomcCre-gp130flox/flox n = 5) or vehicle (WT n = 5; PomcCre-gp130flox/flox n = 4). Double-label IHC for pSTAT3 and ACTH, as a marker of POMC neurons, revealed that LIF increased pSTAT3 immunoreactive (IR) cells per section in the ARC in both WT and PomcCre-gp130flox/flox mice compared with control (WT-LIF, 180.5 ± 12.8 cells/section; WT-vehicle, 72.4 ± 10.9 cells/section; P < 0.001; gp130flox/flox-LIF, 164.3 ± 13.8 cells/section; gp130flox/flox-vehicle, 87.1 ± 14.7 cells/section; P < 0.001). Increased pSTAT3 signal was also observed in POMC neurons in WT mice (LIF, 64.86 ± 1.6%; vehicle, 35.55 ± 3.9%; P < 0.001). Notably, although PomcCre-gp130flox/flox mice also showed an increase in pSTAT3 IR neurons per section after LIF administration, no increase in POMC neurons containing pSTAT3 IR was observed compared with vehicle (LIF, 32.03 ± 2.7%; vehicle, 33.00 ± 2.0%; n.s.) (Fig. 6, A–R). This result indicates that LIF directly stimulates POMC neurons via the LIF-R/gp130 receptor complex. Furthermore, the lack of a response in the transgenic mice provides a functional confirmation of gp130 removal from these neurons.
Figure 6.

LIF-induced pSTAT3 IR in WT and gp130flox/flox POMC neurons. A, E, I, and M, ACTH IR in the ARC of WT and gp130flox/flox mice was used to identify POMC neurons. B and J, pSTAT3 IR is low in vehicle-treated WT (n = 5) and gp130flox/flox (n = 4) mice. C, D, K, and L, Few POMC neurons show pSTAT3 IR in vehicle-treated mice of both genotypes. F and N, LIF treatment (50 ng) increased pSTAT3 IR in WT (n = 6) and gp130flox/flox (n = 5) mice 30 min after i.c.v. injection. G and H, LIF treatment increases pSTAT3 IR in WT POMC neurons. O and P, Despite increased total pSTAT3 IR, few gp130flox/flox POMC neurons show pSTAT3 IR after LIF treatment. D, H, L, and P, Enlargement of C, G, K, and O for clarity (white boxes denote area of enlargement − area chosen to maximize number of POMC neurons in panel). Q, pSTAT3 IR per section is increased by LIF treatment in both genotypes. R, LIF treatment increases the number of POMC neurons containing pSTAT3 IR in WT but not gp130flox/flox mice. Data are expressed as mean ± sem. Statistics calculated by two-way ANOVA followed by a post hoc analysis using a Bonferroni corrected t test (a, P < 0.001 vs. WT/vehicle and gp130flox/flox/vehicle; b, P < 0.001 vs. WT/vehicle, P < 0.01 vs. gp130flox/flox/vehicle; ***, P < 0.001 vs. gp130flox/flox/LIF). Scale bars, 100 mm (A–C, E–G, I–K, M–O) and 50 mm (D, H, L, P). 3V, Third ventricle.
PomcCre-gp130flox/flox hypothalami do not increase α-MSH release in response to LIF
We repeated the hypothalamic explant study in the transgenic mice to determine whether gp130 signaling was necessary for LIF-induced increases in α-MSH release in vitro. Hypothalami were harvested from PomcCre-gp130flox/flox mice and littermate controls and cultured in aCSF with or without LIF (50 nm). Although WT mice again increased α-MSH release in response to LIF, this response was absent in PomcCre-gp130flox/flox hypothalami (WT, 207.70 ± 39.2% control; PomcCre-gp130flox/flox, 120.80 ± 17.8%; P < 0.05) (Fig. 7A). No differences were observed in basal or aCSF-induced α-MSH secretion between genotypes.
Figure 7.

