

Abstract
G-protein coupled receptors (GPCRs) are transmembrane signaling molecules, with a majority of them performing important physiological roles. β2-Adrenergic receptor (β2-AR) is a well-studied GPCRs that mediates natural responses to the hormones adrenaline and noradrenaline. Analysis of the ligand-binding region of β2-AR using the recently solved high-resolution crystal structures revealed a number of highly conserved amino acids that might be involved in ligand binding. However, detailed structure-function studies on some of these residues have not been performed, and their role in ligand binding remains to be elucidated. In this study, we have investigated the structural and functional role of a highly conserved residue valine 114, in hamster β2-AR by site-directed mutagenesis. We replaced V114 in hamster β2-AR with a number of amino acid residues carrying different functional groups. In addition to the complementary substitutions V114I and V114L, the V114C and V114E mutants also showed significant ligand binding and agonist dependent G-protein activation. However, the V114G, V114T, V114S, and V114W mutants failed to bind ligand in a specific manner. Molecular modeling studies were conducted to interpret these results in structural terms. We propose that the replacement of V114 influences not only the interaction of the ethanolamine side-chains but also the aryl-ring of the ligands tested. Results from this study show that the size and orientation of the hydrophobic residue at position V114 in β2-AR affect binding of both agonists and antagonists, but it does not influence the receptor expression or folding.
Keywords: beta2-adrenergic receptor, valine 114, ligand binding, site-directed mutagenesis, molecular modeling
Introduction
G-protein coupled receptors (GPCRs) constitute the largest known family of cell surface receptors. These receptors all contain seven transmembrane (TM) helices (H1–H7) and transduce signals across the cell membranes in response to diverse extracellular stimuli. GPCRs are involved in fundamental biological processes such as sensing taste, light, and odor. β2-Adrenergic receptor (β2-AR) is a well studied member of the GPCR family and mediates physiologic responses to the hormones adrenaline and noradrenaline.1
Significant progress has been made in GPCR structural biology in the past 2 years. Recently, five landmark articles on the crystal structures of class A GPCRs revitalized the GPCR field.2–6 These are the high-resolution crystal structures of β2-AR T4L and β2-AR-Fab bound to the inverse agonist carazolol,2,6 the 2.7 Å crystal structure of turkey β1-AR in complex with the antagonist cyanopindolol,3 the 3.2 Å structure of the bovine opsin bound to synthetic peptide from the carboxyl terminus of the alpha-subunit of the G-protein transducin,5 and the 2.6 Å structure of human A2A adenosine receptor bound to antagonist ZM241385.4 Comparison of the crystal structures of these class A GPCRs reveals that the overall topology is similar, with seven TM helices and an eighth helix that runs parallel to the cytoplasmic face of the membrane. However, significant differences were observed with respect to some important structural features, previously believed to be common among class A GPCRs. These are the second extracellular loop (EL2) and the cytoplasmic loop 2 (CL2), the ionic lock between H3 and H6, and the “rotamer toggle switch” of W6.48 (the nomenclature used to describe amino acid positions in GPCRs follows the convention established by Ballesteros and Weinstein,7 where the helix number is followed by the sequence position relative to the most conserved residue in the helix designated as 50), W265 in rhodopsin, W286 in β2-AR, and W337 in β1-AR. The EL2 in rhodopsin has a secondary structure of a β-hairpin that acts as a lid above the retinal binding pocket in contrast to an α-helix that was found above the pocket in β1-AR and β2-AR, which allows the ligands to diffuse in and out of the ligand binding crevice in β-ARs. The CL2 in β1-AR forms a short α-helix, which is absent in both β2-AR and rhodopsin. Similarly, the ionic lock a network of H-bonds between R3.50 and E6.30, which was believed to be an important feature of the inactive state of GPCRs, was found in rhodopsin but absent in the β-AR structures. Differences in the secondary structures of the EL2 and the CL2 regions are important because these regions are highly conserved between the β-ARs and poorly conserved in rhodopsin.
Analysis of the ligand binding regions of the structures of both turkey β1-AR and human β2-AR reveal that there are relatively few differences in amino acids present in the TM regions that might account for the differences in ligand specificity between β-ARs.3 However, there is a significant difference in primary amino acid sequence of the EL-2 of both β-ARs. Because the EL-2 loop is at the entrance to the ligand binding pocket in β-ARs, it might contribute to the ligand selectivity of the β-AR and other GPCRs, in general.8,9 D113 (3.32) is a highly conserved residue (100%) among β-ARs and is essential for ligand binding.10,11 Another highly conserved residue (98.5%) in the ligand binding pocket is V114 (3.33), but the functional or structural role of this valine was not known until recently, when it was shown that replacement of V114 (3.33) with alanine in hamster β2-AR significantly affected ligand binding.12 Although the crystal structures gave some insights into the mechanism by which this residue might influence the binding to β-AR antagonists and inverse agonists, its role in binding to agonists is still unclear.
