

Abstract
Human age-onset cataracts are believed to be caused by the aggregation of partially unfolded or covalently damaged lens crystallin proteins; however, the exact molecular mechanism remains largely unknown. We have used microseconds of molecular dynamics simulations with explicit solvent to investigate the unfolding process of human lens γD-crystallin protein and its isolated domains. A partially unfolded folding intermediate of γD-crystallin is detected in simulations with its C-terminal domain (C-td) folded and N-terminal domain (N-td) unstructured, in excellent agreement with biochemical experiments. Our simulations strongly indicate that the stability and the folding mechanism of the N-td are regulated by the interdomain interactions, consistent with experimental observations. A hydrophobic folding core was identified within the C-td that is comprised of a and b strands from the Greek key motif 4, the one near the domain interface. Detailed analyses reveal a surprising non-native surface salt-bridge between Glu135 and Arg142 located at the end of the ab folded hairpin turn playing a critical role in stabilizing the folding core. On the other hand, an in silico single E135A substitution that disrupts this non-native Glu135-Arg142 salt-bridge causes significant destabilization to the folding core of the isolated C-td, which, in turn, induces unfolding of the N-td interface. These findings indicate that certain highly conserved charged residues, that is, Glu135 and Arg142, of γD-crystallin are crucial for stabilizing its hydrophobic domain interface in native conformation, and disruption of charges on the γD-crystallin surface might lead to unfolding and subsequent aggregation.
Keywords: non-native interaction, eye cataracts, molecular dynamics, beta-sheet protein folding
Introduction
Human age-onset cataract is an exceptionally common protein deposition disease; it is the major cause of blindness affecting 25 million people worldwide.1,2 The development of novel treatments for cataract is currently hindered by our incomplete understanding of the process(es) of cataract formation in lens cells. Cataracts removed from the aged human eye lens are composed of aggregated states of partially unfolded and/or covalently damaged crystallins.3 These proteins are synthesized in utero, but must remain soluble throughout our lifetime to maintain the transparency of the lens despite the continual UV exposure and potential oxidative stress.4,5 Crystallins are present in very high protein concentration (200–400 mg/mL) in the lens, with β− and γ-crystallins constituting over 50% of total lens proteins. The monomeric γ-crystallins and oligomeric β-crystallins that are comprised of homologous double Greek key domains are mainly structural proteins. All known γ-crystallins from extant vertebrate species have duplicated Greek key domains that are presumably evolved by gene duplication and gene fusion from an ancestral single domain crystallin.6,7 In fact, such single domain crystallins have been identified in the Sea squirt Ciona,8 descendants of lineages that are candidates for the origin of the vertebrates.
The increased stability conferred by the interfacial interactions between the two domains is likely very important for maintaining the long life time of proteins of the lens nucleus, and thus explains the selection for duplicated forms. The crucial role of the domain interface interactions in the folding, stability and oligomerization of both β- and γ-crystallins is also evident from recent experimental results.9–14 In the crowded lens nucleus, those interactions at the domain–domain surface are likely crucial in preventing incorrect protein–protein association leading to cataract formation.
Human γD-crystallin (γD-crys), one of the most abundant γ-crystallins in the lens,15 is comprised of 173 amino acids. As shown in Figure 1(a), the single domains of γD-crystallin, each consisting of two intercalated antiparallel β-sheet Greek key motifs, termed N-td1, N-td2, C-td3, and C-td4, are connected by a six residue linker peptide.5 Each of the Greek key motif consists of four β-strands, namely a, b, c, and d. The ab folded hairpin turn between the first two strands, a and b, is a characteristic feature of the Greek key motif of βγ-crystallins. In γD-crys molecule, the domain interface is composed of (1) a cluster of six hydrophobic residues and (2) two pairs of polar peripheral residues flanking the hydrophobic cluster. The hydrophobic patch is comprised of three amino acids from each domain, which are Met43, Phe56, and Ile81 from the N-terminal domain (N-td) and Val132, Leu145, and Val170 from the C-terminal domain (C-td). Peripheral polar interactions are between Gln54/Gln143 and Arg79/Met147.
Figure 1.
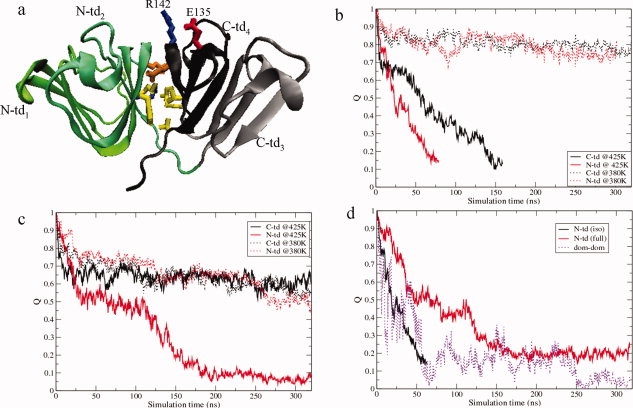
(a) Cartoon representation of γD-crys monomer is shown by specifying the four Greek key motifs (N-td1, N-td2, C-td3, and C-td4) in different colors. The heavy sidechain of the residues at the interdomain surface are shown in ball-stick representation. The predicted substitution site E135 is also shown that forms a salt-bridge interaction with R142 in the wild-type protein during unfolding simulations. Yellow color is used for nonpolar residues, while polar residues are shown in orange. Acidic residues are colored in red and basic residues are colored in blue. (b) Time evolution of the fraction of native contacts, Q, for the single domains in isolation at two different temperatures, 380 K (dashed) and 425 K (solid). (c) Time evolution of the fraction of native contacts, Q, for the single domains within the full monomer at two different temperatures, 380 K (dashed) and 425 K (solid). (d) Time evolution of the fraction of native contacts, Q, of the N-td (solid line) in isolation and in the full monomer and the fraction of native contacts, Q, at the domain interface (dashed line).