gp130flox/flox mice are resistant to both LIF-induced α-MSH release and anorexia. A, LIF-induced α-MSH release from WT and gp130flox/flox murine hypothalamic explants. LIF (50 nm, n = 5) increases α-MSH release from WT hypothalami compared with vehicle (n = 7). LIF does not increase α-MSH release from gp130flox/flox hypothalami (n = 8) compared with vehicle (n = 5). Data shown as mean ± sem and statistics calculated by two-way ANOVA followed by a post hoc analysis using a Bonferroni corrected t test (*, P < 0.05 vs. LIF-treated gp130flox/flox). B, LIF-induced anorexia is attenuated in gp130flox/flox mice. The i.c.v. LIF (50 ng, n = 7) reduced food intake in fasted, ketorolac (40 mg/kg) treated mice at 2 and 4 h after injection compared with vehicle (n = 5). This effect was diminished in gp130flox/flox mice (LIF, n = 6; vehicle, n = 6). Results normalized to percentage of vehicle food intake and expressed as mean ± sem. Statistics calculated by two-way ANOVA followed by a post hoc analysis using a Bonferroni corrected t test (*, P < 0.05; **, P < 0.01 vs. gp130flox/flox). No statistically significant differences between genotypes were found at any time after vehicle treatment (P > 0.05).
PomcCre-gp130flox/flox mice display a diminished anorectic response to LIF
To evaluate whether gp130 expression in POMC neurons is necessary for LIF-induced anorexia, we conducted a feeding study in the PomcCre-gp130flox/flox mice and litter matched controls. LIF-treated (50 ng) WT mice significantly reduced food intake by approximately 30% at 2 and 4 h after injection compared with vehicle, but LIF-treated PomcCre-gp130flox/flox mice showed a blunted anorectic response at both 2 h (P < 0.01 compared with WT, LIF treated) and 4 h after injection (P < 0.05 compared with WT, LIF treated), neither of which was significantly different from vehicle-treated mice. There was no significant difference observed between genotypes at any other time points, although LIF-treated WT mice had cumulatively eaten less than LIF-treated PomcCre-gp130flox/flox mice at all time points to 24 h.
Discussion
In acute and chronic disease, central expression of inflammatory cytokines is significantly elevated. Recent work has demonstrated that CNS amplification of inflammatory signaling is necessary for the induction of disease-associated anorexia (25). Understanding how this process inhibits food intake is essential to develop effective treatment for cachexia. Disease models in animals have been shown to induce LIF expression in the hypothalamus (24,55), and central LIF administration is known to be anorectic both acutely (4) and chronically (37). Before these studies, little was known, however, about which neuronal populations mediate LIF’s anorectic effects. We show here that activation of POMC neurons by LIF is sufficient to inhibit food intake and that POMC neuron-specific ablation of LIF signaling restores food intake.
The results of these experiments represent the first time that the anorectic action of an inflammatory cytokine has been directly linked to the activation of POMC neurons. This work extends previous work in our laboratory, which showed that IL-1β regulates the activity of ARC POMC and NPY/AgRP neurons (21,22), although those studies did not directly link those molecular findings to physiology. We demonstrated that both central IL-1β and peripheral LPS, given in doses that induce an anorexic response, increase hypothalamic LIF mRNA transcripts up to 55-fold over control levels. Although these findings do not confirm that LIF is responsible for reducing food intake in either model, they do support a potential role for LIF in transducing the inflammatory signals within the hypothalamus. We postulate LIF is being expressed and released by cells in the ARC such that it can act in a paracrine or autocrine manner upon POMC neurons there, although the cell type responsible for LIF production remains unidentified. LIF, in contrast to IL-1β or LPS induces a pervasive anorexia when administered chronically, which more closely recapitulates cachexia. Elevations in circulating LIF have been observed cross-sectionally in humans with chronic disease, but it is unknown whether this finding accurately reflects long-term cytokine levels (29,30,31). Our own observations have shown that central LIF exposure itself can potently induce hypothalamic LIF expression (supplemental Fig. S6) suggesting the possibility of positive feedback sustaining central LIF signaling and leading to protracted anorexia in chronic disease. We have demonstrated that the acute anorectic actions of LIF are dependent on central melanocortin signaling, but the role of this system in mediating chronic LIF-induced anorexia has not yet been investigated. Furthermore studies investigating the neurophysiology of chronic LIF exposure are necessary to evaluate the importance of the present findings in cachexia.