In this study, we have further investigated the contribution of V114 in β2-AR ligand binding and efficacy. V114 in hamster β2-AR was replaced with a number of amino acid residues carrying different functional groups, and ligand binding was tested using various agonists in competition experiments with dihydroalprenolol (DHA). To interpret the effects of these mutations on β2-AR structure and function, we created molecular models of hamster β2-AR based on crystal structures and their derivatives as templates. Our results from radioligand binding assays and molecular modeling analysis show that the size and orientation of the hydrophobic residue at position V114 (3.33) in β2-AR affect binding of both agonists and antagonists, but it does not influence the receptor expression or folding. Because V114 is highly conserved in the binding pocket of the amine family, these results are of general significance to the amine family of GPCRs.
Results and Discussion
Expression and subcellular localization
Wild-type (WT) hamster β2-AR and mutants were transiently expressed in COS-1 cells, and 44 hr posttransfection, membranes were prepared and used for radioligand binding assays. Table I shows the saturation binding data for WT, V114I, V114L, V114C, and V114E mutants using the antagonist DHA. Qualitative estimates of the expression levels of the Val114 mutants that bind to DHA were similar to or better than WT β2-AR as quantified by Bmax (Table I). The V114G, V114T, V114S, V114D, and V114W mutants lacked the ability to bind to DHA in a specific manner. Further experiments were carried out to investigate whether this impaired binding is due to insufficient expression levels of receptor, protein misfolding, or due to the inability of the expressed receptor to bind the ligand.
Table I.
Summary of Ligand Binding Properties of Wild-Type β2-AR and V114 Mutant Receptorsa
| Receptor | Kd (nM) | 95% confidence intervals | Bmax (pmol/mg) |
|---|---|---|---|
| Wild-type | 3.0 | 2.35–3.74 | 18 ± 0.6 |
| V114I | 4.1 | 3.73–4.51 | 24 ± 0.4 |
| V114C | 7.8 | 5.51–10.22 | 30 ± 1.7 |
| V114L | 15.5 | 11.53–19.49 | 36 ± 2.5 |
| V114Eb | 35.8 | 20.31–51.42 | 31 ± 4.2 |
The values are expressed as the mean ± S.E (n = 3–5 experiments), and the experiment is performed using [3H] dihydroalprenolol as the radioligand (TRK 649, GE Health care). No significant specific binding detected for the V114G, V114T, V114S, V114D, and V114W mutants under the assay conditions.
High nonspecific binding (15–20% of total binding).
Immunoblotting showed heterogeneous expression of the Val114 mutants in COS-1 cells, as indicated by the presence of three predominant bands in the molecular weight range of 45–65 kDa (Fig. 1). Interestingly, the mutants V114T, V114S, V114G, and V114D, which showed impaired binding to the antagonist DHA (Table I, and Supporting Information Fig. 6), seem to be fully glycosylated and produced the ∼ 65 kDa band (Fig 1). This is in agreement with previous photocrosslinking experiments of hamster β2-AR expressed in COS-1 cells, which showed that the band at ∼ 65 kDa corresponds to the completely glycosylated receptor.13 Confocal immunofluorescence microscopy was used to elucidate whether the expressed mutants were properly folded and transported to the cell surface. Fluorescence images obtained showed that all the Val114 mutants expressed in either COS-1 or HEK293T cells were predominantly localized on the cell surface (Fig. 5).
Figure 1.
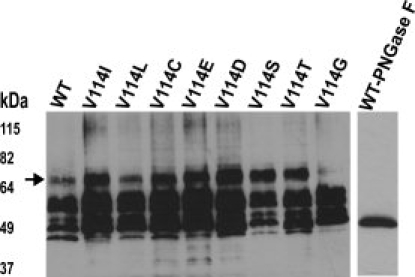
Immunoblot analysis of β2-AR expressed in COS-1 cells using the monoclonal antibody rho-1D4. Immunoblot of membranes expressing the wild-type (WT) β2-AR and Val114 mutants (around 5 μg of solubilized membrane protein was loaded), the single lane shows PNGaseF treated WT β2-AR. The arrow indicates the fully glycosylated receptor. Mobility of molecular weight standards in kilo Daltons is indicated next to the gel.
