

Abstract
PpiD is a periplasmic folding helper protein of Escherichia coli. It consists of an N-terminal helix that anchors PpiD in the inner membrane near the SecYEG translocon, followed by three periplasmic domains. The second domain (residues 264–357) shows homology to parvulin-like prolyl isomerases. This domain is a well folded, stable protein and follows a simple two-state folding mechanism. In its solution structure, as determined by NMR spectroscopy, it resembles most closely the first parvulin domain of the SurA protein, which resides in the periplasm of E. coli as well. A previously reported prolyl isomerase activity of PpiD could not be reproduced when using improved protease-free peptide assays or assays with refolding proteins as substrates. The parvulin domain of PpiD interacts, however, with a proline-containing tetrapeptide, and the binding site, as identified by NMR resonance shift analysis, colocalized with the catalytic sites of other parvulins. In its structure, the parvulin domain of PpiD resembles most closely the inactive first parvulin domain of SurA, which is part of the chaperone unit of this protein and presumably involved in substrate recognition.
Keywords: parvulin, Pin1, prolyl isomerase, SurA, protein maturation, periplasm, SecYEG
Introduction
Proteins destined to the periplasm of bacteria are synthesized in the cytosol, and most of them are exported in an unfolded form through the Sec translocon.1 Folding in the periplasm is assisted by folding helper proteins, such as protein disulfide isomerases, peptidyl-prolyl cis-trans isomerases and chaperones.2–4 Several of these proteins, in particular FkpA, SurA, and PpiD, combine prolyl isomerase domains with chaperone domains.5–8
The SurA protein participates in the maturation of outer membrane proteins. It consists of an N-terminal chaperone domain that is followed by two prolyl isomerase domains of the parvulin type.9 Studies with intact SurA and various truncated forms revealed that only the C-terminal parvulin domain (SurA-P2) is active as a prolyl isomerase.6 SurA mutants are viable but show defects in the folding of outer-membrane proteins.10–14
PpiD was found in a genetic screen as a multicopy suppressor in a surA deletion strain.15 The simultaneous inactivation of both ppiD and surA genes was initially reported to confer synthetic lethality,16 a finding that was recently disputed.17 Both proteins SurA and PpiD bind to peptide substrates with SurA being more specific than PpiD.18
In PpiD, an N-terminal transmembrane helix (residues 16–34) is followed by three domains that face the periplasm.15 The transmembrane domain anchors PpiD in the inner membrane near the SecYEG translocon,19 the first (residues 35–263) or the third domain (residues 358–623) are probably chaperone domains and predicted to contain a high amount of α-helical structure. The second domain (residues 264–357) is a parvulin-like domain.18
Parvulins are ubiquitous globular protein domains of about 100 residues that catalyze the cis/trans isomerization of prolyl peptide bonds,20,21 which are often rate-limiting steps during protein folding.21,22 They fold into a four-stranded antiparallel β-sheet, surrounded by four α-helices (βα3βαβ2), which is called the parvulin-fold.20 At present, structures of eight parvulins are available in the Protein Data Bank.9,23–30 The different subtypes differ in the length and the composition of the loop between the strand β1 and the helix α1. Human Pin1 is the most prominent member of the parvulin family. It shows a strong preference for substrates in which proline is preceded by a negatively charged residue, in particular by phosphorylated serine or threonine.9,23 This specificity of Pin1 is mediated by several positively charged residues in the β1-α1 binding loop. In the first parvulin (P1) domain of SurA, this region contains mainly hydrophobic residues.9,29
Here, we investigated the structure, the stability, and the catalytic prolyl isomerase function of the parvulin domain of PpiD. Its solution structure was solved by NMR spectroscopy and its interaction with a peptide substrate by chemical shift analysis. The parvulin domain of PpiD resembles most closely the first parvulin domain of SurA in its structure as well as in the lack of prolyl isomerase activity.
Results
The parvulin domain of PpiD (264-357) is a well-folded, monomeric, and stable protein
We produced a recombinant version of the parvulin domain of PpiD that comprises residues 264–357 with a hexa-His tag at the carboxy terminus. This isolated domain, denoted as PpiD*, is well folded and monomeric in solution. The circular dichroism spectrum shows a maximum near 195 nm, as expected for a protein with a high content of secondary structure (data not shown). The thermal unfolding transition is cooperative and shows a midpoint (TM) at 57.1°C and an unfolding enthalpy (ΔHD) of 253 kJ mol−1 [Fig. 1(A)]. TM is independent of the protein concentration, and unfolding is about 80% reversible when the unfolded protein is kept in the thermally unfolded form at 70°C for less than 10 min.
Figure 1.
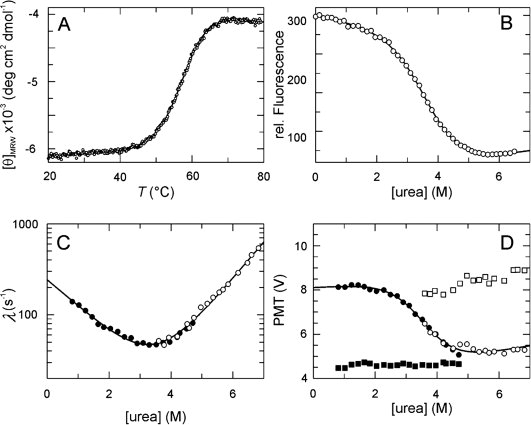
Stability and folding kinetics of PpiD*. (A) Thermal transition of 4 μM PpiD* measured by CD at 222 nm. The two-state analysis (continuous line) results in TM = 57.1 ± 0.1°C and ΔHD = 253 ± 10 kJ mol−1. (B) Urea-induced unfolding transition of 1 μM protein at 25°C measured by protein fluorescence at 340 nm after excitation at 280 nm in 100 mM K phosphate (pH 7.0). The two-state analysis (continuous line) gives values of ΔGD25°C = 15.3 ± 0.6 kJ mol−1, m = 4.2 ± 0.3 kJ mol−1 M−1 and [urea]M = 3.6M. (C) Refolding kinetics (filled symbols) and unfolding kinetics (open symbols). The apparent rate constants λ are shown as a function of the urea concentration. A chevron fitted to the experimental data based on a linear two-state model is shown by the continuous line. The results of the analysis are: knu = 0.99 ± 0.09 s−1, kun = 241 ± 14 s−1, mnu = 0.92 ± 0.02 M−1, mun = − 0.67 ± 0.04 M−1, ΔGD25°C = 14.0 ± 0.4 kJ mol−1, m = 4.0 ± 0.1 kJ mol−1 M−1, [urea]M = 3.5M. (D) Initial (▪,□) and final (•,○) values of the unfolding (open symbols) and refolding (closed symbols) kinetics as a function of the urea concentration. The continuous line indicates the fit of a transition curve to the final values, giving the following parameters: ΔGD25°C = 16.3 ± 2.8 kJ mol−1, m = 4.5 ± 0.7 kJ mol−1 M−1, [urea]M = 3.6M. The folding kinetics were measured after stopped-flow mixing by the change in fluorescence above 320 nm (excitation at 280 nm) in 100 mM K phosphate, pH 7.0 at 25°C.
PpiD* contains one Trp residue, and the fluorescence at 340 nm decreases by 70% between 0 and 6M urea. The urea-induced unfolding transition [Fig. 1(B)] shows a midpoint at 3.6M urea and an m-value of 4.2 kJ mol−1 M−1. The ΔGD values extrapolated from urea-induced unfolding (ΔGD25°C = 15.3 ± 0.6 kJ mol−1) and heat-induced unfolding (ΔGD25°C = 15.7 ± 0.5 kJ mol−1) are very similar, which suggests that PpiD* is a well-folded protein with a two-state equilibrium unfolding transition.
The folding kinetics of PpiD* are well explained by a two-state mechanism
Unfolding and refolding are very rapid, monoexponential reactions at all denaturant concentrations. The logarithms of the microscopic rate constants of refolding and unfolding vary linearly with the concentration of urea, which results in a chevron-type profile for the denaturant dependence of the measured rate constant λ [Fig. 1(C)]. Linear extrapolation to 0M urea gave rate constants of kf = 241 s−1 for refolding and ku = 1.0 s−1 for unfolding.
