

Abstract
Background
Recently, it has been discovered that the human genome contains many transcription start sites for non-coding RNA. Regulatory regions related to transcription of this non-coding RNAs are poorly studied. Some of these regulatory regions may be associated with CpG islands located far from transcription start-sites of any protein coding gene. The human genome contains many such CpG islands; however, until now their properties were not systematically studied.
Results
We studied CpG islands located in different regions of the human genome using methods of bioinformatics and comparative genomics. We have observed that CpG islands have a preference to overlap with exons, including exons located far from transcription start site, but usually extend well into introns. Synonymous substitution rate of CpG-containing codons becomes substantially reduced in regions where CpG islands overlap with protein-coding exons, even if they are located far downstream from transcription start site. CAGE tag analysis displayed frequent transcription start sites in all CpG islands, including those found far from transcription start sites of protein coding genes. Computational prediction and analysis of published ChIP-chip data revealed that CpG islands contain an increased number of sites recognized by Sp1 protein. CpG islands containing more CAGE tags usually also contain more Sp1 binding sites. This is especially relevant for CpG islands located in 3' gene regions. Various examples of transcription, confirmed by mRNAs or ESTs, but with no evidence of protein coding genes, were found in CAGE-enriched CpG islands located far from transcription start site of any known protein coding gene.
Conclusions
CpG islands located far from transcription start sites of protein coding genes have transcription initiation activity and display Sp1 binding properties. In exons, overlapping with these islands, the synonymous substitution rate of CpG containing codons is decreased. This suggests that these CpG islands are involved in transcription initiation, possibly of some non-coding RNAs.
Background
Most mammalian DNA is depleted with CpG dinucleotides [1] whose fraction in a mammalian genome is close to 0.2-0.25 of the value expected from presupposition of random distribution [2]. The shortage of genomic CpG dinucleotides is believed to be the consequence of frequent mutation of mCpG to TpG dinucleotides [1,3-5]. Nevertheless, some mammalian genomic segments called CpG islands (CGIs) [3] possess a high G+C content, with a frequency of CpG close to the expected value. In bioinformatics, CGIs are usually defined as DNA segments that are longer than 200 bp, have above 50% G+C content, and have a CpG frequency of at least 0.6 of that expected assuming letters at each sequence position occurring independently at random with the given composition [3]. The number of CGIs varies substantially in different vertebrate species [4]. There are about 50,200 such CGIs in the human genome, of which approximately 29,000 are in repeat-masked sequences [5].
The increased number of CpG sites in CGIs is often correlated with low methylation of cytosine in CpG dinucleotides [6-9]. This effect is usually explained by postulating protection of these sites from DNA methyltransferase by abundant and commonly utilized DNA binding proteins including Sp1 [10], E2F [11], CTCF [12] and others. The Sp1 protein is particularly strongly implicated in CGI functioning. Gardiner-Garden and Frommer observed [3] that CGIs contain many "G/C boxes", composed of the sequence GGGCGG, demonstrated to act as binding sites for the Sp1 transcription factor [13]. Later, it was found that Sp1 can bind to both methylated and non-methylated variants of this binding site [14], and can protect non-methylated sites from methylation [10].
In his recent study Rozenberg et al. [15] demonstrated that binding sites of several regulatory proteins, including Sp1, contain a CpG pair and play an important role in the formation of sequences of mouse promoters which regulate the expression of housekeeping genes. This suggests that CGIs overlapping with promoters of housekeeping genes are related to their transcription initiation. According to [16] 60% of widely expressed human genes and up to 40% of tissue-specific genes are associated with CpG islands. It has been shown lately that 72% of all promoters have high CpG content, and only 28% are in the class with low CpG content [17].
CGIs located near 5' region of known genes account for only a fraction of all CGIs in the genome (about 25% for CGIs longer than 500 bp in the HOVERGEN compilation [18], and about 50% according to our estimations, see below). Although many non-5' associated CGIs overlap with repeats [18,19], many do not [18,20], but instead are frequently positioned 3' to known genes, overlapping with final transcribed exons [3,20]. Amazingly, CGIs located in these 3' regions have attracted almost no interest, even though these CGIs were mentioned in the publication that initially coined the term "CpG island" [3]. More recently, computational approaches have also identified intragenic CGIs that overlap neither TSS nor final exons [20], although function of these CGIs have not yet been assessed.
CGIs not associated with 5' region of any gene can perform important biological functions. For instance, a C-to-T substitution in CGI encompassing parts of exon 15 and intron 15 of UBA1 affects expression of this gene [21]. A CGI located within intron 10 of KCNQ1 and associated with an oppositely-oriented RNA transcript is involved in imprinting (paternal repression) of its locus [22]. Imprinting of MAP3K12 gene is associated with differential methylation of a CGI located in its last exon [23]. Many CpG islands are located near the 3' ends of genes associated with cancer development [24].
The main objective of this work was to study properties of CGIs located far from TSS of protein coding genes. We demonstrated that substantial selection pressure is applied to CpG pairs in CGIs independently from CGI location in the reference to gene starts locations, which implies functional importance of CpG pairs. We assumed with [15] that most of CGIs are involved in transcription initiation, thus one of our objectives was to study transcriptional activity of CGIs, particularly of CGIs located far from 5' regions of any protein coding gene. To do this we used Cap Analysis Gene Expression (CAGE) tags identified in the FANTOM project [25,26]. We also assessed the representation of binding motifs recognized by regulatory factor Sp1 in CGIs located in 5', 3' and internal gene regions, as well as out of any known genes. In addition, we re-analyzed the published ChIP-chip data on Sp1 binding in chromosomes 21-22 and compared Sp1 binding preferences in DNA not overlapping with CGIs as well as in CGIs located in different gene segments and out of any genes. Fraction of non-5' CGI strongly enriched with CAGE tags was studied with special care; we observed substantial overrepresentation of probably strong Sp1 binding sites in such CGIs and collected known reports of transcription starts sites of long non-coding RNAs associated with such CGIs.
Results
CGIs tend to overlap protein coding exons
Tendency of CGIs to overlap with exons has been observed many times at limited data sets [16,27,28]. As the first step of our study we decided to give a quantitative estimation of this tendency separately for exons and introns located in different gene regions. Exons and introns were categorized according to their location within the gene (see Methods). For exons and introns from each category the total length of overlap with CGIs was calculated. We used Monte Carlo simulations to assess the statistical significance of the observed total overlap length. A round of simulation was performed as follows. Exons (introns) were located as in the human genome and "CGIs" were sampled. Intervals between CGIs were sampled from the interval distribution evaluated from the genome, whereas the lengths of "CGIs" were shuffled length of genuine CGIs. The total overlap between exons (introns) and "CGIs" was calculated. Then the whole procedure was repeated with switched CGI and exon sets, i.e. the annotated CGIs and "simulated exons (introns)" were taken. For each category of gene elements such simulations were repeated 10,000 times and the observed values of exon (intron) overlapping with CGIs were normalized for the average simulated values.
Figure 1 shows that for all categories of exons (except 3' UTR exons) the fraction of their overlapping with CGIs is greater than the similar fraction for corresponding introns. Overlapping with CGIs is greatest for 5'UTRs and first coding exons. This happens because CGIs associated with promoter regions are usually longer than 1 Mb and often extended into 5' UTRs and further downstream into the coding region. Yet, the observed tendency of internal and especially of terminal exons to overlap with CGIs cannot be explained this way.
Figure 1.

The ratio of overlaps of bona fide CGIs and exons (introns) and overlaps of randomly positioned intervals with lengths of exon (intron) and CGI sets. (1) Exon set is fixed, CGI set is sampled. (2) CGI set is fixed, exon set is sampled. 10,000 runs of Monte-Carlo simulation. Length distributions are computed independently for each chromosome.
Frequent overlapping of CGIs with exons cannot be explained as misinterpretation of GC-rich exons as CGIs
It is known that exons are usually more GC-rich than introns [29]. At the same time, the algorithm for CGI computational identification uses the increased C+G content of a test DNA segment as one of the CGI conditions. On the other hand, a CG-rich exon can have an increased number of CpG dinucleotides owing to its specific amino acid composition, e.g. many arginine codons. Therefore, this exon would be misidentified as CGI, and many such events would explain an increased overlapping between CGIs and exons.