Figure 5.
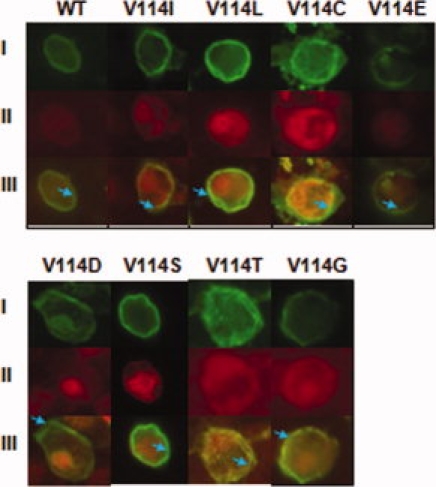
Localization of WT β2-AR and Val114 mutants expressed in COS-1 or HEK293T cells by confocal immunofluorescence microscopy. Confocal microscopy using the mouse rho-1D4 antibody (I) and rabbit anti-calnexin antibody (II) shows the localization of β2-AR in wild-type and mutant COS-1 cells to the cell surface. Mouse rho-1D4 antibody was visualized with anti-mouse-fluorescein isothiocyanate secondary antibody (green), and rabbit anti-calnexin antibody was visualized with anti-rabbit-Texas Red secondary antibody, the overlay is shown in III. Blue arrows are used to localize areas of cell surface membrane.
Taken together, the results from the saturation binding assays, immunoblots, and immunofluorescence microscopy show that all the Val114 mutants were expressed in sufficient amounts, were N-glycosylated and predominantly cell-surface localized. However, only the V114I, V114L, V114C, and V114E mutants were able to bind the ligand in a significant manner.
Ligand binding properties
With the exception of the V114I mutant, which had similar affinity for DHA as the WT hamster β2-AR, the rest of the mutants that bound to DHA showed a decrease in affinity for the ligand in the following order: V114E > V114L > V114C (Table I and Supporting Information Fig. 6). To determine whether the V114 mutants exhibit similar loss in affinity toward both the classical catecholamine agonists and noncatecholamine agonists, such as albuterol (or salbutamol), radioligand displacement assays were performed. The chemical structures of the ligands used in this study are shown in Supporting Information Figure 8.
The data from the competition assays of V114I, V114L, and V114C mutants, which show significant specific binding, are interesting with respect to their ability to bind noncatecholamine and catecholamine agonists with different affinities (Table II). For example, the V114I mutant binds tighter to albuterol with a Ki of 11 μM compared to 25 μM for the WT. In contrast, the V114L and V114C mutants bind with 2- to 4-fold less affinity than WT. However, the binding of these mutants toward the classical catecholamine agonists norepinephrine, and epinephrine is significantly reduced. V114I mutant shows slightly reduced affinities for norepinephrine and epinephrine, whereas V114L and V114C show 10- to 100-fold less affinity than the WT (Table II). These differences in binding are explained using the molecular models of β2-AR bound to different agonists (see later).
Table II.
Summary of Competition Ligand Binding of Wild-Type β2-AR and Mutant Receptorsa
| Ligands (Ki) μM (95% confidence intervals) |
||||
|---|---|---|---|---|
| Receptor | Norepinephrine | Epinephrine | Isoproterenol | Albuterol (salbutamol) |
| Wild-type | 84 (49–144) | 4.9 (3.5–7.0) | 4.2 (2.4–7.5) | 25 (21–30) |
| V114I | 167 (52–535) | 33 (16–68) | 6.6 (3.1–14) | 11 (4.1–31) |
| V114L | 2014 (722–5613) | 486 (241–978) | 92 (48–177) | 82 (42–160) |
| V114C | 960 (434–2123) | 312 (169–572) | 57 (35 to 93) | 101 (57–176) |
| V114Eb | 752 (164–3437) | 2725 (1277–5813) | 1400 (590–3320) | 2129 (954–4748) |
The binding of unlabelled agonists to wild-type β2-AR and mutant receptors was determined by competition with 3 nM [3H] dihydroalprenolol as the radioligand (TRK 649, GE Health care). Data obtained from determinations of three independent transfections and analyzed by nonlinear regression as described under Material and Methods section.
High nonspecific binding of 15–20% observed.