A number of experimental investigations have provided valuable information on the importance of the interdomain interactions governing the complex folding/unfolding of γD-crys.9–14 The individual domains of γD-crys exhibit differential stability with the C-td being more stable than the N-td.16–18 Equilibrium unfolding/refolding experiments of γD-crys at near-physiologic conditions (at neutral pH and 37°C using Guanidinium Chloride [GdmCl] as denaturant) has suggested sequential folding of its domains with the C-td refolding first.16 Furthermore, these experiments suggest the existence of a partially folded intermediate state with its C-td largely folded and N-td fully or partially unstructured.12 This folding intermediate undergoes aggregation below 1.0 M GdmCl that competes with productive refolding.19 Site-specific mutagenesis experiments demonstrated that the domain interface residues nucleate the N-td folding.12–14 Although both isolated domains could refold efficiently by themselves, the N-td exhibited lower stability than the C-td in isolation.20 Stability comparisons of the isolated domains with the full monomer revealed that the domain interface contributes a ΔGH2O of ∼4.2 kcal/mol to the stability of the full γD-crys monomer.20 Nevertheless, the detailed effects of the interdomain interactions on the folding mechanism of the individual domains are not evident from the experiments.
β-sheets are found in more than 80% of the reported crystallographic structures, and misfolding and aggregation of β-sheets have been implicated in many neurodegenerative diseases such as Alzheimer's disease and mad cow disease. The difficulty in deciphering the sequence control of β-sheet folding reflects the importance of interactions between side chains that are distant in the primary amino acid sequence. Although, significant progress has been made in understanding the folding pathways of several β-sheets21–23 and isolated SH3 domain24–28 using experimental and theoretical/simulation techniques, detailed characterization of the folding/unfolding landscape of a biologically important β-sheet protein still remains a challenge.29 For example, even for a small 16-residue β-hairpin (GB1), there are debates over a hydrogen-bond zipping mechanism versus a hydrophobic core collapse mechanism.30–34 In a separate example, even the use of high resolution experimental techniques such as coupling of H/D exchange with 2D NMR only allows detection of intermediate states with almost native-like five-strand β-sheet conformation during the earliest stage of Ubiquitin folding.35,36
Computer simulations of protein models at different resolutions, from simple lattice models (and off-lattice)37–39 to continuum solvent models38,40,41 to all-atom explicit solvent models,42,43 have been used to supplement existing experimental techniques in understanding aspects of protein folding.44,45 In principle, molecular dynamics (MD) simulations with an atomistic description of the protein and solvent molecules should provide the most realistic description of the protein folding landscapes. However, such folding simulations starting from fully extended states are often limited to small peptides,42,43 as the sampling of the huge conformational space of a typical protein is currently inaccessible even with the fastest supercomputers. In this endeavor, unfolding simulations, performed in either chemical denaturing or thermal denaturing conditions, have been widely used to shed light onto the folding scenario. According to the principle of microscopic reversibility, the folding pathway should be reverse of the unfolding pathway under same conditions. In addition, previous studies have shown that unfolding simulations, similar to those performed in this study, can accurately capture crucial details of the folding landscapes of a number of proteins in quantitative agreement with experiments (see Ref. 46 and references therein).
In this study, we elucidate the unfolding mechanism for the isolated domains and the full γD-crys monomer by performing chemical denaturing simulations in 8 M urea solution. Further analyses of the atomistic unfolding simulations provide an in-depth understanding of the differential stability of γD-crys domains, which is consistent with previous experiments. Our simulated unfolding data clearly show that the presence of the interdomain interactions affects both the stability and the folding mechanism of the N-td. In addition, a hydrophobic folding core comprised of a and b β-strands from C-td2 is identified at the domain interface of the C-td. A surprising surface salt-bridge at the end of the ab folded hairpin turn of C-td2, which is absent in the native conformation of γD-crys, is found to play a crucial role in stabilizing the folding core of the C-td. We further show that the disruption of this non-native salt-bridge by a single alanine substitution (E135A) performed in silico dramatically destabilizes the folding nucleus of the C-td, which, in turn, trigger unfolding of the N-td interface. These results underscore the importance of such non-native interactions in folding of β-sheet domains of γ-crystallins that may offer clues to map pathways of crystallin aggregation leading to cataract formation.
System and Methods
The initial structure of the wild-type γD-crystallin protein [see Fig. 1(a)] has been taken from the crystal structure deposited in the Protein Data Bank (PDB ID code 1hk0). The isolated N-td comprised of 85 residues ends at Gly85, while the isolated C-td comprised of 88 residues begins at Ser87. The chemical denaturing simulations were performed in 8 M urea solution (for details of the preparation of immersed protein system within the 8 M urea solution, see Ref. 47). The final molecular system consisted of a single domain or the full monomer of γD-crys in 8 M urea and contained ∼7650 water molecules and ∼1775 urea molecules. This system consisted of a total of ∼40,000 atoms was minimized for 10,000 steps followed by a 1 ns MD equilibration at 310 K and 1 atm. The particle-mesh Ewald (PME) method48 was used for the long-range electrostatic interactions, while the van der Waals interactions were treated with a cutoff distance of 12 Å. The CHARMM32 (c32b1 parameter set) force field49 and TIP3P water model50 was used to run MD. All simulations wee performed using NAMD2 molecular modeling package51,52 with a 2 fs time step in NPT ensemble at 1atm.
At least five MD trajectories were run for each system starting from different initial configurations. For each system, simulations were run at two different temperatures, at 380 K and at 425 K. However, all results mentioned in this article are derived from simulations run at 425 K unless otherwise stated, as at 380 K we did not observe complete unfolding of either isolated domains or individual domains within the full monomer even after >0.5 μs MD simulations. This observation is consistent with the experimental data showing that γD-crys is resistant up to 8 M urea denaturant and an acidic pH is required for completely unfolding.19 However, for a variety of mutant γ-crystallins (such as human V75D γD-crys) that do denature in urea, a partially folded intermediate with the N-td disordered and the C-td folded was populated in urea-induced denaturation/renaturation experiments, similar to the species observed in GdmCl-induced Denaturation (Kate N. Drahos, personal communication). The extent to which the crucial details (such as formation of a non-native salt-bridge) of the unfolding pathway of γD-crys depend on the specific denaturation agent used is currently not clear in experiments. On the other hand, simulations of proteins in a GdmCl solution are particularly challenging due to the less well parameterized GdmCl molecule and the complexity associated with the large number of charges involved.