A more interesting alternative explanation of frequent overlapping of CGIs with exons is that it is caused by the common preferences of both segments to be located in some particular DNA regions. In this case the terminal intron segments that are close to exons would also overlap with CGIs more frequently than internal segments of long introns. To test this, we selected 200 bp intron fragments adjacent to donor and acceptor splice sites. As in the previous section, we used Monte Carlo simulations to assess expectation of the observed overall overlap lengths.
Figure 1 shows the normalized intersection of CGIs with the terminal regions of introns. One can see that the normalized overall overlapping of intron terminal regions with CGIs is more similar to the values for CGI overlapping with exons than to the values for CGI overlapping with the internal segments of introns. Table 1 also shows that CGI overlapping with internal segments of introns is less likely than CGI overlapping with randomly positioned intervals of the same length. Therefore, CGIs have some tendency to avoid being buried within introns. This agrees better with the tendency that both exons and CGIs exhibit a preference to occupy the same DNA regions with yet unknown properties and CGIs overlapping with exons often extend significantly into introns.
Table 1.
Ratio of bona fide CGIs-exons (introns) overlap and "simulated" overlap average
| Gene region | Bona fide overlap length/Simulated overlap average | |
|---|---|---|
| "Simulated" CGIs | "Simulated" exons | |
| 3000 bp flank region | 14.54 | 12.6 |
| exon in 5' UTR | 66.46 | 63.33 |
| intron in 5' UTR (5' 200 bp) | 30.36 | 26.49 |
| intron in 5' UTR | 2.35 | 2.2 |
| intron in 5' UTR (3' 200 bp) | 24.08 | 21.07 |
| initial coding exon | 39.32 | 35.4 |
| initial intron in coding area (5' 200 bp) | 24.16 | 21.77 |
| initial intron in coding area | 1.76 | 1.7 |
| initial intron in coding area (3' 200 bp) | 20.17 | 18.05 |
| internal exon | 7.15 | 14.87 |
| internal intron (5' 200 bp) | 1.81 | 3.25 |
| internal intron | 0.39 | 0.78 |
| internal intron (3' 200 bp) | 1.55 | 2.83 |
| terminal coding exon | 13.01 | 11.75 |
| terminal intron in coding area (5' 200 bp) | 3.51 | 3.22 |
| terminal intron in coding area | 0.64 | 0.62 |
| terminal intron in coding area (3' 200 bp) | 3.05 | 2.77 |
| exon in 3' UTR | 1.93 | 1.82 |
| intron in 3' UTR (5' 200 bp) | 3.99 | 3.26 |
| intron in 3' UTR | 0.61 | 2.25 |
| intron in 3' UTR (3' 200 bp) | 2.8 | 0.56 |
The total length of CGI overlapping with exons and introns in different gene regions normalized for the expectation estimated from overlapping of randomly sampled intervals.
In all gene regions synonymous substitution rates of codons that contain CpG dinucleotides are lower in exons overlapping with CGIs than in exons not overlapping with CGIs
The analysis above demonstrates CGI function is probably carried on at the level of nucleic acids. Therefore CGI presence can affect synonymous substitution rate for codons that overlap with CGIs. To test this, we compared synonymous (dS) and nonsynonymous (dN) substitution rates in human-mouse alignments for codons overlapping and non-overlapping with CGIs. Exons located in different parts of genes were considered separately. The substitution rates were calculated for all codons and separately for codons containing CpG, GpC, ApG and GpA dinucleotides. The results are presented in Table 2 and Figure 2, 3 and 4.
Table 2.
dN, dS and dN/dS
| Codon type | Initial exon | Internal exon | Final exon | ||||||
|---|---|---|---|---|---|---|---|---|---|
| dN | dS | dN/dS | dN | dS | dN/dS | dN | dS | dN/dS | |
| CG containing codons in CGI | 0.131 | 0.512 | 0.257 | 0.097 | 0.910 | 0.106 | 0.100 | 0.800 | 0.125 |
| CG containing codons out of CGI | 0.136 | 0.987 | 0.138 | 0.093 | 1.510 | 0.061 | 0.114 | 1.273 | 0.090 |
| AG containing codons in CGI | 0.146 | 0.485 | 0.302 | 0.101 | 0.644 | 0.157 | 0.109 | 0.599 | 0.181 |
| AG containing codons out of CGI | 0.134 | 0.508 | 0.264 | 0.087 | 0.535 | 0.164 | 0.112 | 0.533 | 0.210 |
| GC containing codons in CGI | 0.130 | 0.381 | 0.342 | 0.098 | 0.534 | 0.183 | 0.101 | 0.503 | 0.201 |
| GC containing codons out of CGI | 0.145 | 0.488 | 0.297 | 0.095 | 0.526 | 0.180 | 0.122 | 0.519 | 0.235 |
| GA containing codons in CGI | 0.120 | 0.381 | 0.314 | 0.084 | 0.531 | 0.159 | 0.091 | 0.463 | 0.197 |
| GA containing codons out of CGI | 0.114 | 0.450 | 0.252 | 0.075 | 0.489 | 0.154 | 0.096 | 0.479 | 0.200 |
| All codons in CGIs | 0.097 | 0.344 | 0.282 | 0.073 | 0.468 | 0.157 | 0.074 | 0.443 | 0.167 |
| All codons out of CGIs | 0.099 | 0.389 | 0.254 | 0.063 | 0.407 | 0.155 | 0.083 | 0.404 | 0.205 |
Rates were calculated separately for various gene elements and various classes of codons.
Figure 2.

dN. Non-synonymous substitution rates calculated for various classes of codons overlapping and not overlapping with CGIs in different gene regions.
Figure 3.

dS. Synonymous substitution rates calculated for various classes of codons overlapping and not overlapping with CGIs in different gene regions.
Figure 4.

dN/dS. Synonymous to non-synonymous substitution rates ratio calculated for various classes of codons overlapping and not overlapping with CGIs in different gene regions.
The nonsynonymous substitution rate for codons containing CpG dinucleotides was very similar to that for other codons and depended only weakly on the overlapping with CGIs (Figure 2). The main factor affecting rates of nonsynonymous substitutions is the codon location near one of the gene termini. Figure 2 shows "V"-shaped dN plots for all the codons outside of CGIs, which indicates that internal exons are less variable than both terminal exons. This effect may be related to the increased protein variability at the N and C termini. At the same time, codons overlapping with CGIs show almost equal dN for the internal and the final exons. Thus, proteins coded by genes having a CGI at their 3' end are generally more conserved at their C end.
In contrast, synonymous substitution rates calculated for codons containing CpG dinucleotides were different from those for other codons and dramatically depended on their overlapping with CGIs (Table 2 and Figure 3). Generally, for codons with CpG dinucleotides overlapping with CGI resulted in dS decrease approximately two-fold (Table 2 and Figure 3). For codons that did not contain CpG the effect of CGI on dSwas much smaller. This effect did not depend on the gene region: a CGI overlapping with a 5', intragenic or 3' exon had a similar effect on dS, reducing the synonymous substitution rates of CpG containing codons by 49%, 40% and 37%, respectively.
Figure 4 shows the dN/dS ratio which reflects the selection pressure at the protein level [30]. For codons that do not contain CpG the dN/dS ratios are almost identical for codons that do overlap and don't overlap with CGIs. Thus, it appears that selection at the protein level for non CpG-containing codons inside or outside of CGIs is practically the same. For CpG-containing codons one can see that the dN/dS ratios calculated for codons overlapping and not overlapping with CGIs are substantially different, and both ratios are much lower (red and light green curve, Figure 4), which indicates a comparatively greater stabilizing selection at such codons at the protein level.
The observation that CpG containing codons have lower dS when they overlap with CGIs gives additional evidence that amino acid composition (e.g. abundance of arginine) cannot explain the abundance of CpG dinucleotides and the frequent overlap of CGIs and exons. Function of CGI indeed seems to be more related to DNA or RNA.
