

Abstract
The Ara h 2 proteins are major determinants of peanut allergens. These proteins have not been fully studied at the molecular level. It has been previously proposed that there are two isoforms of Ara h 2, based on primary structures that were deduced from two reported cDNA sequences. In this report, four isoforms have been purified and characterized individually. Mass spectrometric methods have been used to determine the protein sequences and to define post-translational modifications for all four isoforms. Two pairs of isoforms have been identified, corresponding to a long-chain form and a form that is shorter by 12 amino acids. Each pair is further differentiated by the presence or absence of a two amino acid sequence at the carboxyl terminus of the protein. Modifications that were characterized include site-specific hydroxylation of proline residues, but no glycosylation was found, in contrast to previous reports.
Keywords: peanut antigen, de novo sequencing, tandem mass spectrometry, post-translational modifications
Introduction
Peanut allergies affect ∼3 million people in the United States. Unlike other allergies such as those from milk and eggs, peanut allergy is seldom outgrown after childhood. There are eight allergen proteins in peanut, which are named Ara h 1–8. Ara h 2 is one of the most abundant allergens, present the range of 5.9–9.3% of total protein weight. It is recognized by human Immunoglobulin E in more than 90% of the patients who have peanut allergy.1,2
Two cDNA sequences of Ara h 2 have been reported, Ara h 2.01 and Ara h 2.02 (GenBank Accession Number FJ713110 and AY158467, respectively).3 One protein isoform sequence (corresponding to FJ713110) comprises 160 residues, including the signal peptide (21 amino acids), with glutamic acid and aspartic acid at residues 61 and 151. (In the numbering system used throughout this article the signal peptide is not counted and glutamic acid and aspartic acid appear at residues 40 and 130, respectively). The second isoform is characterized by an insertion of 12 amino acids starting at residue 54 (75 with signal peptide), with residue 40 (61 with signal peptide) as a glycine and residue 142 (163 with signal peptide) as an aspartic acid. The mass added by insertion of this 12 amino acid piece is 1413 Da. A study of the proteins by Yan et al.4 and Viquez et al.5 confirmed that there are amino acids variants at residues 40 and 142. The molecular masses of purified Ara h 2 from raw peanut were measured by MALDI-TOF, which indicated two isoforms of mass 16670 Da and 18050 Da.3 This is consistent with the expression of the protein from two different cDNAs. The mass difference between the measured molecular weights of the two isoforms is 1380 Da, which is close to the 1413 Da proposed by the translated protein sequences based on the two cDNAs.
Previous characterization of Ara h 2 provided the identification of an N-terminal peptide [02–18] and two tryptic peptides [82–94] and [95–110], which were proposed as potential biomarkers for regulatory monitoring.6 A recent study of 2-D gel electrophoretic patterns of proteins from the Virginia type peanut identified fourteen spots that were linked to Ara h 2 by partial peptide mass fingerprinting.7
In a separate study in 1992 focused on potential post-translational modifications (PTM), Burks et al. proposed that Ara h 2 is a glycoprotein, based on periodic acid-Schiff staining. Subsequent analysis reported that this protein contained 20% carbohydrate by mass, with galacturonic acid, arabinose, and xylose as the three most abundant sugars in the putative glycoprotein.8 Their sample was purified from the same runner variety as used in this study, but it was from a different cultivar.
The focus of this study is the detailed elucidation of the primary structure and the disulfide bond pattern of Ara h 2 as the starting point for on-going studies on the effects of food processing, such as roasting the peanuts, on this allergenic protein. In this work, the major isoforms have been characterized using tandem mass spectrometric analysis of peptides generated from enzymatic digests. Microwave acceleration was used to enhance the enzymatic (trypsin) proteolysis of the reduced isoforms.9 Then the peptide products were analyzed using LC-MS/MS. Manual interpretation of the MS/MS spectra allowed de novo sequencing of the primary structures of all the isoforms of the Ara h 2 and identification and location of the PTM. Subsequently, collision induced dissociation (CID) and electron transfer dissociation (ETD) were used to obtain confirmatory sequence information. Database searching was carried out to support both the manual and the confirmatory analyzes.
Results and Discussion
Determination of primary amino acid sequence and modifications
The chromatographic separation of Ara h 2 proteins (Fig. 1) and molecular mass measurement of each fraction (Fig. 1 inset) indicates that there are four isoforms, which have masses measured as 18035, 16662, 17716, and 16344 Da, respectively. These four isoforms were named P1-P4 based on their eluting sequence. The four isoforms may be divided into two pairs based on their molecular masses. P1 and P3 belong to the heavy pair, and P2 and P4 are the light pair. Members of each pair are separated in mass by 319 Da ± 1Da.
Figure 1.