In the folding kinetics, PpiD thus differs from the parvulin 10 (Par10) from E. coli, which was the first parvulin to be discovered.31,32 Par10 shows two refolding reactions, a fast one that represents the direct, conformational folding and a dominant slow reaction that is limited in rate by the trans-to-cis isomerization of the Gly75-Pro76 bond.33 In PpiD, Pro76 is replaced by Val, which apparently simplifies and accelerates folding.
Figure 1(D) shows the initial and final fluorescence values of PpiD* as obtained in the stopped-flow experiments. Identical final values were observed when unfolding and refolding was performed under the same conditions (between 3 and 5M urea). The final values trace the equilibrium unfolding transition of PpiD* [Fig. 1(D)], and the transition midpoint [urea]M determined from this plot (3.6M) is identical with the value obtained from the equilibrium unfolding transition in Figure 1B. Moreover, the equilibrium constants calculated from the ratio of the microscopic rate constants [Fig. 1(C)] agree well with those derived from the equilibrium unfolding transition in Figure 1(B). This confirms that the folding of PpiD* is indeed a reversible two-state reaction.
The solution structure of PpiD* reveals a parvulin fold
The solution structure of PpiD* was determined by NMR spectroscopy. Resonances of 15N-labeled PpiD* were assigned by 15N-NOESY-HSQC, 15N-TOCSY-HSQC, and 1H-NOESY spectra. Using 2357 distance restraints and 162 TALOS34 derived dihedral angle restraints, an ensemble of 50 structures was calculated by ARIA 2.0.35 Experimental restraints and structure statistics for the 10 lowest energy structures are summarized in Table I. The ensemble is well defined between residues 268 and 357 [Fig. 2(A)]. According to the statistics and the quality assessment with PROCHECK-NMR37 the NMR analysis yielded a family of structures of very high quality, with 95% of the backbone dihedral angles residing in the most favored regions of the Ramachandran plot.
Table I.
Statistics of the Structure Calculation
| Experimental restraints | |
| NOEs | 2357 |
| Dihedrals | 164 |
| NOE violations (Å) | |
| >0.5 | 0.00 ± 0.00 |
| >0.3 | 0.00 ± 0.00 |
| >0.1 | 14.80 ± 2.95 |
| Energies (kcal mol−1) | |
| Etot | 160.1 ± 5.8 |
| Ebond | 5.2 ± 0.3 |
| Eangle | 68.1 ± 2.4 |
| Eimproper | 18.7 ± 1.9 |
| ENOE | 28.4 ± 2.2 |
| Ecdih | 39.6 ± 4.5 |
| rms deviation (Å) | |
| Backbone (res. 268–357) | 0.77 ± 0.20 |
| Heavy atom (res. 268–357) | 1.29 ± 0.19 |
| NOE | 0.0155 ± 0.0006 |
| Bond | 0.00181 ± 0.00005 |
| rms deviation (deg) | |
| Angle | 0.394 ± 0.007 |
| Improper | 0.390 ± 0.019 |
| Ramachandran analysisa residue 268–357 (%) | |
| Most favoured | 95.1 |
| Additionally allowed | 4.6 |
| Generously allowed | 0.3 |
| Disallowed | 0.0 |
Analysis by PROCHECK.37
Figure 2.
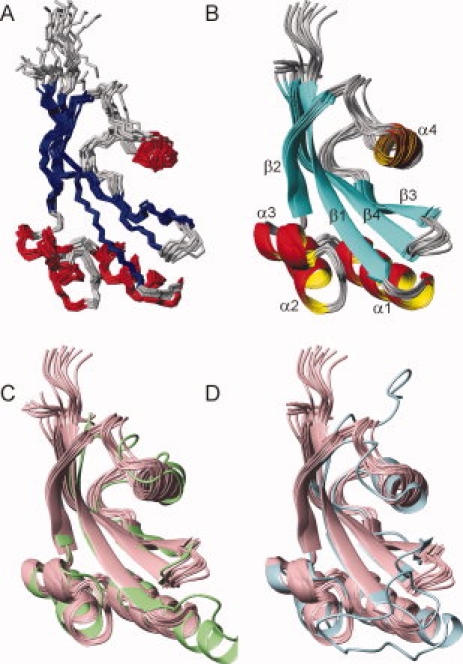
Solution structure of PpiD*. (A) Ensemble of the 10 lowest-energy NMR structures superimposed on the backbone heavy atoms in ordered regions of the protein. (B) Ribbon representation for residues 264–357 in the same orientation as in (A). β strands are colored in blue, α helices in red and coil regions in gray. The α helices and β strands are labeled sequentially from the N- to the C-terminus. (C, D) Superimposition of the NMR structures of PpiD* (pink) and (C) the crystal structure of the first parvulin domain of SurA (2pv2.pdb, green) and (D) the crystal structure of human Pin1 (1pin.pdb, cyan). The figures were prepared with MolMol.36
The PpiD* structure consists of four α-helices (residues 279–291, 295–301, 306–311, and 325–330) and a central four-stranded β-sheet (residues 268–276, 314–319, 338–343, and 348–357) revealing a parvulin fold [Fig. 2(B)].26,29 In its overall structure, PpiD* closely resembles the first parvulin domain of SurA [Fig. 2(C), with a backbone RMSD of 1.5 Å] and human Pin1 [Fig. 2(D)], the best studied parvulin. At the sequence level, the parvulin domain of PpiD shares rather low identities of 34.4% and 26.9%, respectively, with SurA and Pin1 (Supporting Information Figure S4). Within the elements of secondary structure, the backbone atoms of PpiD* and of human Pin1 (as taken from the X-ray structure 1pin.pdb) superimpose with a RMSD of 2.0 Å. The four β strands and helices 1–3 are virtually identical, only helix 4 is slightly re-oriented and shortened in PpiD*. The turns and loops connecting the secondary structure elements α2 with α3, α3 with β2, and α4 with β3 are also similar in PpiD* and Pin1, those connecting β1 with α1, α1 with α2, β2 with α4 and β3 with β4 are different. In particular, the connection between β1 and α1 is strongly shortened in PpiD* [Fig. 2(D)]. In Pin1, this long loop of 17 residues harbors the cluster of positively charged residues, which determines the specificity of Pin1 for phosphorylated Ser/Thr-Pro substrates. Interestingly, the connection between β1 and α1 is also longer in the P1 domain of SurA, which differs in this region from both Pin1 and PpiD* [Fig. 2(C,D)]. These differences are accompanied by different lengths of helix α1, which is extended by one and two turns, respectively, in Pin1 and SurA, relative to PpiD* [Fig. 2(C,D)]. Large variations between the individual structures of the NMR ensemble are observed for the turn between β3 and β4, the loop between β2 and α4 (320–324), the loop between α4 and β3 (331–337), and for helix α4. Whether these deviations in the respective chain regions of PpiD* originate from a high flexibility in solution on a pico-to-nanosecond time scale was elucidated by the following relaxation study.
PpiD is a rigid molecule with a defined structural core and mobile loops
The backbone dynamics of PpiD* on the ps-ns timescale was examined by recording 15N R1 [Fig. 3(A)] and R2 [Fig. 3(B)] relaxation rates and hNOE-values [Fig. 3(C)],39 at two magnetic field strengths (B0 = 14.1 T and B0 = 18.8 T). Based on these relaxation data and an axially symmetric model (axes of inertia 1:0.98:0.84) for overall diffusion of the molecule we calculated order parameters S2 [Fig. 3(D)], internal correlation times (τe), additional exchange terms (Rex) and an overall correlation time (τm) of motion of 7.3 ns40 at 25°C. Only 33 of 91 amides required additional Rex contributions to adequately describe the relaxation rates, 46 required τe terms. They do not cluster over the sequence. This indicates that ps-ns dynamics is described well by a simple model for local motions and no additional dynamics is taking place in this time range.
Figure 3.