Enrichment of CGIs with CAGE tags
In the previous sections we have demonstrated that in exons located far downstream from TSSs and overlapping with CGIs the synonymous substitution rate of CpG-containing codons is reduced. In addition, CGIs found far downstream from TSS often overlap with exons, but such CGIs are unlikely to be the misrecognized exons. Assuming that 5' related CGI are involved into transcription initiation [15,17] we have investigated if CGIs located in other genome regions also participate in transcription initiation. To test this suggestion we have studied association of computationally identified CGIs with transcription start sites as identified by CAGE tagging [25,26]. For our analysis we categorized CGIs into 4 non-overlapping classes: (1) 5' CGIs; (2) intragenic CGIs; (3) 3' CGIs; and (4) intergenic CGIs (see Methods, CGI classes). The number of CGI classes is smaller than the number of gene elements because the same CGI can often overlap with several gene elements, e.g. 5' UTR, the initial coding exon, the first intron, and sometimes other exons as well as downstream located introns.
CAGE tags exhibit a clear tendency to cluster within all classes of CGIs (Table 3). CGIs occupying about 0.7% of the entire genome contain more than 48% of all CAGE tags. About 70% of all CGIs contain at least one CAGE tag. In average 5', intragenic, 3', and intergenic CGIs contain respectively one CAGE tag per 20, 203, 172, and 86 base pairs as compared to the average genome CAGE frequency of 1 tag per 1,891 bp The frequency of CAGE tags in these CGIs is respectively 95-, 9-, 22-, and 11-fold greater than in the genome in average respectively with CGI class. A 5' CGI contains in average 44 CAGE tags; the number of CAGE tags in other classes of CGIs is 7- and 11-fold smaller.
Table 3.
CAGE tags in different CGIs classes
| CGI class | 5prim | intragenic | 3prim | intergenic | total |
|---|---|---|---|---|---|
| #CGIs | 15686 | 3095 | 1808 | 6848 | 27437 |
| Fraction of CGIs, % | 57.17 | 11.28 | 6.59 | 24.96 | 100 |
| Total length of CGIs | 13853661 | 1483283 | 1124521 | 4482821 | 20944286 |
| Average GC-content, % | 66.66 | 65.8 | 66.53 | 66.39 | 66.08 |
| #CGIs with CAGE-tags | 13361 | 1327 | 1005 | 3509 | 19202 |
| Total length of CGIs with CAGE-tags | 12756213 | 784565 | 780040 | 2925761 | 17246579 |
| Fraction of total length of CGIs with CAGE-tags in class, % | 92.08 | 52.89 | 69.37 | 65.27 | 82.35 |
| Total CAGE-tags in CGI class | 698369 | 7300 | 6520 | 52377 | 764566 |
| Fraction of CAGE-tags in CGI class, % | 43.7 | 0.46 | 0.41 | 3.28 | 47.85 |
| CGIs with at least one CAGE-tag, % | 85.18 | 42.88 | 55.59 | 51.24 | 69.99 |
| Average CAGE-tags per CGI | 45 | 2 | 4 | 8 | 28 |
| Average CAGE-tags per CAGE-containing CGI | 52 | 6 | 6 | 15 | 40 |
| Density of CAGE-tags in CGIs, bp-1 | 0.0504 | 0.0049 | 0.0058 | 0.0117 | 0.0365 |
| Density of CAGE-tags in CAGE-containing CGIs, bp-1 | 0.0547 | 0.0093 | 0.0084 | 0.0179 | 0.0443 |
| One CAGE-tag per #bp | 20 | 203 | 172 | 86 | 27 |
Relative number of CGIs of different classes; CAGE tag representation and their frequency in different CGIs.
As it was already reported in [31] CAGE tags tend to form dense clusters in 5' CGIs. CGIs located elsewhere contain much less CAGE tags than 5' CGIs, but, interestingly, some intragenic, 3' or intergenic CGIs contain clusters of CAGE tags with the number and the density of CAGE tags comparable with those found in CAGE clusters in 5' CGIs. Additional file 1 contains intragenic and 3' CGIs that have greater than 40 CAGE tags per a CGI (which approximately corresponds to the average number of CAGE tags per 5' CGI). 3' CGIs usually contain more CAGE tags than intragenic CGIs. In some sense this agrees with the tendency of CGIs to overlap with the final coding exon rather than with internal exons.
Not only 5'CGIs, but also 3', intragenic and intergenic CGIs are enriched with Sp1 binding sites
Authors of [15] reported that CGIs overlapping mouse promoters of housekeeping genes contained an increased number of binding sites for different transcription regulatory factors, in particular Sp1, ETS, and NRF-1. Since binding of Sp1 is well studied with experimental methods, we decided to assess Sp1 binding in CGIs of different localization relative to known genes. We used both bioinformatics methods of identification of Sp1 recognition motifs in DNA sequence as well as re-assessment of the published experimental data.
CGIs were scanned for presence of Sp1 factor binding sites using a positional weight matrix (PWM) constructed from experimental data from the TRANSFAC database. We selected a threshold that identified 90% of Sp1 binding sequences from our experimentally confirmed training set (see Methods). To evaluate the representation of Sp1 binding sites in CGIs, we calculated the P-value (see Methods) for each CGI, i.e. the probability of a random sequence of the same length and the same dinucleotide content to contain at least this number of Sp1 occurrences. This P-value was calculated with the help of the AhoPro program [32]. We compared results obtained for different CGI classes.
Figure 5 shows that for any PWM threshold, there are more Sp1 binding sites found in all types of CGIs including all non 5' CGI groups than in GC-rich control set. Although 5' CGIs contain more Sp1 binding sites than any CGIs, highly significant Sp1 hits (Figure 5, left) are represented to a similar degree in 3' and intergenic CGIs. Intragenic CGIs contain less Sp1 sites. For Sp1 sites of an intermediate quality, intergenic CGIs contain substantially more Sp1 binding sites than CGIs of any other class except for 5' CGIs.
Figure 5.

Statistical significance of the relative occurrence of Sp1 binding sites within different CGI classes and GC-rich shuffled sequences. X-axis: theoretical statistical significance (P-value); Y-axis: the overall fraction of sequences having a statistical significance less or equal than that at the X-axis. A higher statistical significance value reflects more Sp1 sites scoring above the PWM threshold within the selected CGI. CGI classes and GC-rich shuffled sequences are defined in Methods.
It is noteworthy that CGIs containing more than 40 CAGE tags contain a much more high scoring Sp1 recognition motifs than CGIs without evidence of high transcription activity (Figure 5), independently from their localization in relation to genes. Surprisingly, the greatest overrepresentation of high-scoring Sp1 recognition motifs sites is characteristic for 3' CGIs with more than 40 CAGE tags, but not for 5' CGI enriched with CAGE.
ChIP-chip data indicate that Sp1 factors tend to bind within CGIs
For further validation of Sp1 protein binding within CGIs, data on Sp1 transcription factor binding sites, experimentally assessed with ChIP-chip technology and published in [33] were analyzed. Cawley et al. detected frequent Sp1 binding sites far from 5' regions of any gene. We used their data to justify that Sp1 protein binds preferably within CpG islands regardless of their location in relation to genes.
Sp1 binding regions published in [33] are usually longer than 1 kB, which is significantly longer than many CGIs, especially those located far from TSS of genes. The authors of [33] used an extensive filtration procedure, which can lead to a high false negative rate, to limit their results to binding sites frequently occupied with Sp1. Therefore, the raw data were re-analyzed to allow comparison between ChIP signals within CGIs and those in other DNA segments. Additionally ChIP signals within CGIs located in different gene segments were examined.
Figure 6 shows that signals of probes located within all types of CGIs are greater for Sp1 antibody treated samples than for the corresponding signals of control (the untreated input) samples. In contrast it is not possible to observe such a difference in non-CGI DNA. One can see that the control (untreated input) and the Sp1 antibody treated sample signals measured at tags overlapping with different CGI classes correlate, which is probably related to the increase in hybridization specificity with G and C content [34]. The distribution is highly skewed so the average in all cases is higher than the median. However, with one exception of intergenic CGIs, both the mean and the median of Affimetrix perfect match probe (PM) value distributions for Sp1 antibody treated samples are greater than the values of corresponding characteristics for control samples.