Gαs-mediated signaling
To examine the agonist activation of WT hamster β2-AR and mutant receptors, the coupling of the receptors to the Gαs-adenylyl cyclase effector system was measured by cAMP accumulation assay. Analysis of cAMP level was carried out in HEK293T cells, because these cells had lower level of endogenous β2-AR compared with COS-1 cells. The HEK293T cells were transiently transfected with the respective β2-AR mutants and 44 hr after transfection cells were induced with 10 μM isoproterenol and Gαs-mediated cAMP production was measured as described.12,14 Except for V114C mutant, which exhibited a slightly higher level of agonist independent activity compared with WT, the rest of the V114 mutations tested showed no change in the level of agonist independent activity (Fig. 2). The agonist stimulated activity of the V114I, V114L, V114C, and V114E mutations are lower than WT, to varying degrees. This is in agreement with the weaker affinity observed for isoproterenol by these mutations in the competition assay (Table II).
Figure 2.
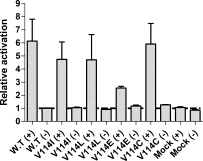
cAMP accumulation assays. Receptor activity was measured by cAMP accumulation assay (see Material and Methods section), and shown are the basal activity (−) and activity after stimulation (+) with 10 μM isoproterenol. The relative activation rates of Val114 β2-AR mutants and mock transfected HEK293T cells are shown. Results are normalized to the basal activity of wild-type receptor and are from at least two to three independent experiments performed in duplicate.
Molecular modeling of antagonist alprenolol-receptor interaction
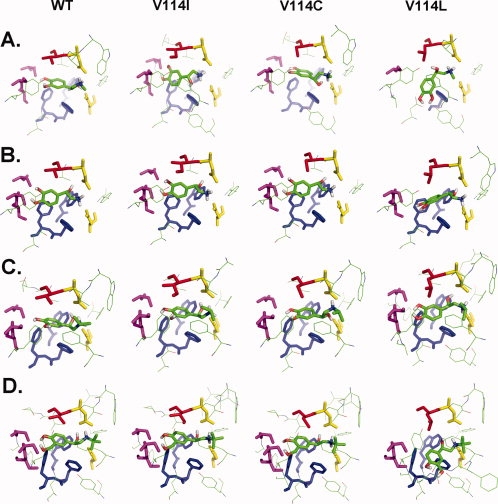
To rationalize the experimental results from the ligand binding studies in view of the structural properties of the receptor, we conducted molecular modeling studies. For investigating antagonist alprenolol-receptor interactions, we generated homology model of hamster β2-AR based on the published crystal structure of human β2-AR.6 Mutations V114 to I, L, or C were introduced using the Pymol (version 0.99) mutagenesis tool.15 Alprenolol was docked using Autodock 4.0 software.16,17 The results are shown in Figure 3. The docked conformation was similar (with a root mean square deviation (RMSD) of <1.2Å) to that of carazolol, the ligand bound to the crystal structure. Analysis of the docked alprenolol structure with respect to amino acids in proximity to the ligand within 4Å showed the interactions expected based on the literature. Thus, the amino group of the ligand is in close proximity to D113 and N312 [Fig. 3(A), colored in yellow], and the head-group is stabilized by W286, F289 and F290 [Fig. 3(A), colored in blue]. These results validate the homology model and docking procedure. Next, we repeated the docking using the mutant structures. In the structures carrying mutations at position 114, we found that the distance of the ligand amine group to the carboxyl group of D113 increased in the sequence V (2.32Å), I (2.51Å), C (2.76Å), and L (2.81Å), which will lead to a corresponding decrease in affinity of the receptor for alprenolol. This result is in good agreement with the KD values (Table I) obtained from saturation binding assays.
Figure 3.

Molecular models of the interaction of alprenolol with hamster β2-AR and its mutants. The predicted binding pocket residues that are within 5 Å distance from alprenolol to WT β2AR and its mutants (B) V114I, (C) V114C, and (D) V114L are shown. The ligand alprenolol is rendered as sticks and colored according to the Cory-Pauling-Kolton scheme. In each case, the residue corresponding to V114 and its mutants are rendered in sticks and colored in red. Residues D113 and N312 are colored in yellow, and W286, F289, and F290 are colored in blue and represented in sticks.