The mRNA levels of another anorectic IL-6 family cytokine, CNTF, were indeed decreased in both inflammatory conditions tested. CNTF is also known to reduce food intake chronically, although at a far higher dose than LIF (56). This distinction is significant because both cytokines have been investigated as potential therapeutics for leptin resistance and obesity, but any effective treatment should avoid replicating the central inflammation seen in cachectic states. The observation that chronic LIF treatment induces cachexia and increased mortality, whereas CNTF and leptin do not (35,36,56) indicates divergent physiologic roles in metabolic regulation. Despite initiating apparently identical intracellular signaling cascades, these three cytokines do not exhibit cross-desensitization; this finding has been attributed to differential induction of phospho-tyrosine phosphatase 1B, which can act as a feedback inhibitor of these cascades (57). Furthermore, exploration of this phenomenon could yield important pathways separating cachexia from weight homeostasis.
Our observation that LIF-induced acute anorexia was almost completely abrogated in the gp130-deficient animals was unexpected. Numerous parallel and intersecting pathways converge to result in the final metabolic response, and it was expected that LIF could act on several pathways in concert to inhibit feeding. Given the dense expression of LIF-R and gp130 in the ARC, it is possible that other ARC neuron populations involved in energy homeostasis, such as those expressing NPY/AgRP play a role as well, but, based on our observations, inhibition of these neurons alone is not sufficient for LIF to induce anorexia. Furthermore, the animals were fasted overnight before LIF was administered, which should increase the activity of NPY/AgRP neurons. We can therefore posit that the increase in melanocortin tone is sufficient to inhibit feeding, even if LIF treatment inhibits NPY/AgRP neurons in parallel. This conclusion may not be that surprising, given that AgRP and NPY knock out mice show a limited metabolic phenotype (58). Recent studies have shown that although NPY and AgRP peptides may not be necessary to maintain food intake, the GABAergic activity of these neurons is essential to prevent starvation. Furthermore, this starvation has been shown to be nonmelanocortin dependent (59,60). The role of this inhibitory tone in cachexia has not yet been evaluated, although the success of melanocortin antagonism in ameliorating anorexia associated with inflammation but not AgRP-neuron ablation suggests disparate pathways. Furthermore, work investigating the role of NPY/AgRP neurons in acute and chronic cachexia is necessary to understand the contribution of these neurons to LIF-induced anorexia.
Taken together, our double-label in situ hybridization data indicate that at least 20% of POMC neurons express LIF-R. This estimate somewhat underrepresents the percentage of POMC neurons found to respond to LIF by IHC (∼30–40%). Given the high background signal in the ARC and the absolute abrogation of pSTAT3 induction by LIF in the PomcCre-gp130flox/flox mice, it is likely that the in situ hybridization assay provides a conservative underestimate of receptor expression. Interestingly, activation of this minority of POMC neurons by LIF is sufficient to inhibit food intake, demonstrating the incredible potency of POMC neuron activation. Having demonstrated a neuroanatomical basis for direct LIF activation of POMC neurons, we then directly assessed the capacity of LIF to increase the release of α-MSH in vitro. Advantages offered by measuring secreted peptide in solution over measuring total peptide in tissue, include the quantitation of released, not stored, peptide, as well as the ability to evaluate the acute effects of our compound on these neurons, which total peptide may not be sensitive enough to detect. We showed that LIF induces a 2- to 2.5-fold increase in α-MSH release over control. This increase in melanocortinergic tone is consistent with the acute inhibition of food intake we observed. Previous studies using this technique have noted increases of similar magnitude in α-MSH secretion after treatment with leptin or IL-1β, suggesting physiological relevance (21,53). We found that hypothalamic explants from mice lacking gp130 in POMC neurons were resistant to increased α-MSH release, supporting our hypothesis that LIF acts directly on POMC neurons to acutely elicit anorexia.