Figure 6.
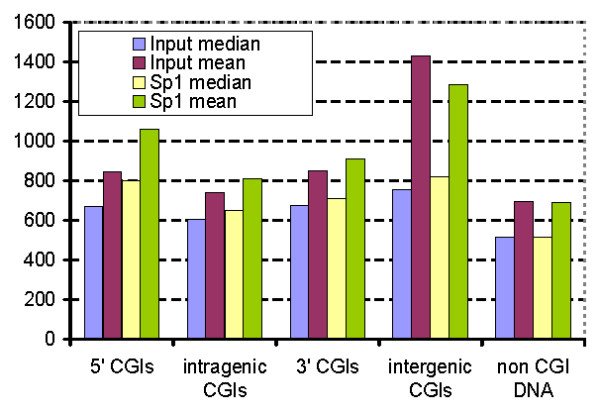
ChIP-chip assessment of Sp1 binding in CGIs in different genome segments. Mean and median intensities for Sp1 and input DNA signal for PM tags located in CGIs from different genome segments.
Figure 7 shows the median of the signal ratios for the treatment and the control calculated for each tag. This value is presented for different CGI classes as well as for non-CGI DNA. All ratios are almost equal to one. As one can see from Figure 7 the binding signal of Sp1 is the greatest in CGIs located near 5' gene region; it is lesser in intergenic and 3' CGIs region and is missing in non-CGI DNA.
Figure 7.
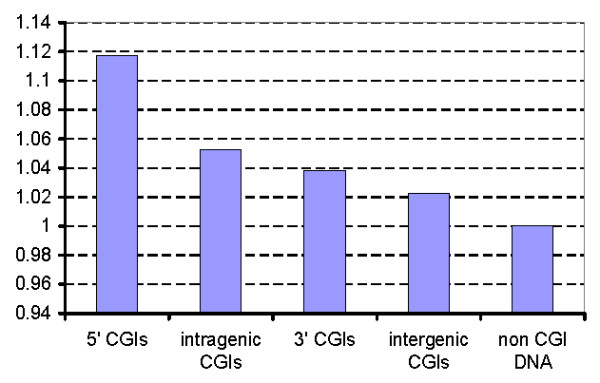
ChIP-chip S/N ratio for Sp1 binding in CGIs in different genome segments. Input/Sp1 signal ratio for PM tags located in CGIs from different genome segments.
Since the difference between medians of hybridization signals for the input and the treated samples was in all cases very small we tested whether this difference was statistically significant using Wilcoxon-Mann-Whitney test statistics. Table 4 shows the P-values of the Wilcoxon-Mann-Whitney test statistics calculated for the input and the treated samples for different classes of CGIs. The test indicates that for all classes of CGIs the distribution of signal values from the Sp1 antibody treaded samples differ significantly (alpha = 5%) from the distribution of signal values of the corresponding control samples. In contrast the difference in non-CGI DNA is not significant.
Table 4.
Statistical significance of Sp1 signal
| Type of the region | ||||
|---|---|---|---|---|
| 5' CGIs | Intragenic CGIs | 3' CGIs | Intergenic CGIs | Non-CGI DNA |
| 2.20E-16 | 4.12E-06 | 0.03978 | 0.0005911 | 0.08426 |
P-values of the Wilcoxon-Mann-Whitney tests performed on signal values from the Sp1 antibody treated sample and the corresponding control DNA sample within different CGI classes.
We also compared Sp1/input ratios between different classes of CGIs using Wilcoxon-Mann-Whitney test statistics. Table 5 shows that signal ratios from tags overlapping with CGIs of all types are significantly different from those in non-CGI DNA.
Table 5.
Statistical significance of Sp1/input ratios
| Type of the region | ||||
|---|---|---|---|---|
| 5' CGIs | Intragenic CGIs | 3' CGIs | Intergenic CGIs | Non-CGI DNA |
| X | <2.2E-16 | 2.75E-06 | 2.08E-05 | <2.2E-16 |
| X | X | 1.44E-05 | <2.2E-16 | <2.2E-16 |
| X | X | X | 3.11E-10 | <2.2E-16 |
| X | X | X | X | <2.2E-16 |
The comparison of of Sp1/input ratios between different types of CGIs using Wilcoxon-Mann-Whitney test statistics.
Non-5' CGIs with multiple CAGE tags are often associated with transcription starts sites of long RNAs for which no encoded proteins are known
We have explored if there are known transcripts associated with non-5'-CGIs enriched with CAGE tags. Table 6 demonstrates that 14 of 22 3' CGIs containing more then 40 CAGE tags are associated with a start of at least one potential coding gene from NCBI Reference Sequences (RefSeq). The corresponding value for intergenic CGIs is only 2 from 30. Other 8 of 22 3' CGIs and 28 of 30 intergenic CGIs also overlap with starts of long transcripts but without any evidence of a coded protein or at least a long ORF. To be exact, 5 3' CGIs and 18 intergenic CGIs contain start sites of mRNAs recorded in GeneBank. It should be mentioned that not all mRNAs in GeneBank are confirmed to code any protein; sometimes such RNAs only demonstrate mRNA properties, like having cap, polyA-tail or splicing. Therefore the mRNA database from GeneBank is likely to contain a fraction of long potentially non-coding RNAs. The remaining 3 3'CGIs and 10 intergenic CGIs contain start sites of spliced or unspliced ESTs. For CGIs containing from 20 to 40 CAGE tags the situation changes dramatically. In this case 29 of 41 3' CGIs contain starts of known long RNAs with no demonstrated protein-coding activity, whereas only 14 3'CGIs contain starts of protein-coding genes maintained in the RefSeq database. From all intragenic CGIs with 20-40 CAGE tags only 1 contains a start of a protein-coding gene and other 43 contain starts of mRNAs (or mRNA-like RNA) and ESTs. Thus, a substantial fraction of CAGE-enriched non 5'CGIs contains TSSs of long RNA showing no evidence of any encoded protein; this is especially true for CGIs with 20-40 CAGE tags.
Table 6.
Transcript starts in non 5' CGI
| Number of CGIs with starts of long transcripts | |||||
|---|---|---|---|---|---|
| Type of CGIs | Unverified RefSeq genes | Gene Bank mRNA and mRNA-like RNA | Spliced and unspliced ESTs | No transcripts found | Total |
| CGIs with more that 40 CAGE-tags per CGI | |||||
| 3' CGI | 14 | 5 | 3 | 0 | 22 |
| Intragenic CGI | 2 | 18 | 10 | 0 | 30 |
| CGIs with 20-40 CAGE-tags per CGI | |||||
| 3' CGI | 12 | 14 | 11 | 4 | 41 |
| intragenic CGI | 1 | 19 | 24 | 0 | 44 |
Number of transcript starts in intragenic and 3' CGIs having more than 40 CAGE tags per a CGI.
The total number of 3' and intragenic CGI with more then 40 CAGE tags is rather small: 22 and 30 respectively. Decreasing the threshold for CAGE tags per CGI to 20-40 leads to 41 3' CGI and 44 intragenic CGIs. However, the number of highly CAGE-enriched non 5' CGIs is not large enough to render a convincing statistical significance value.
Discussion
In this study we tried to systematically assess properties of CpG islands that are found far from transcription start sites of protein coding genes. About 43% of all CGIs belong to this class. Our study of CGIs which overlap with exons demonstrates that stabilizing selection protects CpG pairs located in CGIs from substitutions which do not affect the encoded amino acid sequence. We observed that many CGIs that are found far from TSS overlap with CAGE tags and thus participate in transcription; furthermore, highly CAGE-enriched CGIs are bound by transcription regulatory factor Sp1 with remarkably high significance. Although function of CGIs is still disputed, there is growing evidence that CGIs located near gene starts participate in transcription regulation [15,17,35]. Our finding allowed us to suggest that many CGIs that are found far from the start of any known protein coding gene are also participate in transcription.