We therefore performed the denaturing simulations of γD-crys at an elevated temperature (425 K) in 8 M urea solution to study unfolding. The overall agreement between the current simulated results and our recent experimental data indicates that these simulated unfolding pathways of the γD-crys domains capture the essential features observed in the refolding/unfolding experiments, and therefore are useful in providing further insights into the kinetics of unfolding. It should be noted, that these simulations performed under denaturing conditions solely provide kinetic models for γD-crys unfolding and should not be over extended to measure equilibrium quantities.
Results and Discussions
Figures 1b and c show the time dependence of the fraction of native contacts, Q, for the unfolding of the individual domains in isolation and within the full protein, respectively. Here, a native contact between residues i and j is counted if any heavy atom of residue i is within 6.5 Å of any heavy atom of residue j in the crystal structure of the protein. Time evolution of Q clearly indicates that the isolated N-td unfolded ∼2 times faster than isolated C-td at 425 K, in agreement with experiments (16,20). The N-td denatured faster than the C-td in the full monomer as well, which leads to the formation of a partially unfolded intermediate state [RMSDCα (full protein) ∼10 Å, Q (full protein) ∼0.4] during the full monomer unfolding [Fig. 1(c)]. As revealed from Figure 1(c), this intermediate state has its C-td mostly structured (QC-td >0.5), but the N-td fully or partially unfolded (QN-td <0.1), in agreement with recent time-resolved fluorescence experiments.16–18 The stability of the C-td (both in isolation and in full protein) over the N-td is also manifested by the unfolding simulations at 380 K. Interestingly, a comparison of the time evolution of QN-td in isolation with the same in the full monomer shows that the unfolding of the N-td in full monomer started much later (>100 ns) compared to that of isolated N-td [Fig. 1(d)] at 425 K, suggesting the contribution of the C-td in stabilizing the N-td. The contribution of interdomain interactions becomes clearer, as the domain interface native contacts start to weaken before those within the N-td [Fig. 1(d)]. Therefore, our simulations reveal that the interdomain interactions contribute to the stability of the N-td of γD-crys, in line with the experimental observation that the mutations weakening interdomain interactions lower both the stability and the folding rate of the N-td, while the C-td remains unaffected.12–14
To obtain a more detailed view of the conformational changes during the unfolding processes, we monitored the secondary structure change as a function of time for the isolated domains, as well as of the full monomer (Supporting Information Fig. S1). We find that N-td2, the one closer to the domain interface, of the isolated N-td unfolded first [Fig. S1(a)]. This might be related to the fact that there is no topological equivalent of Arg142 (from motif 4) in motif 2, but there is one in motif 1 (see more discussion below). Surprisingly, this motif remained more structured during the N-td unfolding in the presence of the C-td interface [Fig. S1(b)]. This finding strongly implies that the absence of interdomain surface interactions catalyzes unfolding of N-td2 in the isolated N-td, indicating that the domain interface interactions are most likely needed to stabilize this motif in its native conformation. Taken together, these results clearly demonstrate the importance of interdomain native interactions in determining the stability of the N-td.
During the isolated C-td unfolding, Greek motif 3 C-td3 disrupted much faster than motif 4 C-td4 [Fig. S1(c)]. Within C-td4, strands a and b remain fairly stable until the very end of the simulation despite the fact that RMSDCα grows up to >14–16 Å and the fraction of native contacts decreases to less than 40%. This observation indicates that these two β-strands from C-td4 form a stable folding nucleus in the isolated C-td, which functions as a folding scaffold for the N-td. Our identification of this folding nucleus within the C-td confirms the mutational data,12–14 suggesting population of two major intermediates during γD-crys refolding; the first one most likely has C-td4 folded, while rest of the protein remains unstructured. The second intermediate has the C-td fully folded and the N-td mainly unfolded. In addition, these results obtained from all-atom simulations explain the three-state mechanism observed during earlier equilibrium/kinetic folding/unfolding experiment of the isolated C-td.20
To investigate the physicochemical factors governing the surprisingly high stability of strands a and b of C-td4 observed during our chemical denaturing simulations, we calculated the probabilities of native contact formation for the unfolded conformational ensemble (RMSDCα >10 Å and Q <0.3) populated during isolated C-td unfolding [Fig. 2(a)]. A cluster of native contacts formed between strands a and b from C-td4 that encompass residues 131–135 and residues 141–145 remained unperturbed in the unfolded ensemble, as seen in Figure 2(a). An additional cluster of native contacts was also found in the unfolded ensemble of isolated C-td between strands a and d of C-td4, however, this cluster was much weaker than the one found between a and b strands. Figure 2(b) provides a more precise and dynamic view of unfolding to this point, which shows the time evolution of the probabilities of long-range (|i-j| >6) native contact formation per residue, Qres, for C-td4. This calculation allows us to identify residues that maintained more than 30% of the long range native interactions until the very end of the unfolding simulations. Clearly, these residues are 132–135 and 142–145. Taken together, these results suggest the existence of a folding core encompassing strands a and b of C-td4 that is comprised of mainly hydrophobic residues at the C-td domain interface (V132 and L133 from the a strand, and Y144 and L145 from the b strand).
Figure 2.
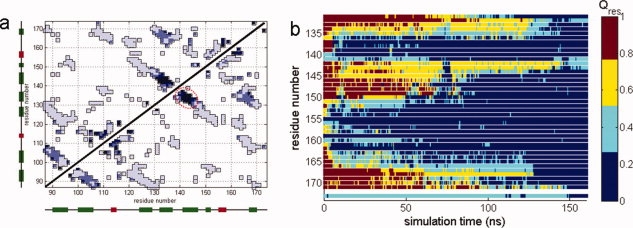
(a) Probabilities of native contact formation for the unfolded conformational ensemble (RMSDCα >10 Å and Q <0.3) of the isolated C-td. Darker shades of blue indicate stronger native contacts. Secondary elements are shown for each residue, β-sheets in green and helices in red. (b) The probabilities of native contact formation per residue, Qres, of C-td4 as a function of simulation time during isolated C-td unfolding. The quantity, Qres is colored according to the colorbar.