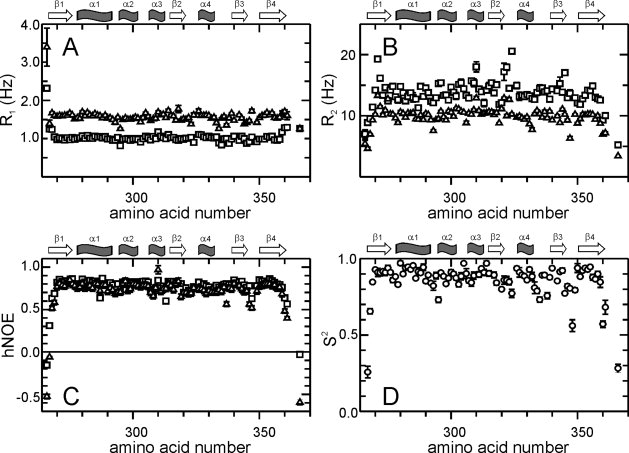
Picoseconds to nanoseconds dynamics of PpiD*. (A) R1 rates, (B) R2 rates, (C) 15N hetero nuclear NOEs and order parameter S2 are plotted as a function of the amino acid sequence. Parameters shown as squares in (A–C) were derived at 800 MHz, those shown as triangles at 600 MHz proton resonance frequency. (D) Order parameters S2 calculated using both datasets in MODELFREE.38 The secondary structure elements are indicated at the top of each panel.
This analysis confirms that the turn between β3 and β4 (residues 344–347), the loops between β2 and α4 (residues 320–324) and between α4 and β3 (residues 331–337) and the chain termini show increased dynamics. This is indicated by a drop of the hNOE values [Fig. 3(C)] below 0.7 (especially for turn β3/β4) and a reduced order parameter (S2) below 0.8 [Fig. 3(D)]. The remaining regions of PpiD* are very rigid (S2 0.8 or higher) on the ps-ns timescale. In its local dynamics, PpiD* resembles the parvulin domain of PrsA of Staphylococcus aureus.25 Slower dynamics on a μs-ms timescale could not be observed by R2-dispersion experiments41,42 (data not shown). The recorded R2eff values show no dependence on the applied field strength by usage of CPMG pulse trains. This holds for all NH vectors.
The protection from H/D exchange43 with the solvent of individual amide NH of PpiD* was used to probe the local stability of folded PpiD* at pH 7.0 and 25°C. 66 NH exchanged with the solvent within the dead time of the experiment, indicating that they show protection factors that are smaller than 100. As expected, all nonhydrogen-bonded regions, the chain termini, and the loops and turns belong to this class of fast exchanging NH. Rapid exchange was also observed for strand β2 and the exposed short helix α3. These structural elements apparently exchange via local unfolding (breathing) and thus do not belong to the cooperatively unfolding core of the molecule. Still, they show a well-ordered structure and do not differ from other elements of structure in ps-ns dynamics.
33 residues exchanged slowly. Nine of them showed protection factors between 100 and 1000 [colored light blue in Fig. 4(B)], and 24 were protected 1000–10,000-fold [colored dark blue in Fig. 4(B)]. Most of them cluster around a value of 2000 [Fig. 4(A)] leading to a ΔGD of about 19 kJ mol−1, which is 3–4 kJ mol−1 higher than the overall value obtained from the thermal and the urea-induced unfolding transitions (Fig. 1).
Figure 4.
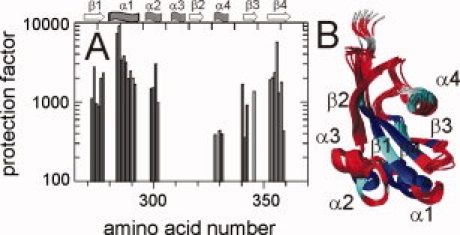
Protection of PpiD* from amide NH exchange. (A) The protection factors derived from H/D exchange experiments in 100 mM K phosphate (pH 7.0) at 25°C are plotted as a function of the amino acid sequence. The secondary structure elements are indicated at the top of the figure. (B) Protection factors higher than 1000 are mapped in blue on the backbone structure, protection factors between 100 and 1000 in cyan, and residues exchanging in the dead time of the experiment are colored in red. Residues lacking an amide proton are colored in gray.
Residues with high protection factors are found predominantly in the central β-strands β1 (residues 268–276) and β4 (residues 348–358), and in helix α1 (residues 279–291), which packs against the center of the β sheet. These elements of secondary structures apparently form a molecular core, which is disrupted only in the course of global unfolding. Several highly protected amide NH were also found in strand β3 (residues 337–339 and 342) and helix 2 (residues 297–300). Both are adjacent to the structural core of PpiD* defined by β1, β4, and α1. With protection factors <500, helix 4 is less protected and, apparently, not linked with this structural core in terms of its exchange dynamics.
In summary, the elements of secondary structure of PpiD* are well defined and superimpose well with the corresponding structures in the homologues Pin1 and SurA. The backbone of PpiD* is rigid on the ps-ns timescale. The peripheral chain regions can undergo local unfolding reactions. Helix α1, the strands β1 and β4 and several adjacent areas define the core of the molecule, which loses its structure only in the course of global unfolding.
PpiD* does not catalyze prolyl isomerization in peptides or proteins
To investigate the prolyl isomerase activity of PpiD and its parvulin domain, we initially used the tetrapeptide succinyl-Ala-Ala-Pro-Phe-4-nitroanilide (Suc-AAPF-pNA) in a protease-coupled assay,44 which exploits the finding that α-chymotrypsin can hydrolyze the 4-nitroanilide amide bond only when the Ala-Pro bond is in the trans conformation. Unlike originally reported,15 we could not detect a prolyl isomerase activity for full-length PpiD or for its parvulin domain. The protease coupled assay can be used only for prolyl isomerases that are protease-resistant. To avoid the coupling with isomer-specific proteolysis, a protease-free assay with improved sensitivity was developed.45,46 For this assay, tetrapeptides are used that carry an aminobenzoyl (Abz) group at the amino terminus and a para nitroanilide (pNA) group at the carboxy terminus. In their general sequence Abz-Ala-Xaa-Pro-Phe-pNA, the position Xaa is occupied by various natural amino acids. The extent of quenching of the Abz fluorescence by the pNA group is sensitive to the isomeric state of the prolyl bond in these peptides. This assay was validated for all three families of prolyl isomerases.46
The prolyl isomerases SlyD47–49 and cyclophilin1850,51 from E. coli (both at 0.5 μM) strongly accelerated the isomerization of the peptide with a Leu-Pro bond [Fig. 5(A)]. In contrast, 10 μM PpiD* could not accelerate this reaction [Fig. 5(A)]. Several prolyl isomerases, in particular those from the FKBP family, show high substrate specificities with respect to the amino acid at position Xaa before the proline.52 To examine, whether PpiD* might have a peculiar substrate specificity as well, we employed additional assay peptides, in which the position before proline was occupied by a negatively charged (Glu), a positively charged (Lys), a small (Ala), or a large hydrophobic (Phe) residue. For all these peptides, a catalysis of prolyl isomerization could not be observed for PpiD*, even when its concentration was increased to 20 μM (Supporting Information Table SI). These data suggest that PpiD* is in fact devoid of prolyl isomerase activity.
Figure 5.
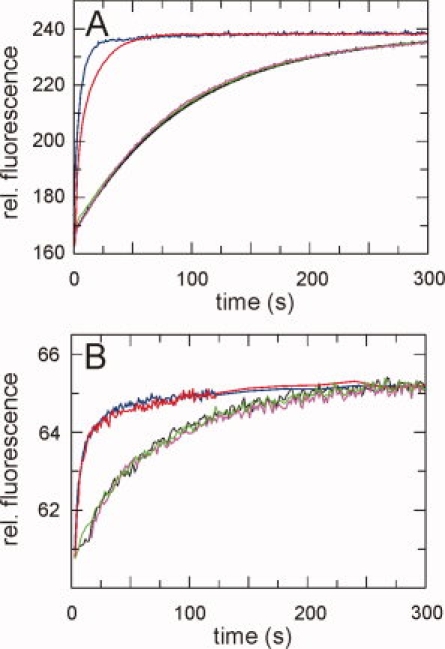
Assays for the prolyl isomerase activity of PpiD. (A) Kinetics of cis/trans isomerization of 5 μM Abz-Ala-Leu-Pro-Phe-pNa followed by fluorescence at 416 nm. (B) Slow refolding of 1 μM of the N2 domain of the gene-3-protein of phage fd, followed by the change in fluorescence at 340 nm after excitation at 280 nm. All kinetics were measured at 15°C in 0.1M K phosphate, pH 7.0. Kinetics without PPIase are shown in black, in the presence of 10 μM PpiD* in green, in the presence of full length PpiD in magenta, in the presence of 0.5 μM SlyD in blue and in the presence of 0.5 μM cyclophilin in red.