As we have demonstrated, many such CGIs often overlap with exons, particularly the terminal gene exons. Many CGIs are located within a gene but far downstream from TSS (see Table 3). The aggregated number of genes with CGIs near their 3' end is estimated at 5 - 10%. Interestingly, it was observed recently [36] that some genes in human T-cells have an uncommon methylation pattern with a decreased methylation level observed near both gene termini.
We have detected many intergenic CGIs (see Table 3). It is known that UCSC browser table Knowngenes contains only highly verified genes, and excludes some genes with low justification scores. Based on our analysis, some CGIs considered in this study as intergenic may be related to these yet unverified genes.
CGIs located far downstream from TSS protect synonymous codon positions from substitutions very similar as do CGIs located near gene starts. A CGI is thought to reduce the CpG mutation rate by protecting DNA from methylation. On the other hand, CGIs probably contain many binding sites for transcription factors that overlap CpG dinucleotides [15]. Such binding can also increase conservation of CpG dinucleotides by applying stabilizing selection at nucleotide level that preserves functional binding sites. The decreased mutation rate and the purifying selective pressure would contribute to reduction of the substitution rate in CpG dinucleotides within CGIs (see Figure 8). This probably explains why the synonymous substitution rate in CpG containing codons in exon segments overlapping with CGIs becomes lower than that in exons not overlapping with CGIs. This effect is observed in all gene regions, 5' as well as 3' or intragenic, which supports the functional role of CGIs located in regions other than gene 5'.
Figure 8.
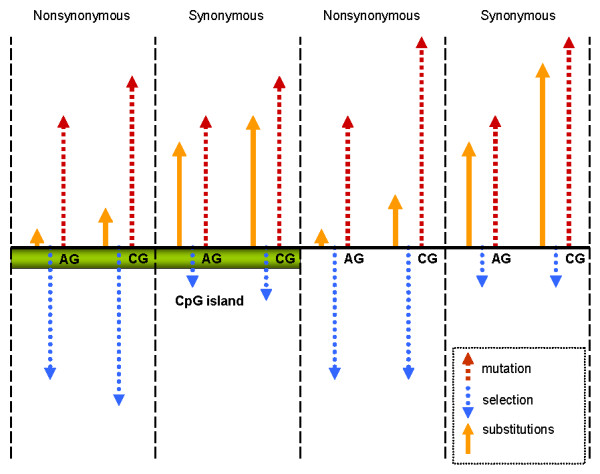
Interaction between mutation process and selection pressure in exons overlapping and non-overlapping with CGIs. In coding exons the substitution rate at synonymous sites is approximately 10-fold greater than at nonsynonymous sites. The mCpG → TG transition rate is about 10-fold greater than AG -> GG transition rate. CpG islands protect CpG dinucleotides from methylation, decreasing the transition rate from CG to TG. CpG dinucleotides in CGIs may be under stronger selection than CpG dinucleotides not overlapping within CGIs.
The selection directed to maintain CGI sequence properties is not strong enough to overcome a strong selection at protein level applied to non-synonymous substitutions. The non-synonymous substitution rate differs only weakly for codons overlapping and non-overlapping with CGIs. However, the dN/dS ratio for CpG containing codons not overlapping with CGIs is much smaller than for codons overlapping with CGIs. This fact indicates that in this case selection at the protein level needs to be stronger to counterbalance the higher mutation rate.
The evolutionary distance between human and mouse is rather large, with the approximate sequence divergence for these species close to 0.5 substitutions for a neutrally evolving site [37]. This value agrees very well with the dS values observed for codons that do not contain CpG. However, dS values for CpG containing codons are about twice as large as those for other codons. This is much less than the approximate ten-fold increase of mutation rate [38]. The possible explanation may come from the effect suggested by Kondrashov et. al. [39]. The idea is that hypermutable CpG dinucleotides [40] at neutral and pseudoneutral sites are likely to be destroyed by mutations and unlikely to be found in the alignment of human and mouse [41-43]. Those CpG dinucleotides that remain aligned in human and mouse genomes are likely to be stabilized by selection pressure of a different nature. Thus, even CpG dinucleotides that do not overlap with CpG islands at synonymous positions may be stabilized with some selection of yet unknown type. Interestingly, Bock et al [44] who specifically identified CGIs related to chromatin epigenetic state observed that about 90% of such islands overlapped with highly conserved DNA elements, including at least 20% of CGIs that did not overlap with TSS.
Although CGI overlap with disproportionally large number of CAGE tags (about a half of total CAGE tags are found within CGIs) many intragenic, intergenic, and gene terminal CGIs overlap with a small number of CAGE tags or with no CAGE tags at all. However, we believe, that FANTOM database can have some functional transcription starts missing.
First, CAGE tags were mapped at the repeat-masked human genome, thus excluding so-called "GC-rich low-complexity regions" and simple repeats such as (CCCCG)n. Many CGIs contain such low complexity regions, and CAGE tags in these regions were excluded from our analysis. It is important to note that even a simple repeat such as (CCCCG)n can probably operate as functional Sp1 site (see Figure 9), and thus may play a role in CGI functioning. It is noteworthy that many computationally identified CGIs overlap with Alu repeats [18], therefore we did not filter out such CGIs, considering that they would only reduce the effect but not create an artifact.
Figure 9.

Sequence logo for identified Sp1 site built using WebLogo [59].
Second, CAGE tags found in FANTOM database are obtained only in a number of tissues. CGIs located in 5' gene regions are usually found at starts of broadly expressed housekeeping genes [15]. Transcripts from TSS tagged in 3' regions of known genes could be tissue specific. Since the number of tissues studied is limited, the number of tagged TSS should be less than that in 5' gene regions.
The suggested role of non-5' CGIs in transcription initiation agrees with the excessive Sp1 binding in CGIs. The observation that Sp1 binding sites are often present in CGIs is not new [10]. It is noteworthy that CGIs enriched with CAGE tags contain more high-scoring Sp1 recognition motifs (Figure 5). Abundant Sp1 binding in gene 3' or intergenic regions was observed in genome-wide site location experiments [33], which reported that 36% of the clusters of Sp1, Myc and p53 binding sites lie within or immediately 3' to well-characterized genes. Authors of [33] assumed that in these cases, non-coding RNA transcription may be initiated.
Recent studies of the mouse genome [31] demonstrated that a large number of ncRNA are initiated in the 3' regions of the genes, with specific enrichment at the 3' terminus of the final exon. There are published reports which show that sometimes long ncRNA are synthesized to open large chromatin segments for subsequent transcription initiation [45,46]. On the other hand a substantial number of these ncRNAs are complementary to known genes as anti-sense strands, which has led to the suggestion of an additional mechanism of gene silencing by natural antisense interference [47]. The authors of [47] also observed that sense-antisense pairing was almost universally associated with candidate imprinted loci. As genetic imprinting is frequently associated with an altered methylation status of CpG islands [48,49], CGIs located in 3' gene regions [23,47] or intragenic CGIs [21,22] may play an important role in this process by regulation of gene expression via inducing antisense-based gene interference.
Conclusions
Abundant non-coding transcripts discovered recently in all parts of a genome allow suggesting that there should be regulatory regions associated with transcription initiation of such RNA types. This agrees with a large number of CGIs not associated with transcription start site of any known protein coding gene. Here we demonstrate that many of such CGIs appear to be related to transcription initiation and at least some of them contain CpG pairs stabilized by natural selection. Expression of RNA controlled by promoters overlapping with these CGIs seems to be regulated by the same transcription factors as expression of protein coding genes, therefore these RNA molecules seem to be involved into the regulatory cascades and cellular processes possibly as non-coding RNA of some function. Additional studies, both experimental and in silico are needed to verify this hypothesis.