Visual inspection and further analyses (see later) of the conformational behavior of the folding core of C-td during unfolding strikingly revealed a salt-bridge interaction between residues Glu135 and Arg142 at the γD-crys surface formed that is maintained until the protein completely unfolds. Surprisingly, this salt-bridge interaction, in which the Oɛ2 atom of Glu135 stays at a distance ∼3.2Å from the Nη2 atom of Arg142 during entire unfolding simulation, is not seen in the wild-type crystal structure. In fact, the sidechain carboxylate oxygens of Glu135 are ∼7 Å away from the sidechain nitrogens of Arg142 in the crystal structure. These results strongly imply that, this surprising non-native salt-bridge interaction near the end of the ab folded hairpin turn of C-td4 protects the two β-strands of the Greek key motif 4 from unfolding, thus promoting the formation of a hydrophobic folding nucleus.1
Such a favorable role of non-native interaction in the formation of protein folding nucleus is in line with previous experimental and theoretical studies, suggesting that specific non-native interactions may accelerate folding by reducing conformational search to the native state.53–63 The identification of a critical salt-bridge in a crystallin folding/unfolding intermediate is another example of non-native interactions crucial for protein folding. Similarly, Presta and Rose59 described helix stop signals that were not necessarily retained in the native state of the folded proteins. Baldwin and coworkers64,65 described ion pairs and helix dipole interactions in the S-peptide of RNase, which were not expected to be maintained in the native state of RNase. Dobson, Schwalbe and coworkers66 observed long-range non-native interactions in lysozyme unfolding in 8 M urea, which were related to a non-native Arg-Trp-Arg sandwich structure, as found by Zhou and coworkers47 with microseconds of MD simulations.
We then performed in silico a single alanine replacement on residue Glu135, a highly conserved residue within γ-crystallin family, to explore the role of this non-native salt-bridge in the unfolding of the isolated C-td and of the N-td in full γD-crys. We expect such a replacement to disrupt the critical Glu135-Arg142 salt-bridge, thereby favoring unfolding of interdomain surface. Indeed, the isolated C-td, as well as the N-td in full γD-crys, of the E135A mutant, unfolded much rapidly compared to the wild-type, as revealed from the time dependence of fraction of native contacts, Q [Fig. 3(a)]. The faster denaturation of both domains in the E135A mutant was a direct consequence of the disruption of Glu135-Arg142 salt-bridge, which is evident from the comparison of the Cα-Cα distance between residue Glu/Ala-135 and Arg142 in the wild-type and mutant protein during unfolding [Fig. 3(b)]. During the wild-type isolated C-td unfolding, those two residues formed the non-native salt-bridge within the first 5 ns of the simulation and maintained as a salt-bridge for the rest of the simulation [Fig. 3(b)]. This persistence of the salt-bridge between Glu135 and Arg142 seems to suggest that they play a key role in the “nucleation process” of the hydrophobic core near the domain interface. In contrast, the distance between Ala135 and Arg142 became larger and fluctuated more in the E135A mutant compared to the wild-type at different stages of unfolding [Fig. 3(b)]. Consequently, the secondary structure of Greek key motif 4 C-td4 of the mutant disrupted more quickly and more drastically, particularly the β-strand near Arg142 is completely lost around 70 ns [Fig. S2(a)]. Other trajectories also indicate early destruction of this β-strand between 45 and 70 ns. In addition, the β-sheets comprising the Greek key motif 2 N-td2 also denature much earlier in the E135A mutant (∼50 to 70 ns) than in the wild-type protein (∼160 ns) [Supporting Information Fig. S2(b)], suggesting the critical role of the Glu135-Arg142 salt-bridge in stabilizing the interdomain surface.
Figure 3.

(a) Time evolution of the fraction of native contacts, Q, for the isolated C-td (solid line) and for the N-td in full protein (dashed line) of the wild-type protein (in black) and the E135A mutant (in red) at 425 K. (b) Cα-Cα Distance between E135 and residue 142 as a function of simulation time in the isolated C-td of the wild-type protein (in black) and the E135A mutant (in red). The time dependence of the distance between the E135-R142 ion-pair in the wild-type isolated C-td is also shown in dashed line. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The effect of the E135A replacement on the tertiary structure loss of the isolated C-td was even more dramatic, as revealed from the detailed comparison of the wild-type unfolding trajectories with those of the mutant (Fig. 4). The ab folded hairpin turn of C-td4 became more flexible at a very early stage of unfolding (∼40 to 80 ns) in the mutant than the wild-type. By ∼80 to 120 ns, the wild-type isolated C-td mostly preserves its overall tertiary structure despite the loss of secondary structures in the Greek Key motif 3 C-td3. On the other hand, by this time the mutant has lost most of its tertiary structure as well as the secondary structures within C-td4, mainly near the mutation site. Finally, at ∼140 ns, both the wild-type and the mutant fully unfold (Q ∼0.1). By this time, strands a and b of C-td4 still maintained their secondary structures in the wild-type C-td. However, all of the tertiary structures, as well as the secondary structures, of C-td4 are completely lost in the mutant at this time, demonstrating the importance of the Glu135-Arg142 ion-pair interaction in preserving the stable folding nucleus of the isolated C-td. This result deserves further validation through direct biochemical experiments.
Figure 4.