We were concerned that LIF may induce nonspecific local CNS inflammation that could ultimately mediate acute anorexia after LIF treatment. In each of the whole-animal physiology studies, high dose ketorolac was administered before LIF treatment to block prostaglandin production. Studies by our laboratory and others have confirmed that prostaglandins have a moderate inhibitory effect on food intake, which may be at least partially melanocortin-dependent (21,61). Nonspecific cyclooxygenase inhibition was used to control for any confounding effect of prostaglandins. Importantly, ketorolac administration alone does not have any effect on food intake or hypothalamic neuropeptide expression levels at the dose used in these experiments (data not shown). We also observed induction of IL-6 mRNA in hypothalami from cachectic animals. Although central IL-6 has been shown to be acutely anorectic, this effect appears to be significantly smaller in magnitude than for LIF (62). Furthermore, we found no evidence of IL-6R mRNA expression in the ARC, which is required for gp130 signaling (63), suggesting that any central IL-6 effects on feeding are mediated via an alternative neural circuit. Furthermore, to minimize nonspecific inflammation, minimal doses of LIF were used to elicit anorexia and neuron activation. Our studies used approximately 20-fold less LIF than previous studies investigating the anorectic effect of class I helical cytokines, such as CNTF and leptin (64,65). If this lower concentration of cytokine is capable of nonselectively activating gp130-deficient POMC neurons, it is possible that the much higher concentrations of CNTF and leptin used in these studies are also nonselectively activating these neurons, possibly through inflammatory pathways. Indeed, it has been shown that the anorectic effects of pharmacologic doses of leptin require IL-1β signaling, whereas physiologic levels do not (66,67).
Our finding that LIF-induced anorexia is mediated by POMC neurons provides the first direct evidence for melanocortin antagonism as a specific cachexia therapy. Treatment with type-4 melanocortin receptor antagonists has been successful in retaining lean mass and increasing food intake in several animal models of cachexia (68,69,70,71,72). Although animals retained mass, it had not been conclusively shown that antagonism at the MC4-R was not simply masking the underlying pathology. As molecular and genetic tools, such as the gp130flox/flox mouse, become more available, the role of melanocortin signaling in other models of cachexia can be better delineated. Also, the successes of melanocortin antagonism in attenuating cachexia suggest that modulation of this system by cytokines such as LIF and IL-1β is of physiologic significance in inflammatory states. We have shown that IL-1β induces the expression of LIF in the hypothalamus. It is possible that LIF mediates some of the acute anorectic effect of IL-1β, although we did not test this in the present study. These two cytokines are known to activate distinct intracellular signaling pathways and likely induce unique inflammatory profiles. Elucidation of these pathways and the downstream mediators of LIF-induced inflammation in chronic models could greatly augment our understanding of the sustained negative energy balance exhibited by cachectic patients and identify other neural populations that contribute to cachexia.
Supplementary Material
Acknowledgments
We would like to thank Drs. P. Enriori and M. Cowley for their help with the α-MSH secretion assay. We also thank Dr. G. Barsh for donation of the PomcCre mouse line and Dr. B. Habecker for donation of the gp130flox/flox mouse line.
Footnotes
This work was supported by National Institutes of Health Grants: National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK) R01 DK 70333, NIDDK F30 DK 84711; AHA Predoctoral Fellowship 0515502Z; and Doernbecher Children’s Hospital Foundation/OHSU Brain Institute Pediatric Neurobiology of Disease Fellowship.
Disclosure Summary: A.J.G., J.M.S., X.X.Z., D.D.B., A.K.B., and T.P.B. have no conflicts of interest to declare. D.L.M. is a consultant for IPSEN, Inc., and Santhera Pharmaceuticals, Inc.
First Published Online December 16, 2009
Abbreviations: AgRP, Agouti-related protein; ARC, arcuate nucleus of the hypothalamus; CFLIR, cFos-like immunoreactivity; CNTF, ciliary neurotrophic factor; EGFP, enhanced green fluorescent protein; i.c.v., intracerebroventricular; IR, immunoreactive; LIF, leukemia inhibitory factor; LPS, lipopolysaccharide; NPY, neuropeptide Y; NTS, solitary tract nucleus; POMC, pro-opiomelanocortin; STAT3, signal transducer and activator of transcription 3; WT, wild type.