Methods
We used a MySQL database and a set of Perl and Ruby scripts as analysis tools. The source code of the scripts, test sequence sets, and lists of genes under putative regulation by non-5' CGIs can be found at http://line.imb.ac.ru/CGI/
DNA Sequence and Annotations
The sequence of the human genome (hg17) and the Knowngene Table were downloaded from http://genome.ucsc.edu/. Redundant copies of genes and multiple copies with the same name but different locations were removed. Genes with less then 3 exons were also excluded. The resulting set amounted to 35,915 entries, derived from an initial 39,368 entries in the Knowngenes Table. Table Cpgislandext (UCSC) was used as the set of CpG islands. We have excluded CGIs with 'random' chromosome location, retaining 27,437 out of 27,801 computationally annotated CGIs. All Knowngenes genes were taken into account when testing if there were any protein coding gene TSS near intergenic or 3' located CGIs. Tables RefSeq genes, Human mRNA, Spliced EST and Human EST (USCS) were used to find starts of potentially protein coding and noncoding genes.
Gene elements definitions
We compiled 11 sets of gene fragments, defined as follows: (1) 5'-flank regions began 3 kb upstream from TSS and extended till first found TSS. Overlaps with any transcribed sequence were excluded. (2) 5' UTR-exon regions contained non-translated 5' exons (or exon segments); overlaps with any translated sequences were excluded. (3) 5' UTR-intron regions contained introns separating non-translated exons or the last non-translated and the initial translated exons; overlaps with any translated sequences were excluded. (4) Initial coding exons contained entire initial coding exons for all gene or exon parts; overlapping exons of different isoforms were merged. (5) Initial introns contained introns separating the 1st and the 2nd coding exons, overlaps with regions included into groups (1-4) were excluded. (6) Internal exons contained all translated internal exons, excluding the initial and the final exons of any gene. (7) Internal introns contained all introns, but not the initial and the final introns and overlaps with any translated sequence. (8) Final exons contained the last translated exons or their parts. (9) Final introns contained the introns separating the final exons and the previous ones, excluding overlaps with any translated sequence. (10) 3' UTR exons contained 3' non-translated exons or their parts, excluding overlaps with any translated sequence. (11) 3' UTR introns contained introns separating the 3' non-translated exons or the 3' non-coding and the final coding exon, excluding overlaps with any translated sequence.
Since introns are usually much longer than exons, we also considered 200 bp intronic segments flanking the donor and the acceptor splice sites. The resulting regions, called "intron terminal regions" are comparable with exons in their length. We used 200 bp intron regions adjacent to exons as an additional control set to exclude the influence of the increased exon GC composition, which can be misinterpreted as CGI during computational identification. All genes elements are available in Additional file 2.
CGI classes
We considered 4 different classes of CpG islands: (1) 5' CGIs that overlapped with gene elements from groups 1-5 above; (2) intragenic CGIs that overlapped with gene elements from groups 6-7; (3) 3' CGIs that overlapped with gene elements from groups 8-11 or with a region 3 kb downstream of any gene; and (4) intergenic CGIs that were located at least 3 Kb from any known gene upstream or downstream. All genes, including single and double-exon genes were taken into account in this case. If a CGI contained at least one bp of a 5' region of any gene it was considered as a 5' CGI regardless of how many additional regions it overlapped. If a CGI contained at least one bp of a 3' region of any gene, but not overlapped with its 5' region, it was considered as a 3' CGI. If a CGI contained at least one bp of a known gene, but not overlapped with its 5' or 3' region, it was considered as an intragenic CGI. A CGI was considered as intergenic if it did not belong to any of these classes. Additionally, we used a control set of CG-rich random sequences with the length and dinucleotide distribution estimated from each of CGIs containing more than 40 CAGE tags. The overall number and length of CGIs of different classes are given in Table 3. Since we took a special care to remove putative 5' CGIs from the other classes, the majority of all CGIs fells into 5' CGI class (Table 3). CGIs of all classes are available in Additional file 3.
Evaluation of the statistical significance of overlaps between interval sets
Given two sets of non-overlapping genome intervals (e.g. CGIs and exons) we used 10,000 Monte-Carlo simulations to compute expected distribution of aggregated overlap length. All length distributions were computed independently on each chromosome. During simulations intervals of one set corresponded to genome coordinates of elements (e.g. CGIs) and the other set contained intervals with lengths corresponding to those of the second set of genome elements but located at random positions in the chromosome. Each run of simulations was repeated twice with a different "fixed" element set (see Figure 1). Program source (Ruby 1.8) and additional details are available in Additional file 4.
Gene segments for substitution rates estimation
To estimate dN and dS we used the EDAS database [50], which contains 28,530 alignments of human and mouse genes. For genes with several isoforms, the longest isoform was taken. Genes with less than three coding exons were excluded. We also excluded genes which had less than 70% identity within protein alignment for any coding exon. The resulting dataset contained exons from 8,775 genes. Six groups of protein coding exon segments were defined: 5', internal, and 3' exons, overlapping and non-overlapping with CGIs. In each of these groups, we selected codons containing a CG dinucleotide; if a CG dinucleotide was split between two adjacent codons, both codons were taken. A similar procedure was performed for codons containing AG, GA, GC dinucleotides. Sequences from each group as well as codons containing CG, AG, GA, GC were concatenated.
Estimation of substitution rates
The transitional to transversional substitution rate ratio (R), as well as the numbers of synonymous substitutions per synonymous site (dS) and nonsynonymous substitutions per nonsynonymous site (dN) were estimated by the Ina method [30]. Unlike maximum likelihood methods, this was effective for very long alignments (~3*106 bp), and was fast enough to allow bootstrap resampling. We used our own implementation of this method (developed in Perl). The 95% confidence intervals for evolutionary parameters were calculated using bootstrap percentiles [51]. 2000 bootstrap replications were used. Sequences of all groups and scripts used for estimation of substitution rates are available in Additional file 5.
CAGE tags
The table of CAGE tags mapped on the RepeatMasked hg17 chromosome-build is available at http://gerg01.gsc.riken.jp/cage/download/hg17prmtr/cage.rep_tag.2005-01-16.chr_all_gff.tar.gz
This table contains 1,597,993 entries. We downloaded alignments of CAGE tag sequence with the genome region with a minimal identity of 0.88.
Identification of Sp1 recognition motifs in DNA sequences
We used a positional weight matrix (PWM) [52,53] as a model. A PWM for Sp1 was constructed by aligning experimental data contained in the TRANSFAC [54] database. Sequences containing binding sites for human Sp1 (mostly footprints), were obtained from the TRANSFAC database (July 2007 release), mapped on the human genome, extracted with genome flanking sequences, and realigned using the SeSiMCMC Gibbs sampler [55] (see Additional file 6 for details). The most frequent sequence length between different SeSiMCMC runs was equal to 9 bp and we accepted that all motifs had this length. A PWM was constructed from the alignment obtained with SeSiMCMC using the formula described in [56]. The resulting alignment included 221 genome sequences (see motif logo in Figure 9).
P-value calculation for Sp1 motif occurrences in sequences
To evaluate the P-value (i.e. to calculate the statistical significance of the observed number of Sp1 sites scoring higher than the fixed threshold in the test sequence) we used AhoPro [32]. For a test sequence containing k possibly overlapping PWM hits scoring higher than threshold T, the P-value was defined as the probability of observing no less than k such (possibly overlapping) hits in the random (i.i.d) sequence with the same nucleotide distribution and length as in the tested CGI.
ChIP-chip data for Sp1 binding
Experiments in [33] were conducted on the Affymetrix GeneChip® Human Tiling 1.0R Array Set. The results were downloaded from http://transcriptome.affymetrix.com/publication/tfbs/. Those chips contain unique 25 base-pair long sequence-tags for human chromosomes 21 and 22. The experiments for Sp1 were performed on two biological samples with three technical replicates for each chip. We used an modified version of TiMAT [57] to re-analyze the published cel-files. To allow comparison between different experiments the probes of each chip were median scaled to a signal value of 500. Additionally quantile-quantile [58] normalization was performed over all chips. The signal values from the two biological samples and the technical replicates were averaged to obtain one value for each probe. Signal values of biological probes of the Sp1 antibody treated and untreated control experiment were collected. As it is recommended in [57] mis-match probes (MM) were excluded and only perfect-match probes (PM) were considered for further investigation. Our aim was to compare the statistical distribution of PM values for tags located far from CGIs with PM values for tags overlapping different classes of CGIs as well as to compare signals for Sp1 antibody treated samples with those for untreated DNA.