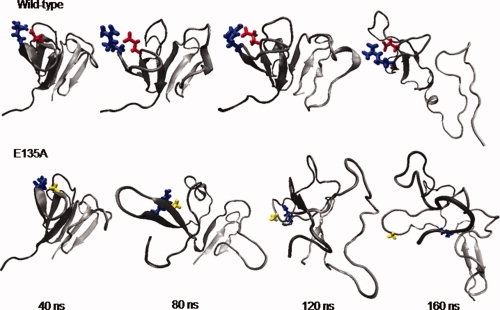
Representative conformations of the isolated C-tds of the wild-type and the E135A mutant during 160 ns unfolding simulations. The cartoon representation of the protein is colored in gray to black from N-terminal to C-terminal. Residues 135 and 142 are shown in ball-stick representation. The colors used to represent different types of residues are similar to that used in Figure 1(a). For the wild-type, the non-native salt-bridge between Glu135 and Arg142 was formed within the first 5 ns of the unfolding simulation and maintained as a salt-bridge for the rest of the simulation. On the other hand, for the E135A mutant, there is no such salt-bridge, and the two residues are separated apart in far distance after about 100 ns simulation. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Based on these observations during kinetic unfolding simulations, we propose a detailed folding mechanism of the β-sheet domains of γD-crys. During the folding of the full γD-crys protein, a partially unfolded intermediate state is populated with its C-td mainly folded and N-td mostly unstructured. We also identify potential intermediates in the formation of double Greek key domains, in which one of the Greek key motifs is folded first, while the other motif remains mainly unstructured. In the full γD-crys monomer, the Greek key motifs that fold first within the individual domains are those near the interdomain surface. Thus, C-td4 that encompasses the C-td interface of γD-crys folds before C-td3. The C-td interface then serves as the folding template for the N-td, thus facilitating the folding of N-td2. Within C-td4, a hydrophobic folding core is formed in the very initial stage of γD-crys folding. This important folding core mainly consists of strands a and b. Surprisingly, a non-native salt-bridge formed between two conserved residues, that is, Glu135 and Arg142, at the end of the ab folded hairpin turn is found to be crucial for the formation of the folding nucleus of the C-td. This non-native salt-bridge interaction allows the early formation of ab folded hairpin turn, thus assisting the folding of strands a and b of C-td4.
It is interesting to note that even though the hydrophobic core comprising strands a and b of C-td4 plays an important role in the nucleation process as explained above, our simulations also showed that the unfolding of the wild-type isolated C-td is not affected much with a few substitutions to alanine for the hydrophobic residues (V132A, Y134A, Q143A, and L145A) of this core. This result is consistent with the experimental mutational data showing that the stability of the C-td was unaffected in the mutants V132A and L145A (12). Thus, our results indicate the importance of the Glu135-Arg142 salt-bridge for the correct formation of the hydrophobic interdomain surface of γD-crys. Interestingly, our recent in vitro thermodynamic and kinetic folding experiments of single mutants of γD-crys suggest that partial unfolding of the C-td is required to induce aggregation.67 Taken together, both experimental and simulation results seem to imply that a partially denatured folding intermediate ensemble of γD-crys with C-td3 mostly unstructured is likely one of the candidates for the aggregation-prone species leading to cataract formation. The importance of salt-bridge interactions in stabilizing γ-crystallins is also evident from the presence of highly polar residues on their surface, as demonstrated by crystallographic data.68 Most of these residues are highly conserved, suggesting their important role in crystallin structure and/or function.69 These polar residues form charge networks on the molecular surface, in which more than 50% of the ionic side chains form ion pairs.68 Such organized charges strongly contribute to the high thermal stability, as well as to the high water solubility, of γ-crystallins within the lens, as the disruption of the charge network by the binding of small molecules to key charged residues leads to the post-translational adduct-mediated unfolding.70 In addition, point mutations associated with congenital cataracts also involve surface polar residues of γ-crystallins.71,72
Taken together, aforementioned experimental observations suggested that the surface charge network, together with the hydrophobic interactions, plays a major role in maintaining the stability of native γD-crys. The greater abundance of acidic and basic residues within the C-td most likely adds to its higher stability over the N-td in the native state. Our current simulations provided more atomic details to these observations and further identified one critical non-native salt-bridge interaction at the protein surface, in addition to the hydrophobic interactions between strands a and b of C-td4. This Glu135-Arg142 salt-bridge helps initiating the folding of the ab folded hairpin turn, resulting in the formation of the C-td surface that is essentially used by the N-td as a folding template. Since a correctly formed interdomain surface of γD-crystallin is likely required to prevent unfolding into aggregation-prone species, disruption of charges on the γD-crystallin surface might lead to aggregation and development of opacity.
Conclusions
The γD-crystallins are synthesized in lens fiber cells in utero and infancy, and then must stay folded and stable within the terminally differentiated and anucleated fiber cells for a lifetime. For these proteins, the stability of the native state is a fundamental biological property. Though it seems reasonable that this stability resides in features of the Greek Key β-sheet fold and its tightly packed hydrophobic core, the molecular basis of the stability has been difficult to unravel. The detailed folding mechanism of the individual double Greek key domains of γD-crys has also resisted elucidation. In this article, we have used large-scale explicit solvent atomistic simulations to investigate the unfolding processes of the isolated domains, as well as of the full monomer, of γD-crys. Our simulations strongly suggest that the stability and the folding mechanism of the N-td are regulated by the interdomain interactions. As hypothesized based on fluorescence spectroscopic data,20 a hydrophobic folding core is identified within the C-td interface of γD-crys that is comprised of strands a and b from Greek motif 4 C-td4. Detailed analyses strikingly reveal that a surface salt-bridge formed between Glu135 and Arg142 initiates folding of the ab folded hairpin turn of C-td4, allowing formation the stable folding core. However, this salt-bridge is not seen in the γD-crys crystal structure. We show the critical role of this non-native salt-bridge in preventing C-td4 from complete unfolding by in silico introduction of a single mutation E135A. This substitution that disrupts the Glu135-Arg142 salt-bridge causes significant destabilization to the stable folding core of the isolated C-td, which, in turn, triggers unfolding of the N-td. Our results thus show that certain non-native interactions formed in the highly conserved polar exterior of γD-crys are crucial for correct folding of its hydrophobic domain interface. Therefore, our simulations in combination to our recent experiments provide a complete molecular picture of the complex unfolding landscape of γD-crystallin that might offer new insights into the development of novel treatments for cataracts.