References
- Tisdale MJ 1997 Biology of cachexia. J Natl Cancer Inst 89:1763–1773 [DOI] [PubMed] [Google Scholar]
- Larkin M 1998 Thwarting the dwindling progression of cachexia. Lancet 351:1336 [DOI] [PubMed] [Google Scholar]
- Plata-Salamán CR, Oomura Y, Kai Y 1988 Tumor necrosis factor and interleukin-1β: suppression of food intake by direct action in the central nervous system. Brain Res 448:106–114 [DOI] [PubMed] [Google Scholar]
- Plata-Salamán CR 1996 Anorexia induced by activators of the signal transducer gp 130. Neuroreport 7:841–844 [DOI] [PubMed] [Google Scholar]
- Sonti G, Ilyin SE, Plata-Salamán CR 1996 Anorexia induced by cytokine interactions at pathophysiological concentrations. Am J Physiol 270:R1394–R1402 [DOI] [PubMed] [Google Scholar]
- Baile CA, Naylor J, McLaughlin CL, Catanzaro CA 1981 Endotoxin-elicited fever and anorexia and elfazepam-stimulated feeding in sheep. Physiol Behav 27:271–277 [DOI] [PubMed] [Google Scholar]
- Cavaillon JM, Haeffner-Cavaillon N 1990 Signals involved in interleukin 1 synthesis and release by lipopolysaccharide-stimulated monocytes/macrophages. Cytokine 2:313–329 [DOI] [PubMed] [Google Scholar]
- Hillhouse EW, Mosley K 1993 Peripheral endotoxin induces hypothalamic immunoreactive interleukin-1β in the rat. Br J Pharmacol 109:289–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray MJ, Murray AB 1979 Anorexia of infection as a mechanism of host defense. Am J Clin Nutr 32:593–596 [DOI] [PubMed] [Google Scholar]
- Plata-Salamán CR, Ilyin SE, Gayle D, Flynn MC 1998 Gram-negative and gram-positive bacterial products induce differential cytokine profiles in the brain: analysis using an integrative molecular-behavioral in vivo model. Int J Mol Med 1:387–397 [DOI] [PubMed] [Google Scholar]
- van Dam AM, Bauer J, Tilders FJ, Berkenbosch F 1995 Endotoxin-induced appearance of immunoreactive interleukin-1β in ramified microglia in rat brain: a light and electron microscopic study. Neuroscience 65:815–826 [DOI] [PubMed] [Google Scholar]
- van Dam AM, Poole S, Schultzberg M, Zavala F, Tilders FJ 1998 Effects of peripheral administration of LPS on the expression of immunoreactive interleukin-1 α, β, and receptor antagonist in rat brain. Ann NY Acad Sci 840:128–138 [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB 1999 From lesions to leptin: hypothalamic control of food intake and body weight. Neuron 22:221–232 [DOI] [PubMed] [Google Scholar]
- Cone RD, Cowley MA, Butler AA, Fan W, Marks DL, Low MJ 2001 The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obes Relat Metab Disord 25 Suppl 5:S63–S67 [DOI] [PubMed] [Google Scholar]
- Cone RD 2005 Anatomy and regulation of the central melanocortin system. Nat Neurosci 8:571–578 [DOI] [PubMed] [Google Scholar]
- Broberger C, Johansen J, Johansson C, Schalling M, Hökfelt T 1998 The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci USA 95:15043–15048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD 1997 Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385:165–168 [DOI] [PubMed] [Google Scholar]
- Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS 1997 Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278:135–138 [DOI] [PubMed] [Google Scholar]
- Castro MG, Morrison E 1997 Post-translational processing of proopiomelanocortin in the pituitary and in the brain. Crit Rev Neurobiol 11:35–57 [DOI] [PubMed] [Google Scholar]
- Appleyard SM, Hayward M, Young JI, Butler AA, Cone RD, Rubinstein M, Low MJ 2003 A role for the endogenous opioid β-endorphin in energy homeostasis. Endocrinology 144:1753–1760 [DOI] [PubMed] [Google Scholar]
- Scarlett JM, Jobst EE, Enriori PJ, Bowe DD, Batra AK, Grant WF, Cowley MA, Marks DL 2007 Regulation of central melanocortin signaling by interleukin-1β. Endocrinology 148:4217–4225 [DOI] [PubMed] [Google Scholar]