Abbreviations
CGI: CpG island; TSS: transcription start site; CAGE: cap analysis gene expression; ncRNA: non-coding RNA; EST: Expressed sequence tag; PM: Affimetrix perfect match probe; MM: Affimetrix mis-match probe
Authors' contributions
YM participated in data preparation, carried out the data analysis, and contributed to results interpretation and the writing of the manuscript. MF contributed to data analyses and results interpretation. NO contributed to data preparation and analysis. DM contributed to data preparation for comparative genomics analysis. EE contributed to estimation of substitution rates. IK carried Monte Carlo simulations of statistical values and analysis of Sp1 binding sites. AH performed the analysis of ChIP-chip data. VM contributed to work coordination, results interpretation and writing of the manuscript. All authors read and approved the final manuscript.
Supplementary Material
CAGE enriched non 5' CGI. Tables contain lists of genes with CAGE-enriched CGIs in 3' and intragenic regions separately.
Gene elements. The archive contains row data used for statistical significance of gene elements and CGIs overlap. See Table 1 and Figure 1 in the manuscript.
CGI classes. The archive contains classes of CGIs used for calculation of CAGE tag frequency.
Monte Carlo Simulations. The archive contains Ruby scripts used to evaluate statistical significance of overlapping of gene segments and CGIs. Results of simulations with different "fixed" elements (see Methods) are also included.
Comparative genomics. The archive contains row data and Perl scripts to perform substitution rates calculation.
Sp1. This folder contains the data used for Sp1 binding sites prediction and detailed description of the procedure.
Contributor Information
Yulia A Medvedeva, Email: ju.medvedeva@gmail.com.
Marina V Fridman, Email: marina-free@mail.ru.
Nina J Oparina, Email: oparina@gmail.com.
Dmitry B Malko, Email: dmitry.malko@gmail.com.
Ekaterina O Ermakova, Email: ermakova@iitp.ru.
Ivan V Kulakovskiy, Email: ivan.kulakovskiy@gmail.com.
Andreas Heinzel, Email: mail@aheinzel.at.
Vsevolod J Makeev, Email: vsevolod.makeev@gmail.com.
Acknowledgements
The authors are pleased to thank Erica Golemis for valuable comments on the manuscript. We thank Biobase GmBH for providing free access to the Transfac database and personally Alexander Kel for his interest to this work and many discussions. The authors are also pleased to acknowledge Shamil Sunyaev, Andrei Mironov, Inna Pertsovskaya, Alexander Favorov, Sandy Der and Thomas Stranzl for fruitful discussions and Vadim Radionov for help with figures preparation. This study was partially supported with INTAS project 05-1000008-8028 (YAM, MVF, DBM, EOE, VJM), Russian Foundation of Basic Research projects 07-04-01584 (YAM, MVF, VJM), 07-04-01623 (DBM, IVK), 09-04-92745 (EOE), 07-04-12243 (NJO), International Science & Technology Center project 3737 (NJO), Russian Academy of Sciences Presidium program "Molecular and Cellular Biology" (EOE, NJO, VJM), Russian Academy of Sciences Presidium program "Basic Sciences for Medicine" (EOE), HHMI 55005610 (EOE), and Russian Federal Agency for Science and Innovation State Contracts 02.531.11.9003 (YAM, MVF, DBM, IVK, VJM) and 02.740.11.5008 (YAM, MVF, DBM, IVK, VJM).
References
- Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8(7):1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58(23):5489–5494. [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196(2):261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Han L, Su B, Li WH, Zhao Z. CpG island density and its correlations with genomic features in mammalian genomes. Genome Biol. 2008;9(5):R79. doi: 10.1186/gb-2008-9-5-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Heisler LE, Torti D, Boutros PC, Watson J, Chan C, Winegarden N, Takahashi M, Yau P, Huang TH, Farnham PJ, Jurisica I, Woodgett JR, Bremner R, Penn LZ, Der SD. CpG Island microarray probe sequences derived from a physical library are representative of CpG Islands annotated on the human genome. Nucleic Acids Res. 2005;33(9):2952–2961. doi: 10.1093/nar/gki582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SH, Charlton JA, Nan X, Bird AP. Purification of CpG islands using a methylated DNA binding column. Nat Genet. 1994;6(3):236–244. doi: 10.1038/ng0394-236. [DOI] [PubMed] [Google Scholar]
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38(12):1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Watanabe H, Miura F, Soejima H, Uchiyama M, Iwasaka T, Mukai T, Sakaki Y, Ito T. A comprehensive analysis of allelic methylation status of CpG islands on human chromosome 21q. Genome Res. 2004;14(2):247–266. doi: 10.1101/gr.1351604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod D, Charlton J, Mullins J, Bird AP. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994;8(19):2282–2292. doi: 10.1101/gad.8.19.2282. [DOI] [PubMed] [Google Scholar]
- Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 2002;16(2):235–244. doi: 10.1101/gad.943102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recillas-Targa F, De La Rosa-Velazquez IA, Soto-Reyes E, Benitez-Bribiesca L. Epigenetic boundaries of tumour suppressor gene promoters: the CTCF connection and its role in carcinogenesis. J Cell Mol Med. 2006;10(3):554–568. doi: 10.1111/j.1582-4934.2006.tb00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs MR, Kadonaga JT, Bell SP, Tjian R. Purification and biochemical characterization of the promoter-specific transcription factor, Sp1. Science. 1986;234(4772):47–52. doi: 10.1126/science.3529394. [DOI] [PubMed] [Google Scholar]
- Holler M, Westin G, Jiricny J, Schaffner W. Sp1 transcription factor binds DNA and activates transcription even when the binding site is CpG methylated. Genes Dev. 1988;2(9):1127–1135. doi: 10.1101/gad.2.9.1127. [DOI] [PubMed] [Google Scholar]
- Rozenberg JM, Shlyakhtenko A, Glass K, Rishi V, Myakishev MV, FitzGerald PC, Vinson C. All and only CpG containing sequences are enriched in promoters abundantly bound by RNA polymerase II in multiple tissues. BMC Genomics. 2008;9:67. doi: 10.1186/1471-2164-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen F, Gundersen G, Lopez R, Prydz H. CpG islands as gene markers in the human genome. Genomics. 1992;13(4):1095–1107. doi: 10.1016/0888-7543(92)90024-M. [DOI] [PubMed] [Google Scholar]
- Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA. 2006;103(5):1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponger L, Duret L, Mouchiroud D. Determinants of CpG islands: expression in early embryo and isochore structure. Genome Res. 2001;11(11):1854–1860. doi: 10.1101/gr.174501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JR, Herman JG, Myohanen S, Baylin SB, Vertino PM. Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J Biol Chem. 1997;272(35):22322–22329. doi: 10.1074/jbc.272.35.22322. [DOI] [PubMed] [Google Scholar]
- Hackenberg M, Previti C, Luque-Escamilla PL, Carpena P, Martinez-Aroza J, Oliver JL. CpGcluster: a distance-based algorithm for CpG-island detection. BMC Bioinformatics. 2006;7:446. doi: 10.1186/1471-2105-7-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramser J, Ahearn ME, Lenski C, Yariz KO, Hellebrand H, von Rhein M, Clark RD, Schmutzler RK, Lichtner P, Hoffman EP, Meindl A, Baumbach-Reardon L. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am J Hum Genet. 2008;82(1):188–193. doi: 10.1016/j.ajhg.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, Smallwood AC, Joyce JA, Schofield PN, Reik W, Nicholls RD, Weksberg R, Driscoll DJ, Maher ER, Shows TB, Higgins MJ. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci USA. 1999;96(14):8064–8069. doi: 10.1073/pnas.96.14.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada S, Tevendale M, Baker J, Georgiades P, Campbell E, Freeman T, Johnson MH, Paulsen M, Ferguson-Smith AC. Delta-like and gtl2 are reciprocally expressed, differentially methylated linked imprinted genes on mouse chromosome 12. Curr Biol. 2000;10(18):1135–1138. doi: 10.1016/S0960-9822(00)00704-1. [DOI] [PubMed] [Google Scholar]