We also examined whether PpiD* is able to catalyze prolyl isomerization in the course of protein folding and measured its effect on the proline-limited folding reactions of two proteins, the reduced and carboxymethylated form of the S54G/P55N variant of ribonuclease T1 (RCM-T1)53 and the N2 domain of the gene-3-protein of the phage fd.54 Again, a catalysis by PpiD* could not be detected [Fig. 5(B) and Figure S1, Supporting Information]. Full length PpiD was equally inactive in the peptide and the protein folding assays [Fig. 5(A,B)].
PpiD* has the same peptide binding site as other parvulins
The lack of prolyl isomerase activity might originate from impaired binding of PpiD* to its substrate. To examine whether PpiD* is able to bind to proline-containing peptides, we titrated 0.59 mM15N labeled PpiD* with Suc-Ala-Leu-Pro-Phe-pNA, a highly soluble peptide substrate, which is commonly used in prolyl isomerase assays.21 Adding 2.29 mM of this peptide led to shifts stronger than 0.035 ppm for the NH resonances of 12 residues [Fig. 6(A)]. In all cases, the resonances changed their positions continuously during the titration, indicating that binding and dissociation occurred in the fast exchange limit of the NMR chemical shift timescale (about 100 ms). Saturation could not be reached in these titration experiments, indicating that the dissociation constant of the complex between the peptide and PpiD* is in the range of 1 mM. Similar values were observed previously for other parvulins25,30 and for cyclophilin18.55–57
Figure 6.
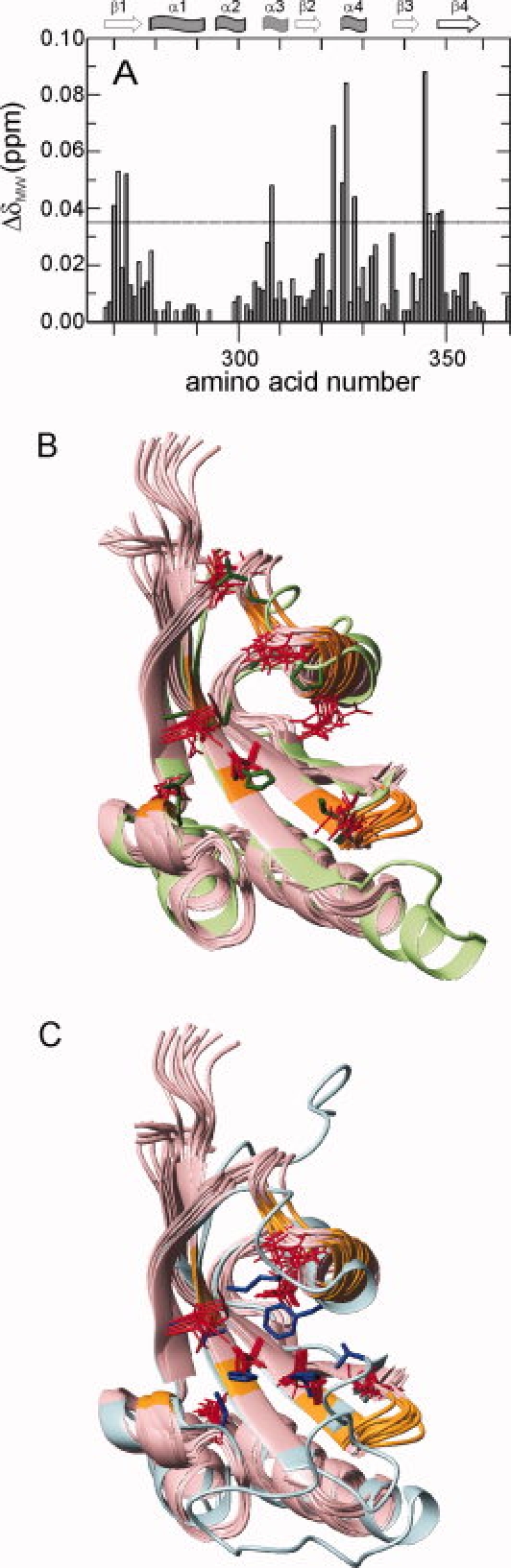
Peptide binding site of PpiD*. (A) The change in the averaged weighted chemical shift ΔδMW, caused by titration with Suc-Ala-Leu-Pro-Phe-pNA) at a ratio of 4:1 (2.29 mM peptide, 0.59 mM PpiD*) at 25°C, 0.1M K phosphate and pH 7.0 is plotted as a function of the amino acid sequence. Secondary structure elements are indicated on top. The threshold of 0.035 ppm is indicated by the horizontal line. The backbone of residues of PpiD* with a ΔδMW larger than this threshold (R270, Y271, I273, S308, I323, D325, E326, K328, S345, V346, F348, L349) are colored in orange in (B) and (C). (B) Overlay of the solution structure of PpiD* (in pink) and the crystal structure of the first parvulin domain of SurA (2pv2.pdb, in light green) in ribbon representation. The side chains of the active site residues of SurA (H178, Q223, M231, E238, L239, P240, V263) are shown in green, the corresponding side chains of PpiD* (I273, S308, M315, T322, P324, E326, V346) in red. (C) Overlay of the solution structure of PpiD* (in pink) and the crystal structure of human Pin1 (1pin.pdb, in cyan) in ribbon representation. The side chains of the active site residues of Pin1 (H59, C113, L121, M130, F134, T152, H157) are shown in blue, the corresponding side chains of PpiD* (I273, D305, M315, P324, L327, S344, L349) in red.
Figure 6(B,C) shows the backbone positions of the 12 residues for which the NH chemical shifts changed by more than 0.035 ppm upon peptide binding (R270, Y271, I273, S308, I323, D325, E326, K328, S345, V346, F348, L349) in comparison with the first parvulin domain of Sur A [Fig. 6(B)] and the parvulin domain of Pin1 [Fig. 6(C)]. The residues involved in peptide binding are all located along the rim of a groove in the PpiD* structure, which, in the homologous parvulins (the first parvulin domain of SurA, human Par14,29 Pin1At from Arabidopsis thaliana27 and PrsA from Bacillus subtilis30) forms the substrate binding site. The residues that are assumed to participate in substrate binding and catalysis (in SurA: H178, Q223, M231, E238, L239, P240, V263; in Pin1: H59, C113, L121, M130, F134, T152, H157) are shown in green and blue in Figure 6(B,C), respectively. The respective side chains of PpiD* in fact occupy similar positions as the side chains of the corresponding residues H178, M231, E238, and V263 of SurA and H59, C113, L121, and H157 of Pin1. The chemical nature of these side chains is, however, different, which probably is correlated with the lack of prolyl isomerase activity of PpiD*.
Prolyl isomerase activity could not be generated by substitutions at the peptide binding site of PpiD*
In its architecture, the binding site for proline-containing peptides in PpiD* resembles the catalytic prolyl isomerase site of Pin1, but PpiD* is apparently inactive. To examine whether a possibly latent prolyl isomerase function in PpiD* can be activated by mutations towards the Pin1 sequence at the seven sites that were suggested to be involved in the prolyl isomerase activity of Pin1 [Fig. 6(C)],23 we replaced the corresponding amino acids of PpiD* with those present in Pin1, first individually in seven single mutants (I273H, D305C, M315L, P324M, L327F, S344T, L349H) and then in various combinations.