- Scarlett JM, Zhu X, Enriori PJ, Bowe DD, Batra AK, Levasseur PR, Grant WF, Meguid MM, Cowley MA, Marks DL 2008 Regulation of agouti-related protein messenger ribonucleic acid transcription and peptide secretion by acute and chronic inflammation. Endocrinology 149:4837–4845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layé S, Gheusi G, Cremona S, Combe C, Kelley K, Dantzer R, Parnet P 2000 Endogenous brain IL-1 mediates LPS-induced anorexia and hypothalamic cytokine expression. Am J Physiol Regul Integr Comp Physiol 279:R93–R98 [DOI] [PubMed] [Google Scholar]
- Layé S, Parnet P, Goujon E, Dantzer R 1994 Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res Mol Brain Res 27:157–162 [DOI] [PubMed] [Google Scholar]
- Wisse BE, Ogimoto K, Tang J, Harris MK, Raines EW, Schwartz MW 2007 Evidence that lipopolysaccharide-induced anorexia depends upon central, rather than peripheral, inflammatory signals. Endocrinology 148:5230–5237 [DOI] [PubMed] [Google Scholar]
- Ishizaki S, Murase T, Sugimura Y, Banno R, Arima H, Miura Y, Oiso Y 2004 Leukemia inhibitory factor stimulates vasopressin release in rats. Neurosci Lett 359:77–80 [DOI] [PubMed] [Google Scholar]
- Kariagina A, Romanenko D, Ren SG, Chesnokova V 2004 Hypothalamic-pituitary cytokine network. Endocrinology 145:104–112 [DOI] [PubMed] [Google Scholar]
- Auernhammer CJ, Melmed S 2000 Leukemia-inhibitory factor-neuroimmune modulator of endocrine function. Endocr Rev 21:313–345 [DOI] [PubMed] [Google Scholar]
- Ren SG, Seliktar J, Li X, Braunstein GD, Melmed S 1998 Measurement of leukemia inhibitory factor in biological fluids by radioimmunoassay. J Clin Endocrinol Metab 83:1275–1283 [DOI] [PubMed] [Google Scholar]
- Gouin F, Heymann D, Raher S, De Groote D, Passuti N, Daculsi G, Godard A 1998 Increased levels of leukaemia inhibitory factor (LIF) in urine and tissue culture supernatant from human primary bone tumours. Cytokine 10:110–114 [DOI] [PubMed] [Google Scholar]
- Lorgeot V, Praloran V, Turlure P, Denizot Y 1997 Concentrations of serum leukemia inhibitory factor (LIF) in patients with hematologic malignancies. Leukemia 11:311–312 [DOI] [PubMed] [Google Scholar]
- Villers D, Dao T, Nguyen JM, Bironneau E, Godard A, Moreau M, De Groote D, Nicolas F, Soulillou JP, Anegon I 1995 Increased plasma levels of human interleukin for DA1.a cells/leukemia inhibitory factor in sepsis correlate with shock and poor prognosis. J Infect Dis 171:232–236 [DOI] [PubMed] [Google Scholar]
- Carlson CD, Bai Y, Jonakait GM, Hart RP 1996 Interleukin-1 β increases leukemia inhibitory factor mRNA levels through transient stimulation of transcription rate. Glia 18:141–151 [DOI] [PubMed] [Google Scholar]
- Gayle D, Ilyin SE, Flynn MC, Plata-Salamán CR 1998 Lipopolysaccharide (LPS)- and muramyl dipeptide (MDP)-induced anorexia during refeeding following acute fasting: characterization of brain cytokine and neuropeptide systems mRNAs. Brain Res 795:77–86 [DOI] [PubMed] [Google Scholar]
- Metcalf D, Gearing DP 1989 Fatal syndrome in mice engrafted with cells producing high levels of the leukemia inhibitory factor. Proc Natl Acad Sci USA 86:5948–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Yamaguchi K, Honda S, Nagasaki K, Ueda M, Abe O, Abe K 1991 Cancer cachexia syndrome developed in nude mice bearing melanoma cells producing leukemia-inhibitory factor. Cancer Res 51:6656–6659 [PubMed] [Google Scholar]