Acknowledgments
The authors thank Eric Kronstadt for the help of initiating this project on Blue Gene/L. They thank Bruce Berne, Ajay Royyuru for helpful discussions. They also thank Sameer Kumar for numerous help with porting NAMD2 onto IBM BlueGene/L. They also acknowledge the contributions of the BlueGene/L hardware, system software, and science application teams whose efforts and assistance made it possible to use the BlueGene/L supercomputer at the IBM Watson Center.
Footnotes
The presence of an Arginine in Greek key motifs at a topological equivalent position of Arg142 likely contributes to their folding and stability. For example, N-td2 that lacks topological equivalent of Arg142 unfolds before N-td1 (do have the topological equivalent of Arg142) in the isolated N-td. Consistently, C-td3 that lacks topological equivalent of Arg142 unfolds before C-td3 in the isolated C-td.
References
- 1.Resnikoff S, Pascolini D, Etya'ale D, Kocur I, Pararajasegaram R, Pokharel GP, Mariotti SP. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004;82:844–851. [PMC free article] [PubMed] [Google Scholar]
- 2.Pandey SK, Apple DJ, Werner L, Maloof AJ, Milverton EJ. Posterior capsule opacification: a review of the aetiopathogenesis, experimental and clinical studies and factors for prevention. Indian J Ophthalmol. 2004;52:99–112. [PubMed] [Google Scholar]
- 3.Benedek GB. Cataract as a protein condensation disease: the Proctor Lecture. Invest Ophthalmol Vis Sci. 1997;38:1911–1921. [PubMed] [Google Scholar]
- 4.Aarts HJM, Lubsen NH, Schoenmakers JGG. Crystallin gene-expression during rat lens development. Eur J Biochem. 1989;183:31–36. doi: 10.1111/j.1432-1033.1989.tb14892.x. [DOI] [PubMed] [Google Scholar]
- 5.Lampi KJ, Shih M, Ueda Y, Shearer TR, David LL. Lens proteomics: analysis of rat crystallin sequences and two-dimensional electrophoresis map. Invest Ophthalmol Visual Sci. 2002;43:216–224. [PubMed] [Google Scholar]
- 6.Lubsen NH, Aarts HJM, Schoenmakers JGG. The evolution of lenticular proteins—the beta-crystallin and gamma-crystallin super gene family. Prog Biophys Mol Biol. 1988;51:47–76. doi: 10.1016/0079-6107(88)90010-7. [DOI] [PubMed] [Google Scholar]
- 7.Piatigorsky J. Crystallin genes: specialization by changes in gene regulation may precede gene duplication. J Struct Funct Genomics. 2003;3:131–137. [PubMed] [Google Scholar]
- 8.Shimeld SM, Purkiss AG, Dirks RPH, Bateman OA, Slingsby C, Lubsen NH. Urochordate beta gamma-crystallin and the evolutionary origin of the vertebrate eye lens. Curr Biol. 2005;15:1684–1689. doi: 10.1016/j.cub.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 9.Hope JN, Chen HC, Hejtmancik JF. Aggregation of beta-A3-crystallin is independent of the specific sequence of the domain connecting peptide. J Biol Chem. 1994;269:21141–21145. [PubMed] [Google Scholar]
- 10.Mayr EM, Jaenicke R, Glockshuber R. Domain interactions and connecting peptides in lens crystallins. J Mol Biol. 1994;235:84–88. doi: 10.1016/s0022-2836(05)80017-8. [DOI] [PubMed] [Google Scholar]
- 11.Palme S, Slingsby C, Jaenicke R. Mutational analysis of hydrophobic domain interactions in gamma B-crystallin from bovine eye lens. Protein Sci. 1997;6:1529–1536. doi: 10.1002/pro.5560060717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flaugh SL, Kosinski-Collins MS, King J. Interdomain side-chain interactions in human gamma D crystallin influencing folding and stability. Protein Sci. 2005;14:2030–2043. doi: 10.1110/ps.051460505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flaugh SL, Kosinski-Collins MS, King J. Contributions of hydrophobic domain interface interactions to the folding and stability of human gamma D-crystallin. Protein Sci. 2005;14:571–581. doi: 10.1110/ps.041111405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flaugh SL, Mills IA, King J. Glutamine deamidation destabilizes human gamma D-crystallin and lowers the kinetic barrier to unfolding. J Biol Chem. 2006;281:30782–30793. doi: 10.1074/jbc.M603882200. [DOI] [PubMed] [Google Scholar]
- 15.Lampi KJ, Ma ZX, Shih M, Shearer TR, Smith JB, Smith DL, David LL. Sequence analysis of beta A3, beta B3, and beta A4 crystallins completes the identification of the major proteins in young human lens. J Biol Chem. 1997;272:2268–2275. doi: 10.1074/jbc.272.4.2268. [DOI] [PubMed] [Google Scholar]
- 16.Kosinski-Collins MS, Flaugh SL, King J. Probing folding and fluorescence quenching in human gamma D crystallin Greek key domains using triple tryptophan mutant proteins. Protein Sci. 2004;13:2223–2235. doi: 10.1110/ps.04627004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Toptygin D, Brand L, King J. Mechanism of the efficient tryptophan fluorescence quenching in human gammaD-crystallin studied by time-resolved fluorescence. Biochemistry. 2008;47:10705–10721. doi: 10.1021/bi800499k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Callis PR, King J. Mechanism of the very efficient quenching of tryptophan fluorescence in human gamma D- and gamma S-crystallins: the gamma-crystallin fold may have evolved to protect tryptophan residues from ultraviolet photodamage. Biochemistry. 2009;48:3708–3716. doi: 10.1021/bi802177g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosinski-Collins MS, King J. In vitro unfolding, refolding, and polymerization of human gamma D crystallin, a protein involved in cataract formation. Protein Sci. 2003;12:480–490. doi: 10.1110/ps.0225503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishara AM, Flaugh LS, Kosinski-Collins MS, King JA. Folding and stability of the isolated Greek key domains of the long-lived human lens proteins gamma D-crystallin and gamma S-crystallin. Protein Sci. 2007;16:2427–2444. doi: 10.1110/ps.072970207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Alba E, Santoro J, Rico M, Jimenez MA. De novo design of a monomeric three-stranded antiparallel beta-sheet. Protein Sci. 1999;8:854–865. doi: 10.1110/ps.8.4.