Next, we analyzed the effect of the mutation at position 114 on the interactions between the binding pocket and the aromatic ring of the alprenolol ligand. This includes V114 and the amino acids V117, Y199, W286, F289, and F290. By estimating their contact areas in WT and the mutants using ICMBrowserPro analysis,18 we find that V114 has the maximum contact area with the ligand in the WT, followed by V117, F289, F290, W286, and Y199 (Supporting Information Table III). However, when the D113 was also considered in the ICMBrowserPro analysis, it had the largest contact area with alprenolol, followed by V114 and N312. This supports a greater role of the propanolamine side chain in stabilization of ligand binding in the WT. In contrast, in the V114I and V114C mutants, D113 ranked only second, with either N312 or the cysteine or isoleucine at position 114 ranking first. Thus, the substitution at position 114 actually stabilizes the interaction with the ring (as indicated by the greater contact area), but because of the increased steric hindrance exerted on the ligand by the mutant residues, the ligand position is shifted, explaining the above described effect of increasing distance to the propanolamine side chain. Finally, in the V114L mutant, D113 contributed less to the alprenolol binding than V114L, F290, and N312, further decreasing its role in ligand stabilization. We conclude that the experimentally observed difference in the affinity of alprenolol for the different mutants is primarily an effect on the interaction of the propanolamine portion of alprenolol. Although there are clear changes in interactions between the aromatic ring portion of alprenolol, because these would be expected to increase rather than decrease the affinity to the ligand, these aromatic ring interactions seem to indirectly influence the interaction with the propanolamine portion. Specifically, we propose that changing the interactions with the ring group forces the salt-bridge to weaken by increasing the distance between the positive charge on the ligand and the negative charge on D113.
Binding pocket analysis for agonists
As the published crystal structures of human β2-AR6 and turkey β1-AR3 are in the presence of an inverse agonist and antagonist, respectively, homology models for hamster β2-AR are not suitable for docking of agonist molecules. Therefore, we investigated alternative models that take the agonist induced conformational changes into account. To study all four ligands, we generated homology models based on the recently published theoretical human β2-AR models by the Abagyan group,19 who kindly shared their models with us. In these models, the serines were found to interact with all four ligands, isoproterenol, epinephrine, norepinephrine, and salbutamol. For all models, the corresponding mutations of V114 to I, L, or C were introduced using the Pymol mutagenesis tool15 as earlier. Ligands were docked using Autodock 4.0 software.16,17 The results are shown in Figure 4. The difference in binding of the various ligands to the WT receptor are described later, for detailed discussion of the effect of mutation on each ligand please refer to the Supporting Information.
Figure 4.
Comparison of the binding pockets for β2-AR agonist ligands in the wild-type (WT) and the three mutants V114I, V114C, and V114L (from left to right). Residues within 5 Å of the ligand are shown. Highlighted as sticks are the following entities: the ligands are shown in Cory-Pauling-Kolton coloring scheme. The residue at position 114 is shown in red. Residues D113 and N312 are colored in yellow, and W286, F289, and F290 are colored in blue and represented in sticks. S203, S204, and S207 are colored in magenta. (A) Epinephrine, (B) norepinephrine, (C) isoproterenol, and (D) Salbutamol.
We investigated in detail the two amino acid residues D113 and N312 that make important stabilizing polar contacts with the ethanolamine side chain of the agonists, by estimating their contact areas in WT and the mutants using ICMBrowserPro software.18 The contribution of the residues D113 and N312 to agonist binding are as follows (Supporting Information Table III). Overall, D113 was found to be consistently ranked at the top, contributing the largest contact area for the binding of any ligand to the hamster β2-AR WT structures. The contact area made by D113 with the ligand decreased in the order of ligands, isoproterenol, epinephrine, salbutamol, and norepinephrine, in agreement with the decrease in Ki values experimentally observed for these ligands (Table II). In the case of two agonists, isoproterenol and epinephrine, the residues D113 and N312 contributed the most to the contact area when compared with any other residue found in the predicted binding pocket, possibly explaining why the earlier two ligands compete with DHA better than salbutamol and norepinephrine. In the case of norepinephrine, a set of hydrophobic residues in the order, V114, V117, F289, and F290 were ranked higher when compared with N312, indicating a stronger hydrophobic interaction as compared to the polar interaction made by N312. These predictions imply that the stronger the polar interactions made by the amino acid residues D113 and N312 with the agonist ligands, the better the competition with DHA.
Ligand binding pocket in β-ARs and Val114
The differences in the binding pocket between the β-ARs are relatively small, with a recent MD simulation study giving an RMSD of 1.2Å for both WT β1-AR and β2-AR.20 Based on the crystal structure of β1-AR, it was hypothesized that the ligand binding site will contract 2–3 Å on activation to enable the agonist adrenaline to make contacts with residues on H5 and H3/H7.3
Our results show that the size and orientation of the hydrophobic residue at position V114 (3.33) in β2-AR affects binding of both agonists and antagonists. In the case of alprenolol, V114 is important for the interaction for the WT, but the mutations cause a decrease in surface area and a loss in affinity. In agonist binding, V114 is not important in the WT receptor but becomes increasingly important in the mutants in the following sequence V114I, V114C, and V114L. Thus, the interactions made here may be more suitable for stabilizing the inactive conformation of the receptor, thus decreasing the affinity for activating ligands. These studies suggest that both the propanolamine and the aryl moiety are influenced by the introduced mutation at position 114.