- Beretta E, Dhillon H, Kalra PS, Kalra SP 2002 Central LIF gene therapy suppresses food intake, body weight, serum leptin and insulin for extended periods. Peptides 23:975–984 [DOI] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F 2003 Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem J 374:1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio MA 2007 gp130 receptor ligands as potential therapeutic targets for obesity. J Clin Invest 117:841–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DW, Ren SG, Melmed S 1996 Leukemia inhibitory factor (LIF) stimulates proopiomelanocortin (POMC) expression in a corticotroph cell line. Role of STAT pathway. J Clin Invest 97:1852–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbud RA, Kelleher R, Melmed S 2004 Cell-specific pituitary gene expression profiles after treatment with leukemia inhibitory factor reveal novel modulators for proopiomelanocortin expression. Endocrinology 145:867–880 [DOI] [PubMed] [Google Scholar]
- Ray DW, Ren SG, Melmed S 1998 Leukemia inhibitory factor regulates proopiomelanocortin transcription. Ann NY Acad Sci 840:162–173 [DOI] [PubMed] [Google Scholar]
- Auernhammer CJ, Chesnokova V, Bousquet C, Melmed S 1998 Pituitary corticotroph SOCS-3: novel intracellular regulation of leukemia-inhibitory factor-mediated proopiomelanocortin gene expression and adrenocorticotropin secretion. Mol Endocrinol 12:954–961 [DOI] [PubMed] [Google Scholar]
- Bousquet C, Melmed S 1999 Critical role for STAT3 in murine pituitary adrenocorticotropin hormone leukemia inhibitory factor signaling. J Biol Chem 274:10723–10730 [DOI] [PubMed] [Google Scholar]
- Bousquet C, Zatelli MC, Melmed S 2000 Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J Clin Invest 106:1417–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M, Mortrud M, Liu B, Low MJ 1993 Rat and mouse proopiomelanocortin gene sequences target tissue-specific expression to the pituitary gland but not to the hypothalamus of transgenic mice. Neuroendocrinology 58:373–380 [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ 2001 Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411:480–484 [DOI] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS 2005 PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest 115:951–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz UA, Bloch W, van den Broek M, Yoshida K, Taga T, Kishimoto T, Addicks K, Rajewsky K, Müller W 1998 Postnatally induced inactivation of gp130 in mice results in neurological, cardiac, hematopoietic, immunological, hepatic, and pulmonary defects. J Exp Med 188:1955–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS 2006 Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab 4:123–132 [DOI] [PubMed] [Google Scholar]
- Streetz KL, Wüstefeld T, Klein C, Kallen KJ, Tronche F, Betz UA, Schütz G, Manns MP, Müller W, Trautwein C 2003 Lack of gp130 expression in hepatocytes promotes liver injury. Gastroenterology 125:532–543 [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G 1997 The mouse brain in stereotactic coordinates. San Diego: Academic Press [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA 2007 Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab 5:181–194 [DOI] [PubMed] [Google Scholar]
- Morgan JI, Curran T 1986 Role of ion flux in the control of c-fos expression. Nature 322:552–555 [DOI] [PubMed] [Google Scholar]
- Wang Z, Ren SG, Melmed S 1996 Hypothalamic and pituitary leukemia inhibitory factor gene expression in vivo: a novel endotoxin-inducible neuro-endocrine interface. Endocrinology 137:2947–2953 [DOI] [PubMed] [Google Scholar]
- Prima V, Tennant M, Gorbatyuk OS, Muzyczka N, Scarpace PJ, Zolotukhin S 2004 Differential modulation of energy balance by leptin, ciliary neurotrophic factor, and leukemia inhibitory factor gene delivery: microarray deoxyribonucleic acid-chip analysis of gene expression. Endocrinology 145:2035–2045 [DOI] [PubMed] [Google Scholar]