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrara P, Caflisch A. Folding simulations of a three-stranded antiparallel beta-sheet peptide. Proc Natl Acad Sci USA. 2000;97:10780–10785. doi: 10.1073/pnas.190324897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deechongkit S, Nguyen H, Jager M, Powers ET, Gruebele M, Kelly JW. Beta-Sheet folding mechanisms from perturbation energetics. Curr Opin Struct Biol. 2006;16:94–101. doi: 10.1016/j.sbi.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 24.Viguera AR, Martinez JC, Filimonov VV, Mateo PL, Serrano L. Thermodynamic and kinetic-analysis of the SH3 domain of spectrin shows a 2-state folding transition. Biochemistry. 1994;33:2142–2150. doi: 10.1021/bi00174a022. [DOI] [PubMed] [Google Scholar]
- 25.Plaxco KW, Guijarro JI, Morton CJ, Pitkeathly M, Campbell ID, Dobson CM. The folding kinetics and thermodynamics of the Fyn-SH3 domain. Biochemistry. 1998;37:2529–2537. doi: 10.1021/bi972075u. [DOI] [PubMed] [Google Scholar]
- 26.Grantcharova VP, Baker D. Folding dynamics of the src SH3 domain. Biochemistry. 1997;36:15685–15692. doi: 10.1021/bi971786p. [DOI] [PubMed] [Google Scholar]
- 27.Ding F, Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Molecular dynamics simulation of the SH3 domain aggregation suggests a generic amyloidogenesis mechanism. J Mol Biol. 2002;324:851–857. doi: 10.1016/s0022-2836(02)01112-9. [DOI] [PubMed] [Google Scholar]
- 28.Settanni G, Gsponer J, Caflisch A. Formation of the folding nucleus of an SH3 domain investigated by loosely coupled molecular dynamics simulations. Biophys J. 2004;86:1691–1701. doi: 10.1016/S0006-3495(04)74238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roder H, Maki K, Cheng H. Early events in protein folding explored by rapid mixing methods. Chem Rev. 2006;106:1836–1861. doi: 10.1021/cr040430y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munoz V, Thompson PA, Hofrichter J, Eaton WA. Folding dynamics and mechanism of beta-hairpin formation. Nature. 1997;390:196–199. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 31.Klimov DK, Thirumalai D. Mechanisms and kinetics of beta-hairpin formation. Proc Natl Acad Sci USA. 2000;97:2544–2549. doi: 10.1073/pnas.97.6.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zagrovic B, Sorin EJ, Pande V. Beta-hairpin folding simulations in atomistic detail using an implicit solvent model. J Mol Biol. 2001;313:151–169. doi: 10.1006/jmbi.2001.5033. [DOI] [PubMed] [Google Scholar]
- 33.Zhou R, Berne BJ, Germain R. The free energy landscape for beta hairpin folding in explicit water. Proc Natl Acad Sci USA. 2001;98:14931–14936. doi: 10.1073/pnas.201543998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Felts AK, Harano Y, Gallicchio E, Levy RM. Free energy surfaces of beta-hairpin and alpha-helical peptides generated by replica exchange molecular dynamics with the AGBNP implicit solvent model. Proteins. 2004;56:310–321. doi: 10.1002/prot.20104. [DOI] [PubMed] [Google Scholar]
- 35.Meier S, Strohmeier M, Blackledge M, Grzesiek S. Direct observation of dipolar couplings and hydrogen bonds across a beta-hairpin in 8 M urea. J Am Chem Soc. 2007;129:754–755. doi: 10.1021/ja067522k. [DOI] [PubMed] [Google Scholar]
- 36.Meier S, Grzesiek S, Blackledge M. Mapping the conformational landscape of urea-denatured ubiquitin using residual dipolar couplings. J Am Chem Soc. 2007;129:9799–9807. doi: 10.1021/ja0724339. [DOI] [PubMed] [Google Scholar]
- 37.Wolynes PG, Onuchic JN, Thirumalai D. Navigating the folding routes. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 38.Kang SG, Saven JG. Computational protein design: structure, function and combinatorial diversity. Curr Opin Chem Biol. 2007;11:329–334. doi: 10.1016/j.cbpa.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 39.Dill KA, Bromberg S, Yue K, Chan HS, Ftebig KM, Yee DP, Thomas PD. Principles of protein folding—a perspective from simple exact models. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou R, Berne BJ. Can a continuum solvent model reproduce the free energy landscape of a beta-hairpin folding in water? Proc Natl Acad Sci USA. 2002;99:12777–12782. doi: 10.1073/pnas.142430099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feig M, Brooks CL. Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr Opin Struct Biol. 2004;14:217–224. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 42.Simmerling C, Strockbine B, Roitberg AE. All-atom structure prediction and folding simulations of a stable protein. J Am Chem Soc. 2002;124:11258–11259. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- 43.Snow CD, Nguyen H, Pande VS, Gruebele M. Absolute comparison of simulated and experimental protein-folding dynamics. Nature. 2002;420:102–106. doi: 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- 44.Warshel A. Molecular dynamics simulations of biological reactions. Acc Chem Res. 2002;35:385–395. doi: 10.1021/ar010033z. [DOI] [PubMed] [Google Scholar]
- 45.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Mol Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 46.Daggett V. Protein folding-simulation. Chem Rev. 2006;106:1898–1916. doi: 10.1021/cr0404242. [DOI] [PubMed] [Google Scholar]