- Shiraishi M, Sekiguchi A, Terry MJ, Oates AJ, Miyamoto Y, Chuu YH, Munakata M, Sekiya T. A comprehensive catalog of CpG islands methylated in human lung adenocarcinomas for the identification of tumor suppressor genes. Oncogene. 2002;21(23):3804–3813. doi: 10.1038/sj.onc.1205454. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H. et al. The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Kawaji H, Kasukawa T, Fukuda S, Katayama S, Kai C, Kawai J, Carninci P, Hayashizaki Y. CAGE Basic/Analysis Databases: the CAGE resource for comprehensive promoter analysis. Nucleic Acids Res. 2006. pp. D632–636. [DOI] [PMC free article] [PubMed]
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002;99(6):3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biol. 2003;3(3):235–240. [PubMed] [Google Scholar]
- Aissani B, D'Onofrio G, Mouchiroud D, Gardiner K, Gautier C, Bernardi G. The compositional properties of human genes. J Mol Evol. 1991;32(6):493–503. doi: 10.1007/BF02102651. [DOI] [PubMed] [Google Scholar]
- Ina Y. New methods for estimating the numbers of synonymous and nonsynonymous substitutions. J Mol Evol. 1995;40(2):190–226. doi: 10.1007/BF00167113. [DOI] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, Forrest AR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori-Katayama M, Kitazume Y, Kawaji H, Kai C, Nakamura M, Konno H, Nakano K, Mottagui-Tabar S, Arner P, Chesi A, Gustincich S, Persichetti F. et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38(6):626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- Boeva V, Clement J, Regnier M, Roytberg MA, Makeev VJ. Exact p-value calculation for heterotypic clusters of regulatory motifs and its application in computational annotation of cis-regulatory modules. Algorithms Mol Biol. 2007;2:13. doi: 10.1186/1748-7188-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116(4):499–509. doi: 10.1016/S0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- Koltai H, Weingarten-Baror C. Specificity of DNA microarray hybridization: characterization, effectors and approaches for data correction. Nucleic Acids Res. 2008;36(7):2395–2405. doi: 10.1093/nar/gkn087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JH, Bell AC, Felsenfeld G. Characterization of the chicken beta-globin insulator. Proc Natl Acad Sci USA. 1997;94(2):575–580. doi: 10.1073/pnas.94.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JK, Bae JB, Lyu J, Kim TY, Kim YJ. Nucleosome deposition and DNA methylation at coding region boundaries. Genome Biol. 2009;10(9):R89. doi: 10.1186/gb-2009-10-9-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Subramanian S. Mutation rates in mammalian genomes. Proc Natl Acad Sci USA. 2002;99(2):803–808. doi: 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Shimotake N, Ohki I, Chiba T, Saya H, Shirakawa M, Nakao M. Mechanism of transcriptional regulation by methyl-CpG binding protein MBD1. Mol Cell Biol. 2000;20(14):5107–5118. doi: 10.1128/MCB.20.14.5107-5118.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA, Ogurtsov AY, Kondrashov AS. Selection in favor of nucleotides G and C diversifies evolution rates and levels of polymorphism at mammalian synonymous sites. J Theor Biol. 2006;240(4):616–626. doi: 10.1016/j.jtbi.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Arndt PF, Burge CB, Hwa T. DNA sequence evolution with neighbor-dependent mutation. J Comput Biol. 2003;10(3-4):313–322. doi: 10.1089/10665270360688039. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Kumar S. Neutral substitutions occur at a faster rate in exons than in noncoding DNA in primate genomes. Genome Res. 2003;13(5):838–844. doi: 10.1101/gr.1152803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Kumar S. Evolutionary anatomies of positions and types of disease-associated and neutral amino acid mutations in the human genome. BMC Genomics. 2006;7:306. doi: 10.1186/1471-2164-7-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Kumar S. Higher intensity of purifying selection on >90% of the human genes revealed by the intrinsic replacement mutation rates. Mol Biol Evol. 2006;23(12):2283–2287. doi: 10.1093/molbev/msl123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Walter J, Paulsen M, Lengauer T. CpG island mapping by epigenome prediction. PLoS Comput Biol. 2007;3(6):e110. doi: 10.1371/journal.pcbi.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Elefant F, Liebhaber SA, Cooke NE. Locus control region transcription plays an active role in long-range gene activation. Mol Cell. 2006;23(3):365–375. doi: 10.1016/j.molcel.2006.05.041. [DOI] [PubMed] [Google Scholar]
- Gribnau J, Diderich K, Pruzina S, Calzolari R, Fraser P. Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Mol Cell. 2000;5(2):377–386. doi: 10.1016/S1097-2765(00)80432-3. [DOI] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, Wells C, Frith M, Ravasi T, Pang KC, Hallinan J, Mattick J, Hume DA, Lipovich L, Batalov S, Engstrom PG, Mizuno Y, Faghihi MA, Sandelin A, Chalk AM, Mottagui-Tabar S, Liang Z, Lenhard B. et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309(5740):1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- Teodoridis JM, Strathdee G, Brown R. Epigenetic silencing mediated by CpG island methylation: potential as a therapeutic target and as a biomarker. Drug Resist Updat. 2004;7(4-5):267–278. doi: 10.1016/j.drup.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Hirota T, Ieiri I, Takane H, Maegawa S, Hosokawa M, Kobayashi K, Chiba K, Nanba E, Oshimura M, Sato T, Higuchi S, Otsubo K. Allelic expression imbalance of the human CYP3A4 gene and individual phenotypic status. Hum Mol Genet. 2004;13(23):2959–2969. doi: 10.1093/hmg/ddh313. [DOI] [PubMed] [Google Scholar]
- Nurtdinov RN, Neverov AD, Mal'ko DB, Kosmodem'ianskii IA, Ermakova EO, Ramenskii VE, Mironov AA, Gel'fand MS. [EDAS, databases of alternatively spliced human genes] Biofizika. 2006;51(4):589–592. [PubMed] [Google Scholar]
- Efron B, Tibshirani R. An Introduction to the Bootstrap. New York: Chapman & Hall/CRC; 1994. [Google Scholar]
- Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. J Mol Biol. 1987;193(4):723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- Stormo GD, Schneider TD, Gold L. Quantitative analysis of the relationship between nucleotide sequence and functional activity. Nucleic Acids Res. 1986;14(16):6661–6679. doi: 10.1093/nar/14.16.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006. pp. D108–110. [DOI] [PMC free article] [PubMed]
- Favorov AV, Gelfand MS, Gerasimova AV, Ravcheev DA, Mironov AA, Makeev VJ. A Gibbs sampler for identification of symmetrically structured, spaced DNA motifs with improved estimation of the signal length. Bioinformatics. 2005;21(10):2240–2245. doi: 10.1093/bioinformatics/bti336. [DOI] [PubMed] [Google Scholar]
- Lifanov AP, Makeev VJ, Nazina AG, Papatsenko DA. Homotypic regulatory clusters in Drosophila. Genome Res. 2003;13(4):579–588. doi: 10.1101/gr.668403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biemar F, Nix DA, Piel J, Peterson B, Ronshaugen M, Sementchenko V, Bell I, Manak JR, Levine MS. Comprehensive identification of Drosophila dorsal-ventral patterning genes using a whole-genome tiling array. Proc Natl Acad Sci USA. 2006;103(34):12763–12768. doi: 10.1073/pnas.0604484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CAGE enriched non 5' CGI. Tables contain lists of genes with CAGE-enriched CGIs in 3' and intragenic regions separately.
Gene elements. The archive contains row data used for statistical significance of gene elements and CGIs overlap. See Table 1 and Figure 1 in the manuscript.
CGI classes. The archive contains classes of CGIs used for calculation of CAGE tag frequency.
Monte Carlo Simulations. The archive contains Ruby scripts used to evaluate statistical significance of overlapping of gene segments and CGIs. Results of simulations with different "fixed" elements (see Methods) are also included.
Comparative genomics. The archive contains row data and Perl scripts to perform substitution rates calculation.
Sp1. This folder contains the data used for Sp1 binding sites prediction and detailed description of the procedure.