Materials and Methods
Materials
The detergent n-dodecyl-β-d-maltoside (DM) was purchased from Anatrace (Maumee, OH, USA). The monoclonal antibody, rho-1D4, was prepared by the Cell Culture Center (Minneapolis, MN, USA) from a cell line provided by R.S. Molday (University of British Columbia, Vancouver, Canada). Fetal Bovine Serum and Dulbecco's Modified Eagle Medium high glucose were purchased from Sigma and Invitrogen. The β2-AR antagonist [3H] DHA was purchased from GE Healthcare (GE Healthcare Biosciences, Little Chalfont, UK). Alprenolol, formoterol, procaterol, isoproterenol, epinephrine, norepinephrine, and salbutamol were purchased from Sigma (St. Louis, MO, USA). Protease inhibitors and common chemicals were purchased either from Fisher or Sigma. Synthetic oligonucleotides were purchased from Invitrogen (Carlsbad, CA, USA).
Buffers used were as follows: phosphate-buffered saline (PBS) buffer: 137 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4, and 10 mM Na2HPO4 (pH 7.4); buffer A (lysis buffer), 10 mM Tris-HCl, pH 7.4, containing protease inhibitors (1 mM ethylenediaminetetraacetic acid (EDTA), 10 μg/ml benzamidine, 10 μg/ml leupeptin, 20 μg/ml soybean trypsin inhibitor, 5 μg/ml aprotinin, and 0.2 mM phenylmethylsulfonyl fluoride); buffer B (storage buffer) 50 mM Tris-HCl, pH 7.4, 12.5 mM MgCl2, and containing protease inhibitors as in buffer A; buffer C (binding buffer), 75 mM Tris-HCl, pH 7.4, 12.5 mM MgCl2, and containing protease inhibitors as in buffer A; buffer D, 20 mM Tris-HCl, pH 7.4, containing 100 mM NaCl, and 1 mM EDTA.
Construction of mutant β2-AR genes
Amino acid substitutions were introduced into the synthetic hamster β2-AR gene carried by the pMT4 expression vector as described previously12 or synthesized commercially (Genewiz Inc, NJ, USA). DNA sequences of all the mutated genes were verified by automated DNA sequencing (MICB DNA Sequencing facility, Winnipeg).
Cell culture and immunoblot analysis
The WT hamster β2-AR and Val114 mutant genes were expressed in COS-1 cells using a Diethylaminoethyl cellulose (DEAE-dextran) based transient transfection method.13,21 Briefly, 106 COS-1 cells were plated into 10-cm tissue culture treated plates and plasmid DNA (10 μg) was transfected using the DEAE-dextran based transient transfection method.13,21 For transient transfection of HEK293T cells using the plasmid pMT4, lipofectamine 2000 (Invitrogen)-mediated transfection was used as described by the manufacturer. Membranes were prepared as described previously.22 The protein concentration in the resuspended membrane pellet was determined using a modified DC protein assay kit from Bio-Rad Laboratories (Hercules, CA, USA). A total of 2.5 or 5 μg of the solubilized protein were resolved by 10% SDS-PAGE (sodium dodecyl sulphate-polyacrylamide gel electrophoresis) and electroblotted onto a nitrocellulose membrane. β2-AR was visualized by immunodetection with the monoclonal antibody, rho-1D4.22 Cell membranes expressing β2-AR were solubilized with 0.5% DM and were treated with PNGaseF and incubated for 1 hr at 37°C. The reaction products are separated by SDS-PAGE and visualized by immunodetection.
Radioligand binding assays
Saturation binding assays were carried out using the radioligand [3H] DHA as described earlier.22 Competition binding assays were performed using 3 nM [3H] DHA and different concentrations of unlabeled agonists (10−2 to 10−9M). Briefly, different concentrations of the unlabelled agonists were added to COS-1 cell membranes expressing the WT hamster β2-AR or the mutants followed by addition of [3H] DHA, the components thoroughly mixed for three times by pipetting, and the reactions were carried out for 2 hr at room temperature (RT). Binding was terminated by filtering under vacuum on GF/A filters (Millipore). Filter-bound radioactivity was measured using a liquid scintillation counter. Equilibrium dissociation constants (KD) were determined from saturation isotherms. Radioligand binding data obtained from competition curves were analyzed by nonlinear regression analysis to determine the EC50 values and Ki values using PRISM software version 4.03 (GraphPad Software Inc, San Diego, CA, USA).