- 47.Zhou R, Eleftheriou M, Royyuru AK, Berne BJ. Destruction of long-range interactions by a single mutation in lysozyme. Proc Natl Acad Sci USA. 2007;104:5824–5829. doi: 10.1073/pnas.0701249104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 49.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 50.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 51.Kumar S, Huang C, Almasi G, Kale LV. Achieving Strong Scaling with NAMD on Blue Gene/L. In Proceedings of IEEE International Parallel and Distributed Processing Symposium; April 2006; Rhodes Island, Greece. 2006. [Google Scholar]
- 52.Kumar S, Huang C, Zheng G, Bohm E, Bhatele A, Phillips JC, Yu H, Kale LV. Scalable molecular dynamics with NAMD on blue gene/L. IBM J Res Dev. 2008;52:177–188. [Google Scholar]
- 53.Blanco FJ, Ortiz AR, Serrano L. Role of a nonnative interaction in the folding of the protein G B1 domain as inferred from the conformational analysis of the alpha-helix fragment. Folding Des. 1997;2:123–133. doi: 10.1016/s1359-0278(97)00017-5. [DOI] [PubMed] [Google Scholar]
- 54.Clementi C, Plotkin SS. The effects of nonnative interactions on protein folding rates: theory and simulation. Protein Sci. 2004;13:1750–1766. doi: 10.1110/ps.03580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Nardo AA, Korzhnev DM, Stogios PJ, Zarrine-Afsar A, Kay LE, Davidson AR. Dramatic acceleration of protein folding by stabilization of a nonnative backbone conformation. Proc Natl Acad Sci USA. 2004;101:7954–7959. doi: 10.1073/pnas.0400550101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Mirny LA, Shakhnovich EI. Kinetics, thermodynamics and evolution of non-native interactions in a protein folding nucleus. Nat Struc Biol. 2000;7:336–342. doi: 10.1038/74111. [DOI] [PubMed] [Google Scholar]
- 57.Morton VL, Friel CT, Allen LR, Paci E, Radford SE. The effect of increasing the stability of non-native interactions on the folding landscape of the bacterial immunity protein Im9. J Mol Biol. 2007;371:554–568. doi: 10.1016/j.jmb.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 58.Neudecker P, Zarrine-Afsar A, Choy WY, Muhandiram DR, Davidson AR, Kay LE. Identification of a collapsed intermediate with non-native long-range interactions on the folding pathway of a pair of Fyn SH3 domain mutants by NMR relaxation dispersion spectroscopy. J Mol Biol. 2006;363:958–976. doi: 10.1016/j.jmb.2006.08.047. [DOI] [PubMed] [Google Scholar]
- 59.Presta LG, Rose GD. Helix signals in proteins. Science. 1988;240:1632–1641. doi: 10.1126/science.2837824. [DOI] [PubMed] [Google Scholar]
- 60.Treptow WL, Barbosa MAA, Garcia LG, de Araujo AFP. Non-native interactions, effective contact order, and protein folding: a mutational investigation with the energetically frustrated hydrophobic model. Proteins. 2002;49:167–180. doi: 10.1002/prot.10166. [DOI] [PubMed] [Google Scholar]
- 61.Vanhove M, Lejeune A, Guillaume G, Virden R, Pain RH, Schmid FX, Frere JM. A collapsed intermediate with nonnative packing of hydrophobic residues in the folding of TEM-1 beta-lactamase. Biochemistry. 1998;37:1941–1950. doi: 10.1021/bi972143c. [DOI] [PubMed] [Google Scholar]
- 62.Viguera AR, Vega C, Serrano L. Unspecific hydrophobic stabilization of folding transition states. Proc Natl Acad Sci USA. 2002;99:5349–5354. doi: 10.1073/pnas.072387799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zarrine-Afsart A, Wallin S, Neculai AM, Neudecker P, Howell PL, Davidson AR, Chan HS. Theoretical and experimental demonstration of the importance of specific nonnative interactions in protein folding. Proc Natl Acad Sci USA. 2008;105:9999–10004. doi: 10.1073/pnas.0801874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bierzynski A, Kim PS, Baldwin RL. A salt bridge stabilizes the helix formed by isolated C-peptide of RNase A. Proc Natl Acad Sci USA. 1982;79:2470–2474. doi: 10.1073/pnas.79.8.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mayo SL, Baldwin RL. Guanidinium chloride induction of partial unfolding in amide proton exchange in RNase A. Science. 1993;262:873–876. doi: 10.1126/science.8235609. [DOI] [PubMed] [Google Scholar]
- 66.Klein-Seetharaman J, Oikawa M, Grimshaw SB, Wirmer J, Duchardt E, Ueda T, Imoto T, Smith LJ, Dobson CM, Schwalbe H. Long-range interactions within a nonnative protein. Science. 2002;295:1719–1722. doi: 10.1126/science.1067680. [DOI] [PubMed] [Google Scholar]
- 67.Drahos K, King JA. Hydrophobic core mutations associated with cataract development in mice destabilize human rD-crystallin. J Biol Chem. 2009;284 doi: 10.1074/jbc.M109.031344. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wistow G, Turnell B, Summers L, Slingsby C, Moss D, Miller L, Lindley P, Blundell T. X-ray-analysis of the eye lens protein gamma-II crystallin at 1.9 Å resolution. J Mol Biol. 1983;170:175–202. doi: 10.1016/s0022-2836(83)80232-0. [DOI] [PubMed] [Google Scholar]
- 69.Chirgadze Y, Nevskaya N, Vernoslova E, Nikonov S, Sergeev Y, Brazhnikov E, Fomenkova N, Lunin V, Urzhumtsev A. Crystal-structure of calf eye lens gamma-crystallin-IIIB at 2.5-Å resolution—its relation to function. Exper Eye Res. 1991;53:295–304. doi: 10.1016/0014-4835(91)90233-5. [DOI] [PubMed] [Google Scholar]
- 70.Martin S, Harding JJ. Site of carbamoylation of bovine gamma-II-crystallin by potassium[C-14] cyanate. Biochem J. 1989;262:909–915. doi: 10.1042/bj2620909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pande A, Pande J, Asherie N, Lomakin A, Ogun O, King J, Benedek GB. Crystal cataracts: human genetic cataract caused by protein crystallization. Proc Natl Acad Sci USA. 2001;98:6116–6120. doi: 10.1073/pnas.101124798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Talla V, Narayanan C, Srinivasan N, Balasubramanian D. Mutation causing self-aggregation in human gamma C-crystallin leading to congenital cataract. Invest Ophthalmol Vis Sci. 2006;47:5212–5217. doi: 10.1167/iovs.06-0427. [DOI] [PubMed] [Google Scholar]