- Benomar Y, Berthou F, Vacher CM, Bailleux V, Gertler A, Djiane J, Taouis M 2009 Leptin but not ciliary neurotrophic factor (CNTF) induces phosphotyrosine phosphatase-1B expression in human neuronal cells (SH-SY5Y): putative explanation of CNTF efficacy in leptin-resistant state. Endocrinology 150:1182–1191 [DOI] [PubMed] [Google Scholar]
- Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, Chen A, Camacho RE, Shearman LP, Gopal-Truter S, MacNeil DJ, Van der Ploeg LH, Marsh DJ 2002 Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol 22:5027–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Howell MP, Cowley MA, Palmiter RD 2008 Starvation after AgRP neuron ablation is independent of melanocortin signaling. Proc Natl Acad Sci USA 105:2687–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Boyle MP, Palmiter RD 2009 Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell 137:1225–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohinata K, Yoshikawa M 2008 Central prostaglandins in food intake regulation. Nutrition 24:798–801 [DOI] [PubMed] [Google Scholar]
- Plata-Salamán CR, Sonti G, Borkoski JP, Wilson CD, French-Mullen JM 1996 Anorexia induced by chronic central administration of cytokines at estimated pathophysiological concentrations. Physiol Behav 60:867–875 [PubMed] [Google Scholar]
- Taga T, Kishimoto T 1997 Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 15:797–819 [DOI] [PubMed] [Google Scholar]
- Janoschek R, Plum L, Koch L, Münzberg H, Diano S, Shanabrough M, Müller W, Horvath TL, Brüning JC 2006 gp130 signaling in proopiomelanocortin neurons mediates the acute anorectic response to centrally applied ciliary neurotrophic factor. Proc Natl Acad Sci USA 103:10707–10712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley RJ, van Dijk G, Campfield LA, Smith FJ, Burn P, Nelligan JA, Bell SM, Baskin DG, Woods SC, Schwartz MW 1996 Intraventricular leptin reduces food intake and body weight of lean rats but not obese Zucker rats. Horm Metab Res 28:664–668 [DOI] [PubMed] [Google Scholar]
- Wisse BE, Ogimoto K, Morton GJ, Williams DL, Schwartz MW 2007 Central interleukin-1 (IL1) signaling is required for pharmacological, but not physiological, effects of leptin on energy balance. Brain Res 1144:101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luheshi GN, Gardner JD, Rushforth DA, Loudon AS, Rothwell NJ 1999 Leptin actions on food intake and body temperature are mediated by IL-1. Proc Natl Acad Sci USA 96:7047–7052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WW, Kuo HJ, Markison S, Chen C, Foster AC, Marks DL, Mak RH 2007 Peripheral administration of the melanocortin-4 receptor antagonist NBI-12i ameliorates uremia-associated cachexia in mice. J Am Soc Nephrol 18:2517–2524 [DOI] [PubMed] [Google Scholar]
- Joppa MA, Gogas KR, Foster AC, Markison S 2007 Central infusion of the melanocortin receptor antagonist agouti-related peptide (AgRP(83-132)) prevents cachexia-related symptoms induced by radiation and colon-26 tumors in mice. Peptides 28:636–642 [DOI] [PubMed] [Google Scholar]
- Markison S, Foster AC, Chen C, Brookhart GB, Hesse A, Hoare SR, Fleck BA, Brown BT, Marks DL 2005 The regulation of feeding and metabolic rate and the prevention of murine cancer cachexia with a small-molecule melanocortin-4 receptor antagonist. Endocrinology 146:2766–2773 [DOI] [PubMed] [Google Scholar]
- Marks DL, Cone RD 2001 Central melanocortins and the regulation of weight during acute and chronic disease. Recent Prog Horm Res 56:359–375 [DOI] [PubMed] [Google Scholar]
- Wisse BE, Frayo RS, Schwartz MW, Cummings DE 2001 Reversal of cancer anorexia by blockade of central melanocortin receptors in rats. Endocrinology 142:3292–3301 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.