To examine the consequences of these substitutions for the stability of PpiD*, we measured thermal unfolding transitions for all variants (Supporting Information Figure S1A). Two substitutions (M315L, S344T) were found to be stabilizing, three were mildly destabilizing (P324M, L327F, D305C) and two (I273H and L349H) were strongly destabilizing. They decreased the melting temperature TM by 8.6 and 14.5 degrees, respectively, indicating that these two residues are important for protein stability. This is consistent with results obtained for other parvulins. In PrsA from B. subtilis the mutation H122A (corresponding to I273 in PpiD*) leads to unfolding,30 and in Pin1, mutations at position 157 (corresponding to position L349 in PpiD*) are also strongly destabilizing.58
The potential prolyl isomerase activities of the seven variants with single substitutions were assayed by using five different tetrapeptide substrates and two proline-limited protein folding reactions. Catalytic activities could not be detected, even when PpiD* concentrations as high as 20 μM were employed (Supporting Information Table SI).
Next, the seven single substitutions were combined in various ways, leading to eight variants with two to seven substitutions (Supporting Information Table SI). All variants were completely folded at 15°C, where the prolyl isomerase assays were performed and showed circular dichroism spectra similar to the wild-type protein (data not shown). The effect on the stability of the amino acid replacements were almost additive (Supporting Information Figure S1B) and the seven-fold variant showed a TM value of 40.9°C, which is 16.4 degrees lower than the TM of wild-type PpiD*.
All PpiD* variants, including the sevenfold variant, were inactive in our prolyl isomerase assays (Supporting Information Figure S2, Table SI). Grafting all residues that have been implicated to form the active site of Pin1 onto the conserved scaffold of PpiD* is apparently not sufficient to create a prolyl isomerase activity or to activate a latent activity in this protein.
Discussion
The parvulin domain of PpiD (PpiD*) is a well-folded protein with a high stability and a simple two-state folding mechanism. The analysis by NMR spectroscopy resulted in a well determined structure with a parvulin fold, which shows the highest similarity to the first parvulin domain of SurA.
Dartigalongue and Raina reported very high prolyl isomerase activities of up to 3 × 109 M−1 s−1 for PpiD in a protease-coupled peptide assay.15 We were unable to confirm this very high prolyl isomerase activity for the isolated parvulin domain by using the same protease-coupled PPIase assay. We extended this investigation to full-length PpiD and used a panel of five different peptides in a protease-free assay with improved sensitivity.46 In addition, a protein folding assay with refolding proteins as substrates was employed. With all these assays, we could not detect prolyl isomerase activity, even at high PpiD concentration. The origin for this discrepancy remains unclear. All attempts to activate PpiD* by transplanting seven key residues of the catalytically active homolog Pin1, individually or in various combinations, to PpiD* remained unsuccessful. It is thus unlikely that PpiD* possesses a latent prolyl isomerase function that can be activated by variations in the substrate or mutations in PpiD*. PpiD* interacts, however, with a standard tetrapeptide as used for the assay, and the binding site, as identified by NMR resonance shift analysis, colocalized with the catalytic sites of other parvulins.
The natural function of PpiD is not known. It is ubiquitous in Gram-negative bacteria and the residues of the substrate binding site of the parvulin domain are highly conserved (Supporting Information Figure S3). This domain is probably important for the biological function of PpiD. Dartigalongue and Raina used a screen based on the ability to induce the σE response as well as hypersensitivity to novobiocin15 to identify residues that are critical for PpiD function in vivo. Four of the isolated point mutations mapped to the parvulin domain. The NMR analysis of PpiD* provides a structural explanation for the effects of the inactivating mutations G312R and G313R. Both are located in the loop that connects helix α3 with strand β2. These positions are sterically demanding and residues other than glycine probably interfere with the proper folding of PpiD*. The other two positions identified, G347 and I350, are located at the peptide binding site, suggesting that substrate binding to the parvulin domain is important for the function of PpiD.
Originally, PpiD was identified as a multicopy suppressor in a surA deletion strain,15 and in fact, the parvulin domain of PpiD shows the closest structural similarity to the first parvulin domain of SurA. SurA consists of three globular domains and a carboxyterminal helix.9 The N-terminal domain, which is also α-helical, the first parvulin domain and the C-terminal helix form a compact structural entity, which is thought to function as a chaperone. The second parvulin domain protrudes from this entity, to which it is tethered by two long and presumably flexible linkers.9 It is active as a prolyl isomerase, but apparently dispensable for the function of SurA in vivo.6,9 The first parvulin domain, which is structurally similar to the parvulin domain of PpiD, is devoid of prolyl isomerase activity as well, both in intact SurA and as an isolated domain.6 As a part of the chaperone entity, it seems to cooperate with the N-terminal domain of SurA. It was suggested that this domain acts as a binding module for unfolded proteins and confers specificity for unfolded outer-membrane proteins.6,13,59,60
The similarity between PpiD and SurA extends also to the helical aminoterminal domains of the two proteins. The 86–191 region of PpiD shows 31% sequence identity and 50% similarity with the 65–168 region of SurA, which comprises the major part of the chaperone entity of this protein (residues 21–168).9 In the 54–85 region of PpiD, the similarity to SurA is less pronounced, but this region is predicted to form two short β-sheets and a long α-helix as the corresponding 21–64 region of SurA. The long helix α1 is involved in substrate binding in SurA9 and possibly also in PpiD. These similarities in sequence and structure suggest that the aminoterminal regions of the two proteins share an identical fold. Together with the also highly conserved inactive parvulin domain, they probably form homologous chaperone entities with similar architecture and function.
The binding interface of the PpiD parvulin domain is nonpolar, and similar to the corresponding binding site of the inactive parvulin domain of SurA. This explains the finding that these domains bind to similar peptide substrates and that binding is sensitive to the presence of Triton X-100. Using a variety of peptide substrates, Stymest and Klappa found, that the interaction with PpiD usually requires longer peptide chains compared with SurA, and that PpiD seems to be less specific than SurA.18 This difference might reflect the different biological functions of the two proteins. PpiD is anchored in the inner membrane, near the SecYEG translocon19 and can be crosslinked to translocated proteins after their release into the periplasm. PpiD is thus possibly a periplasmic gatekeeper at the exit site of the Sec translocon and involved in the folding of many exported proteins. SurA is a soluble periplasmic protein and presumably participates in the maturation of proteins destined to the outer membrane.15
The folding helper proteins trigger factor in the cytosol and SurA and PpiD in the periplasm of E. coli show interesting common design principles. Trigger factor is targeted to the exit site for newly synthesized protein chains by a ribosome-binding domain, PpiD is located next to the exit site of the SecYEG translocon by a membrane-anchor helix. The chaperone domains of trigger factor, SurA and, presumably, also PpiD share a common helical fold. In trigger factor and in SurA, a prolyl isomerase domain is inserted into a long loop of the chaperone domain. This prolyl isomerase module is an FKBP domain in trigger factor but a parvulin domain in SurA. PpiD lacks such a flexibly tethered active prolyl isomerase domain, but it shares with SurA an inactive parvulin domain, which presumably forms an integral part of the chaperone unit in both proteins. Clearly, further structural and functional studies, in particular of the other domains of PpiD, are needed to elucidate the common functions of this family of chaperones and their structural basis.
Materials and Methods
Expression and purification of variants of PpiD*
For the expression of the parvulin domain of PpiD* (residues 264–357 of mature PpiD, plus a C-terminal Ala2His6 extension) the gene fragments were PCR-amplified from E. coli XL1-Blue. The fragments were cloned into the expression plasmid pET11a (Novagen, Madison, Wisconsin) via its NdeI and BamHI restriction sites, and the proteins were overproduced in E. coli BL21(DE3) ΔslyD (gift from B. Eckert). Site-directed mutagenesis of PpiD* was performed by Quik-Change (Stratagene).