Immunofluorescence microscopy
HEK293 or COS-1 cells were seeded into six-well tissue culture plates containing sterilized poly-L-lysine (Sigma)-treated glass coverslips and transiently transfected with 3.0 μg/ml WT or mutant hamster β2-AR DNA according to the aforementioned transfection protocol. Cells were fixed in 3.7% formaldehyde/1× PBS for 15 min at RT. The cells were then permeablized with 0.05% triton X-100/1× PBS for 30 min at RT. The permeablization buffer was then washed out with 1× PBS, and the cells were blocked with 1× PBS containing 2% bovine serum albumin (IgG and protease free) for 1 hr at RT. β2-AR was labeled using mouse-anti-rho-1D4 monoclonal antibody (C-terminal tagged β2-AR), 1:1000 dilution, and 1:250 dilution of rabbit-anti-calnexin polyclonal antibody (endoplasmic reticulum marker) for 90 min at RT, washed, and incubated with a fluorescent coupled secondary antibody, 1:500 dilution of anti-mouse-fluorescein isothiocyanate, and anti-rabbit-Texas Red for 60 min at RT in the dark. The coverslip were washed twice, mounted with Prolong-antifade-gold for 15 min at RT, and edges sealed with nail-polish. Representative cells were selected and visualized using Olympus BX61 confocal microscope for cytoplasmic or plasma membrane localization.
Determination of receptor activation by cAMP assays
Functional characterization of β2-AR was carried out using a commercially available cAMP assay system (TRK 432, GE healthcare biosciences) following the directions supplied by the manufacturer. Details are as described before.12,14 The assays were carried out using HEK293T cells and cAMP values were normalized to the total membrane protein in each assay. One difference between the assay reported here and our previously reported assay12 is the use of HEK29T cells. Previously we used the HEK293S cells for cAMP assays, and as our expression vector pMT4 does not express the T-antigen, it required cotransfection with a plasmid that carried the T-antigen. In contrast, the HEK293T cells harbour the T-antigen and do not require cotransfection, however, we noticed a decrease in the relative activation rate for the WT β2-AR compared with our previous assay.12
Molecular modeling of the β2-adrenoreceptor
Three-dimensional models of the hamster β2-AR (swissprot entry: P04274) were built by homology modeling using the MODELLER software.23 The inactive homology model of hamster β2-AR was generated based on the crystal structure of human β2-AR antagonist bound model (PDB ID: 2RH1).6 In addition to this, specific homology models were also generated for epinephrine, norepinephrine, isoproterenol, and salbutamol based on recently published theoretical models.19 The sequence alignment between the hamster β2-AR (swissprot entry: P04274) and human β2-AR (swissprot entry; P07550) was performed using clustalW and is shown in Supporting Information Figure 7.25 All models were validated using MolProbity.26 The mutants at position 114 in each model were generated using the mutagenesis tool provided by PyMol visualization software (version 0.99).15 The mutation was introduced by selecting a backbone dependent rotamer conformation that does not clash with other residues in the pocket.
Computational docking of ligands to receptor structures
A set of five ligands alprenolol, epinephrine, norepinephrine, isoproterenol, and salbutamol were docked to all of the homology models generated using the Lamarckian genetic algorithm provided by the Autodock 4.0 software.16,17 The docking procedure used was similar to the studies performed earlier,24 except for the following changes. A cubic box was built around the protein with 126 × 126 × 126 points with a spacing of 0.375 Å between the grid points was used. A total of 25 genetic algorithm runs were considered in each case with an initial population of 300 and a maximum number of 5,000,000 energy evaluations. The top 25 resulting orientations that have less than or equal to 0.5 Å RMSD were clustered together. The best ligand bound receptor structure in each case was chosen based on the position of the propanolamine side chain of the ligand in proximity to D113 of the receptor and on lowest energy and maximum number of conformations in a cluster.
The ligands were docked to different receptor models because previous studies indicated that the structures will be different for pharmacologically different ligands.19 For docking of the antagonist alprenolol, the hamster homology model based on the β2-AR crystal structure (pdb id 2RH1) was used. For each of the other ligands (salbutamol epinephrine, norepinephrine, and isoproterenol), a separate homology model was built based on the respective structures for each ligand generated by Abhagyan's group.19
Acknowledgments
We thank Ms. Thi Le for technical assistance. This research used Computational Resources provided by the PittGrid (www.pittgrid.pitt.edu). M. A. and P. C. thank Profs. Nishimura Fusanori and Anthony Iacopino for financial support to MA.