After lysis of the cells in 50 mM Tris/HCl, 50 mM NaCl, pH 8.0, 40 mM imidazol with a microfluidizer and centrifugation, all PpiD*-variants were found in the supernatant. The proteins were purified by immobilized metal-affinity chromatography on a Ni-NTA column (elution with 250 mM imidazole) and then subjected to size-exclusion chromatography in 100 mM K phosphate, pH 7.0 on a Superdex HiLoad column (GE Healthcare). The protein-containing fractions were pooled and concentrated in Amicon Ultra units (Millipore). Yields were about 5–30 mg L−1. Isotopically labeled 15N-NMR-samples were produced using M9 minimal medium containing 15NH4Cl as the source of 15N and supplemented with vitamin mix. The isolated N2 domain variants and RCM-T1 were expressed and purified as described.61–63
For the expression of PpiD (residues 37–623 of mature PpiD, without the N-terminal transmembrane helix), the gene fragment was PCR-amplified from E. coli XL1-Blue. The fragment was cloned into the expression plasmid pET11a (Novagen, Madison, WI) via its NdeI and BamHI restriction sites, and the protein was overproduced in E. coli BL21(DE3) (Stratagene, La Jolla, CA). The cells were lysed as described above, and the supernatant was applied to a Fractogel EMD DEAE-650(M) column (2.5 × 20 cm2) equilibrated with 50 mM Tris/HCl, 50 mM NaCl. Using a linear 0–1M NaCl gradient (500 mL), the protein was eluted, concentrated and subjected to size-exclusion chromatography in 50 mM Tris (pH 8.0) on a Superdex HiLoad column (GE Healthcare). The protein-containing fractions were pooled and subjected to a second anion-exchange chromatography on a MonoQ HR5/5 column and eluted with a linear 0–1M NaCl gradient in 50 mM Tris (pH 8.0). The protein-containing fractions were pooled and concentrated in an Amicon Ultra unit (Millipore). The yield was about 100 mg L−1.
Equilibrium unfolding transitions
Samples of PpiD* (1.0 μM) were incubated for 1 h at 25°C in 100 mM K phosphate, pH 7.0 and varying concentrations of urea. The fluorescence of the samples was measured in 1-cm cuvettes at 340 nm (10-nm band width) after excitation at 280 nm (5-nm band width) in a Hitachi F4010 fluorescence spectrometer. The experimental data were analyzed according to a two-state model by assuming that ΔGD as well as the fluorescence emissions of the folded and the unfolded form depend linearly on the urea concentration. A nonlinear least-squares fit with proportional weighting of the experimental data was used to obtain ΔGD as a function of the urea concentration.64
The heat induced unfolding transitions were measured in a Jasco J-600A spectropolarimeter equipped with a PTC 348 WI peltier element at a protein concentration of 4 μM in 100 mM K phosphate, 1 mM EDTA, pH 7.0 at a heating rate of 1 °C min−1. The transitions were monitored by the increase of the CD signal at 222 nm with 1 nm band width and 10-mm path length. The experimental data were analyzed on the basis of the two-state approximation,63 with a calculated heat capacity change ΔCp of 6000 J mol−1 K−1.65
Folding experiments
A DX.17MV stopped flow spectrometer from Applied Photophysics (Leatherhead, UK) was employed to follow the urea-induced unfolding and refolding kinetics. All experiments were performed in 100 mM K phosphate, pH 7.0 at 25°C. The native or the unfolded (in 8.8M urea) protein was diluted 11-fold with urea solutions of varying concentrations. The kinetics were followed by the change in fluorescence above 320 nm after excitation at 280 nm (10-nm bandwidth) in an observation cell with 2 mm path length. A 0.5-cm cell with acetone was placed between the observation chamber and the photomultiplier to absorb scattered light from the excitation beam. The kinetics were measured at least eight times under identical conditions and averaged.
Prolyl isomerase activity assays
The prolyl isomerase activities of the PpiD* variants were measured by using tetrapeptides with the general formula Abz-Ala-Xaa-Pro-Phe-pNA. They show an aminobenzoyl (Abz) group at the aminoterminus and a 4-nitroanilide (pNA) group at the carboxyterminus, Position Xaa before proline was occupied by Glu, Lys, Ala, or Phe. For the assay, the peptide substrates Abz-Ala-Xaa-Pro-Phe-pNa (3 mM) were dissolved in trifluoroethanol containing 0.5M LiCl. Under these conditions, about 50% of the peptide molecules are in the cis conformation. Upon 600-fold dilution into aqueous buffer the cis content decreases to about 10%. The kinetics of the decrease in cis content was measured by the change in fluorescence at 416 nm (5-nm bandwidth) after excitation at 316 nm (3-nm bandwidth) in a Jasco FP-6500 fluorescence spectrophotometer. The assays were carried out in 50 mM Hepes/NaOH, (pH 7.8) at 15°C. Under these conditions, the cis-to-trans isomerization of the prolyl bond was a mono-exponential process.46 The folding experiments with RCM-T1 and the variants of N2 as substrates were performed as described.62,66
NMR measurements and NMR structure calculations
All spectra were measured in 100 mM K phosphate, pH 7.0 at 25°C, including 10% (v/v) D2O, processed with NMRPipe67 and analyzed with NMRView.68 For resonance assignment and structure calculation 3D 15N-NOESY-HSQC (120 ms mixing time), 3D 15N-TOCSY-HSQC (80 ms mixing time) and 2D 1H1H-NOESY (120 ms mixing time) spectra of a 5 mM15N PipD* sample were acquired at a Bruker AvanceIII 800 NMR spectrometer equipped with an inverse triple-resonance cryoprobe. A complete assignment of all 15N resonances and 1H without Lysine Hζ, Methionine Hɛ and residues of the histidine tag could be achieved. A final ensemble of 50 structures was calculated with ARIA 2.035 using 2357 NOESY derived ambiguous distance restraints and 162 TALOS34 derived dihedral angle restraints (Table I). The final 10 lowest energy structures were water refined. Their stereochemistry was analyzed with PROCHECK37 and structure ensembles were aligned according to their secondary structure elements and analyzed using MOLMOL.36 Final energies, r.m.s. deviations from the averaged structure and from ideal geometry as well as the Ramachandran analysis are given in Table I. The backbone superposition of the NMR structure of PpiD*, the first parvulin domain of SurA (2pv2.pdb) and human Pin1 (1pin.pdb) was performed on the basis of the lowest energy NMR structure of PpiD*.
Dynamics, H/D exchange, and substrate binding
Spectra for the analysis of the nanosecond-to-picosecond dynamics of PpiD*, for following the H/D exchange and for substrate binding were acquired at a Bruker AvanceIII 600 NMR spectrometer equipped with a triple-resonance probe. Measurements of the dynamics were complemented with spectra acquired at a Bruker AvanceIII 800. R1 rate constants, R2 rate constants and 1H-15N heteronuclear NOEs39 were analyzed using MODELFREE38 and TENSOR 2.0.28 We have chosen an axially symmetric model for overall diffusion of the molecule (principal axes of inertia are 1: 0.98:0.84). H/D exchange was started by exchanging the aqueous buffer against D2O buffer (pD 6.6, pH-meter reading) using a PD MiniTrap G-25 column (GE Healthcare) and followed by a series of 30 15N-HSQC spectra. Protection factors were calculated assuming an EX2 exchange mechanism and using exchange rates from reference peptides.43 Suc-Ala-Leu-Pro-Phe-pNA was titrated in 10 steps from an 8 mM stock solution to 820 μM15N labeled PpiD* up to a fourfold excess (2.29 mM peptide, 0.59 mM PpiD*). Changes in the chemical shifts were followed by 15N-HSQC spectra and quantified by calculating the averaged weighted chemical shift ΔδMW(ppm).69 A cutoff value of ΔδMW (ppm) = 0.035 was used for structural interpretations.
Data deposition
The atomic coordinates have been deposited in the RCSB Protein Data Bank and are available under the accession code 2KGJ. Resonance assignments and restraints are deposited in the BMRB (16211).
Acknowledgments
The authors thank G. Zoldák and T. Aumüller for a generous gift of the labeled substrate peptide for the prolyl isomerase assay and the members of our groups for many fruitful discussions.
Glossary
Abbreviations:
- PpiD
periplasmic folding helper protein from Escherichia coli;
- PpiD*
parvulin-like domain of PpiD (residues 264–357)
- PPIase
peptidyl-prolyl cis/trans isomerase
- CD
circular dichroism
- TM
midpoint of a thermal unfolding transition
- ΔHD
vańt Hoff enthalpy of denaturation at TM
- ΔGD
Gibbs free energy of denaturation
- m
cooperativity value of a denaturant-induced equilibrium unfolding transition
- Δτ
time constant of a folding reaction
- ku
microscopic rate constant and mu (= ∂lnku/∂[urea]), kinetic m-value of unfolding
- kf, mf
microscopic rate constant and kinetic m-value of refolding
- NOESY
nuclear Overhauser enhancement and exchange spectroscopy
- HSQC
hetero single quantum coherence
- hNOE
15N hetero nuclear NOE
- HX
hydrogen exchange
- NH
amide proton
- RCM-T1
reduced and carboxymethylated form of the S54G/P55N double mutant of ribonuclease T1
- N2
N2 domain of the gene 3 protein of phage fd.
References
- 1.Driessen AJ, Manting EH, van der Does C. The structural basis of protein targeting and translocation in bacteria. Nat Struct Biol. 2001;8:492–498. doi: 10.1038/88549. [DOI] [PubMed] [Google Scholar]
- 2.Rietsch A, Beckwith J. The genetics of disulfide bond metabolism. Annu Rev Genet. 1998;32:163–184. doi: 10.1146/annurev.genet.32.1.163. [DOI] [PubMed] [Google Scholar]
- 3.Ito K, Inaba K. The disulfide bond formation (Dsb) system. Curr Opin Struct Biol. 2008;18:450–458. doi: 10.1016/j.sbi.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Kleinschmidt JH. Membrane protein folding on the example of outer membrane protein A of Escherichia coli. Cell Mol Life Sci. 2003;60:1547–1558. doi: 10.1007/s00018-003-3170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behrens S. Periplasmic chaperones—new structural and functional insights. Structure. 2002;10:1469–1471. doi: 10.1016/s0969-2126(02)00893-6. [DOI] [PubMed] [Google Scholar]
- 6.Behrens S, Maier R, de Cock H, Schmid FX, Gross CA. The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO J. 2001;20:285–294. doi: 10.1093/emboj/20.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramm K, Plückthun A. The periplasmic Escherichia coli peptidylprolyl cis,trans-isomerase FkpA. II. Isomerase-independent chaperone activity in vitro. J Biol Chem. 2000;275:17106–17113. doi: 10.1074/jbc.M910234199. [DOI] [PubMed] [Google Scholar]
- 8.Ramm K, Plückthun A. High enzymatic activity and chaperone function are mechanistically related features of the dimeric E. coli peptidyl-prolyl-isomerase FkpA. J Mol Biol. 2001;310:485–498. doi: 10.1006/jmbi.2001.4747. [DOI] [PubMed] [Google Scholar]
- 9.Bitto E, McKay DB. Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure. 2002;10:1489–1498. doi: 10.1016/s0969-2126(02)00877-8. [DOI] [PubMed] [Google Scholar]
- 10.Tormo A, Almiron M, Kolter R. surA, an Escherichia coli gene essential for survival in stationary phase. J Bacteriol. 1990;172:4339–4347. doi: 10.1128/jb.172.8.4339-4347.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rouviere PE, Gross CA. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Gene Develop. 1996;10:3170–3182. doi: 10.1101/gad.10.24.3170. [DOI] [PubMed] [Google Scholar]
- 12.Lazar SW, Kolter R. SurA assists the folding of Escherichia coli outer membrane proteins. J Bacteriol. 1996;178:1770–1773. doi: 10.1128/jb.178.6.1770-1773.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bitto E, McKay DB. Binding of phage-display-selected peptides to the periplasmic chaperone protein SurA mimics binding of unfolded outer membrane proteins. FEBS Lett. 2004;568:94–98. doi: 10.1016/j.febslet.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 14.Lazar SW, Almiron M, Tormo A, Kolter R. Role of the Escherichia coli SurA protein in stationary-phase survival. J Bacteriol. 1998;180:5704–5711. doi: 10.1128/jb.180.21.5704-5711.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dartigalongue C, Raina S. A new heat-shock gene, Ppid, encodes a peptidyl-prolyl isomerase required for folding of outer membrane proteins in Escherichia coli. EMBO J. 1998;17:3968–3980. doi: 10.1093/emboj/17.14.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizzitello AE, Harper JR, Silhavy TJ. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J Bacteriol. 2001;183:6794–6800. doi: 10.1128/JB.183.23.6794-6800.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Justice SS, Hunstad DA, Harper JR, Duguay AR, Pinkner JS, Bann J, Frieden C, Silhavy TJ, Hultgren SJ. Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J Bacteriol. 2005;187:7680–7686. doi: 10.1128/JB.187.22.7680-7686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stymest KH, Klappa P. The periplasmic peptidyl prolyl cis-trans isomerases PpiD and SurA have partially overlapping substrate specificities. FEBS J. 2008;275:3470–3479. doi: 10.1111/j.1742-4658.2008.06493.x. [DOI] [PubMed] [Google Scholar]
- 19.Antonoaea R, Furst M, Nishiyama K, Müller M. The periplasmic chaperone PpiD interacts with secretory proteins exiting from the SecYEG translocon. Biochemistry. 2008;47:5649–5656. doi: 10.1021/bi800233w. [DOI] [PubMed] [Google Scholar]
- 20.Fanghänel J, Fischer G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front Biosci. 2004;9:3453–3478. doi: 10.2741/1494. [DOI] [PubMed] [Google Scholar]
- 21.Fischer G, Tradler T, Zarnt T. The mode of action of peptidyl prolyl cis/trans isomerases in vivo: binding vs catalysis. FEBS Lett. 1998;426:17–20. doi: 10.1016/s0014-5793(98)00242-7. [DOI] [PubMed] [Google Scholar]
- 22.Balbach J, Schmid FX. Prolyl isomerization and its catalysis in protein folding. In: Pain RH, editor. Mechanisms of protein folding. Oxford: Oxford University Press; 2000. pp. 212–237. [Google Scholar]
- 23.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 24.Bayer E, Goettsch S, Mueller JW, Griewel B, Guiberman E, Mayr LM, Bayer P. Structural analysis of the mitotic regulator hPin1 in solution:insights into domain architecture and substrate binding. J Biol Chem. 2003;278:26183–26193. doi: 10.1074/jbc.M300721200. [DOI] [PubMed] [Google Scholar]
- 25.Heikkinen O, Seppala R, Tossavainen H, Heikkinen S, Koskela H, Permi P, Kilpelainen I. Solution structure of the parvulin-type PPIase domain of Staphylococcus aureus PrsA—implications for the catalytic mechanism of parvulins. BMC Struct Biol. 2009;9:17. doi: 10.1186/1472-6807-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhlewein A, Voll G, Hernandez Alvarez B, Kessler H, Fischer G, Rahfeld JU, Gemmecker G. Solution structure of Escherichia coli Par10:The prototypic member of the Parvulin family of peptidyl-prolyl cis/trans isomerases. Protein Sci. 2004;13:2378–2387. doi: 10.1110/ps.04756704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landrieu I, Wieruszeski JM, Wintjens R, Inze D, Lippens G. Solution structure of the single-domain prolyl cis/trans isomerase PIN1At from Arabidopsis thaliana. J Mol Biol. 2002;320:321–332. doi: 10.1016/S0022-2836(02)00429-1. [DOI] [PubMed] [Google Scholar]
- 28.Daragan VA, Ilyina EE, Fields CG, Fields GB, Mayo KH. Backbone and side-chain dynamics of residues in a partially folded beta-sheet peptide from platelet factor-4. Protein Sci. 1997;6:355–363. doi: 10.1002/pro.5560060211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sekerina E, Rahfeld JU, Müller J, Fanghanel J, Rascher C, Fischer G, Bayer P. NMR solution structure of hPar14 reveals similarity to the peptidyl prolyl cis/trans isomerase domain of the mitotic regulator hPin1 but indicates a different functionality of the protein. J Mol Biol. 2000;301:1003–1017. doi: 10.1006/jmbi.2000.4013. [DOI] [PubMed] [Google Scholar]
- 30.Tossavainen H, Permi P, Purhonen SL, Sarvas M, Kilpelainen I, Seppala R. NMR solution structure and characterization of substrate binding site of the PPIase domain of PrsA protein from Bacillus subtilis. FEBS Lett. 2006;580:1822–1826. doi: 10.1016/j.febslet.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 31.Rahfeld JU, Rucknagel KP, Schelbert B, Ludwig B, Hacker J, Mann K, Fischer G. Confirmation of the existence of a third family among peptidyl–prolyl cis/trans isomerases—amino acid sequence and recombinant production of parvulin. FEBS Lett. 1994;352:180–184. doi: 10.1016/0014-5793(94)00932-5. [DOI] [PubMed] [Google Scholar]