Glossary
Abbreviations:
- Bmax
binding maximum of the ligand for the receptor
- β2-AR
β2-adrenergic receptor
- CL
cytoplasmic loop
- COS-1
monkey kidney cells
- [3H] DHA
tritium-labeled dihydroalprenolol
- EL
extracellular loop
- GPCR
G-protein coupled receptor
- HEK293
human embryonic kidney cells
- KD
equilibrium dissociation constant of the ligand
- TM
transmembrane.
References
- 1.Lefkowitz RJ. Seven transmembrane receptors: a brief personal retrospective. Biochim Biophys Acta. 2007;1768:748–755. doi: 10.1016/j.bbamem.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 3.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 7.Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 8.Ahuja S, Crocker E, Eilers M, Hornak V, Hirshfeld A, Ziliox M, Syrett N, Reeves PJ, Khorana HG, Sheves M, Smith SO. Location of the retinal chromophore in the activated state of rhodopsin*. J Biol Chem. 2009;284:10190–10201. doi: 10.1074/jbc.M805725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, Ziliox M, Sakmar TP, Sheves M, Reeves PJ, Smith SO, Eilers M. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat Struct Mol Biol. 2009;16:168–175. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strader CD, Sigal IS, Register RB, Candelore MR, Rands E, Dixon RAF. Identification of residues required for ligand binding to the β-adrenergic receptor. Proc Natl Acad Sci USA. 1987;84:4384–4388. doi: 10.1073/pnas.84.13.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strader CD, Sigal IS, Candelore MR, Rands E, Hill WS, Dixon RA. Conserved aspartic acid residues 79 and 113 of the beta-adrenergic receptor have different roles in receptor function. J Biol Chem. 1988;263:10267–10271. [PubMed] [Google Scholar]
- 12.Chelikani P, Hornak V, Eilers M, Reeves PJ, Smith SO, RajBhandary UL, Khorana HG. Role of group-conserved residues in the helical core of beta2-adrenergic receptor. Proc Natl Acad Sci USA. 2007;104:7027–7032. doi: 10.1073/pnas.0702024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noda K, Saad Y, Graham RM, Karnik SS. The high affinity state of the beta 2-adrenergic receptor requires unique interaction between conserved and non-conserved extracellular loop cysteines. J Biol Chem. 1994;269:6743–6752. [PubMed] [Google Scholar]
- 14.Kim JM, Hwa J, Garriga P, Reeves PJ, RajBhandary UL, Khorana HG. Light-driven activation of beta 2-adrenergic receptor signaling by a chimeric rhodopsin containing the beta 2-adrenergic receptor cytoplasmic loops. Biochemistry. 2005;44:2284–2292. doi: 10.1021/bi048328i. [DOI] [PubMed] [Google Scholar]
- 15.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 16.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 17.Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008 doi: 10.1002/0471250953.bi0814s24. Chapter 8:Unit 8 14. [DOI] [PubMed] [Google Scholar]
- 18.Abagyan RA, Raush E, Budagyan L, Totrov M. ICM Browser Pro. Version 3.6. La Jolla, CA: MolSoft LLC; 2007. [Google Scholar]
- 19.Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, Abagyan R. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J Mol Recognit. 2009;22:307–318. doi: 10.1002/jmr.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanni S, Neri M, Tavernelli I, Rothlisberger U. Observation of “ionic lock” formation in molecular dynamics simulations of wild-type beta(1) and beta(2) adrenergic receptors. Biochemistry. 2009;48:4789–4797. doi: 10.1021/bi900299f. [DOI] [PubMed] [Google Scholar]
- 21.Oprian DD, Molday RS, Kaufman RJ, Khorana HG. Expression of a synthetic bovine rhodopsin gene in monkey kidney cells. Proc Natl Acad Sci USA. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chelikani P, Reeves PJ, Rajbhandary UL, Khorana HG. The synthesis and high-level expression of a beta2-adrenergic receptor gene in a tetracycline-inducible stable mammalian cell line. Protein Sci. 2006;15:1433–1440. doi: 10.1110/ps.062080006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2:Unit 2.9. [DOI] [PubMed] [Google Scholar]
- 24.Yanamala N, Tirupula KC, Klein-Seetharaman J. Preferential binding of allosteric modulators to active and inactive conformational states of metabotropic glutamate receptors. BMC Bioinformatics. 2008;9:S16. doi: 10.1186/1471-2105-9-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. (2007) Clustal W and Clustal X version 2.0. Bioinformatics. 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 26.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]