- 32.Rahfeld J-U, Schierhorn A, Mann K-H, Fischer G. A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett. 1994;343:65–69. doi: 10.1016/0014-5793(94)80608-x. [DOI] [PubMed] [Google Scholar]
- 33.Scholz C, Rahfeld J, Fischer G, Schmid FX. Catalysis of protein folding by parvulin. J Mol Biol. 1997;273:752–762. doi: 10.1006/jmbi.1997.1301. [DOI] [PubMed] [Google Scholar]
- 34.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 35.Linge JP, Habeck M, Rieping W, Nilges M. ARIA:automated NOE assignment and NMR structure calculation. Bioinformatics. 2003;19:315–316. doi: 10.1093/bioinformatics/19.2.315. [DOI] [PubMed] [Google Scholar]
- 36.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 38.Palmer AG, III, Rance M, Wright PE. Intramolecular motions of a zinc finger DNA-binding domain from xfin characterized by proton detected natural abundance C-13 heteronuclear NMR spectroscopy. J Am Chem Soc. 1991;113:4371–4380. [Google Scholar]
- 39.Dayie K, Wagner G. Relaxation-rate measurements for 15N-1H groups with pulsed-field gradients and preservation of coherence pathways. J Magn Reson A. 1994;111:121–126. [Google Scholar]
- 40.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magentic resonance relaxation in macromolecules. II. Analysis of experimental results. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 41.Loria JP, Rance M, Palmer AG., III A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J Biomol NMR. 1999;15:151–155. doi: 10.1023/a:1008355631073. [DOI] [PubMed] [Google Scholar]
- 42.Skrynnikov NR, Mulder FA, Hon B, Dahlquist FW, Kay LE. Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: application to methionine residues in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2001;123:4556–4566. doi: 10.1021/ja004179p. [DOI] [PubMed] [Google Scholar]
- 43.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer G, Bang H, Mech C. Nachweis einer Enzymkatalyse für die cis-trans-Isomerisierung der Peptidbindung in prolinhaltigen Peptiden. Biomed Biochim Acta. 1984;43:1101–1111. [PubMed] [Google Scholar]
- 45.Garcia-Echeverria C, Kofron JL, Kuzmic P, Kishore V, Rich DH. Continuous fluorimetric direct (uncoupled) assay for peptidyl prolyl cis-trans-isomerases. J Am Chem Soc. 1992;114:2758–2759. [Google Scholar]
- 46.Zoldák G, Aumüller T, Lücke C, Hritz J, Oostenbrink C, Fischer G, Schmid FX. A library of fluorescent peptides for exploring the substrate specificities of prolyl isomerases. Biochemistry. 2009;48:10423–10436. doi: 10.1021/bi9014242. [DOI] [PubMed] [Google Scholar]
- 47.Scholz C, Eckert B, Hagn F, Schaarschmidt P, Balbach J, Schmid FX. SlyD proteins from different species exhibit high prolyl isomerase and chaperone activities. Biochemistry. 2006;45:20–33. doi: 10.1021/bi051922n. [DOI] [PubMed] [Google Scholar]
- 48.Weininger U, Haupt C, Schweimer K, Graubner W, Kovermann M, Brüser T, Scholz C, Schaarschmidt P, Zoldak G, Schmid FX, Balbach J. NMR solution structure of SlyD from Escherichia coli: spatial separation of prolyl isomerase and chaperone function. J Mol Biol. 2009;387:295–305. doi: 10.1016/j.jmb.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 49.Hottenrott S, Schumann T, Plückthun A, Fischer G, Rahfeld JU. The Escherichia coli SlyD is a metal ion-regulated peptidyl-prolyl cis/trans-isomerase. J Biol Chem. 1997;272:15697–15701. doi: 10.1074/jbc.272.25.15697. [DOI] [PubMed] [Google Scholar]
- 50.Konno M, Ito M, Hayano T, Takahashi N. The substrate-binding site in Escherichia coli cyclophilin A preferably recognizes a cis-proline isomer or a highly distorted form of the trans isomer. J Mol Biol. 1996;256:897–908. doi: 10.1006/jmbi.1996.0136. [DOI] [PubMed] [Google Scholar]
- 51.Compton LA, Davis JM, Macdonald JR, Bächinger HP. Structural and functional characterization of Escherichia coli peptidyl- prolyl cis-trans isomerases. Eur J Biochem. 1992;206:927–934. doi: 10.1111/j.1432-1033.1992.tb17002.x. [DOI] [PubMed] [Google Scholar]
- 52.Stein RL. Mechanism of enzymatic and nonenzymatic prolyl cis- trans isomerization. Adv Prot Chem. 1993;44:1–24. doi: 10.1016/s0065-3233(08)60562-8. [DOI] [PubMed] [Google Scholar]
- 53.Mücke M, Schmid FX. Folding mechanism of ribonuclease T1 in the absence of the disulfide bonds. Biochemistry. 1994;33:14608–14619. doi: 10.1021/bi00252a029. [DOI] [PubMed] [Google Scholar]
- 54.Knappe TA, Eckert B, Schaarschmidt P, Scholz C, Schmid FX. Insertion of a chaperone domain converts FKBP12 into a powerful catalyst of protein folding. J Mol Biol. 2007;368:1458–1468. doi: 10.1016/j.jmb.2007.02.097. [DOI] [PubMed] [Google Scholar]
- 55.Kern D, Kern G, Scherer G, Fischer G, Drakenberg T. Kinetic analysis of cyclophilin-catalyzed prolyl cis/trans isomerization by dynamic NMR spectroscopy. Biochemistry. 1995;34:13594–13602. doi: 10.1021/bi00041a039. [DOI] [PubMed] [Google Scholar]
- 56.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 57.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 58.Bailey ML, Shilton BH, Brandl CJ, Litchfield DW. The dual histidine motif in the active site of Pin1 has a structural rather than catalytic role. Biochemistry. 2008;47:11481–11489. doi: 10.1021/bi800964q. [DOI] [PubMed] [Google Scholar]
- 59.Webb HM, Ruddock LW, Marchant RJ, Jonas K, Klappa P. Interaction of the periplasmic peptidylprolyl cis-trans isomerase SurA with model peptides. The N-terminal region of SurA is essential and sufficient for peptide binding. J Biol Chem. 2001;276:45622–45627. doi: 10.1074/jbc.M107508200. [DOI] [PubMed] [Google Scholar]
- 60.Xu X, Wang S, Hu YX, McKay DB. The periplasmic bacterial molecular chaperone SurA adapts its structure to bind peptides in different conformations to assert a sequence preference for aromatic residues. J Mol Biol. 2007;373:367–381. doi: 10.1016/j.jmb.2007.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jakob RP, Schmid FX. Molecular determinants of a native-state prolyl isomerization. J Mol Biol. 2009;387:1017–1031. doi: 10.1016/j.jmb.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 62.Jakob RP, Schmid FX. Energetic coupling between native-state prolyl isomerization and conformational protein folding. J Mol Biol. 2008;377:1560–1575. doi: 10.1016/j.jmb.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 63.Mayr LM, Landt O, Hahn U, Schmid FX. Stability and folding kinetics of ribonuclease T1 are strongly altered by the replacement of cis-proline 39 with alanine. J Mol Biol. 1993;231:897–912. doi: 10.1006/jmbi.1993.1336. [DOI] [PubMed] [Google Scholar]
- 64.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method. I. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 65.Privalov PL, Gill SJ. Stability of protein structure and hydrophobic interaction. Adv Prot Chem. 1988;39:191–234. doi: 10.1016/s0065-3233(08)60377-0. [DOI] [PubMed] [Google Scholar]
- 66.Schmid FX. Prolyl isomerase—enzymatic catalysis of slow protein-folding reactions. Annu Rev Biophys Biomol Struct. 1993;22:123–143. doi: 10.1146/annurev.bb.22.060193.001011. [DOI] [PubMed] [Google Scholar]
- 67.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 68.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 69.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. 1996;35:10256–10261. doi: 10.1021/bi9611164. Biochemistry. [DOI] [PubMed] [Google Scholar]