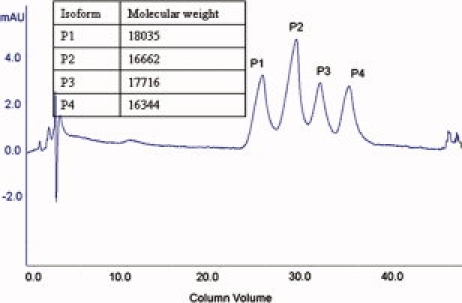
Chromatogram of Ara h 2 isoform separation by anion exchange column and molecular weight of each Ara h 2 isoform which were measured by Q-TOF mass spectrometer.
Detailed primary sequence characterization of the four isoforms was conducted as discussed later. The amino acid sequences for the four isoforms, as determined here, are shown in Figure 2. As expected, the signal peptide is not present. The primary amino acid sequences observed here are largely in concert with those predicted by translation of the two reported cDNA sequences. However, significant modifications are observed, as discussed later.
Figure 2.

Diagram of the primary structure of four isoforms of peanut allergen protein Ara h 2. Hydroxyprolines are highlighted in grey. Figure formed using CLC free workbench 3.
In Figure 3 (a), (c), and (d), CID spectra of C-terminal peptides of isoforms P1, P3, and P4, respectively, are displayed. However, CID of the C-terminal peptide of P2 [114–139] did not provide information as useful as that produced by ETD. The inability to obtain useful information from the CID spectrum of the P2 peptide was unexpected, since the C-terminal peptides of P1 and P2 only differ by the replacement of glutamic acid by aspartic acid at position 142; the reasons for this behavior are under investigation. In any case, ETD of the C-terminal peptide from P2 provides twelve c ions and seven z ions, as shown in Figure 3(b). The CID and ETD spectra allow for assignment of the C-terminal sequence for each of the isoforms. A previously reported amino acid variation at residue 1423,4 is confirmed here. The b17 fragment ion in Figure 3(a) indicates that there is a glutamic acid at residue 142 in the P1 isoform. Corresponding fragment ions in Figures 3(b), (c), and (d) indicate that P3 also has a glutamic acid at residue 142, while P2 and P4 have aspartic acid. The results also show that the two sets of isoforms vary by two amino acids at the end of the sequence. The primary structures of the C-termini of P1 and P2 are consistent with the protein sequences deduced from the two cDNAs. However, P3 and P4 carry two fewer amino acids, Arg and Tyr, at the carboxyl termini, compared to P1 and P2 (Figs. 2 and 3). An artifactual origin of the truncated proteins during the commercial isolation seems less likely, because the same family of proteins has now been detected in extracts of raw peanuts of the same variety made in the authors' laboratory.
Figure 3.
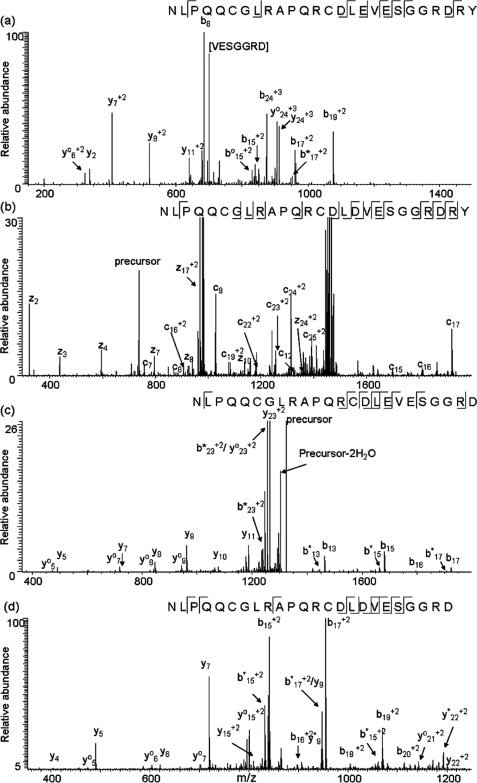
MS/MS spectra acquired on a LTQ mass spectrometer of C-terminal peptides of each isoform of peanut allergen protein. (a) CID spectrum of C-terminal peptide [126–151] from the P1 isoform, quadruply charged ion at m/z 741 was selected as the precursor ion; (b) ETD spectrum of C-terminal peptide [114–139] from the P2 isoform, quadruply charged ion at m/z 738 was selected as the precursor ion; (c) CID spectrum of C-terminal peptide [126–149] from the P3 isoform, doubly charged ion at m/z 1322 was selected as the precursor ion; (d) CID spectrum of C-terminal peptide [114–137] from the P4 isoform, triply charged ion at m/z 877 was selected as the precursor ion. “°” indicates loss of H2O, “*” indicates loss of NH3.
Further analysis of the protein isoforms confirmed the sequences shown in Figure 2 and elucidated details about modifications not predicted by the cDNA. The extended tryptic digestion of P1 (60 min) produced smaller peptides with fewer missed cleavages. From this digestion, PTM were identified in peptides from the central region of the protein [Figs. 4 (a,b)]. In Figure 4(b) the CID spectrum of peptide [42–59] from P1 fragmentation reveals cleavages at 15 of 17 possible backbone sites. It can be seen that the masses of prolines 46 and 53 are increased by 16 Da, while prolines 43, 50, and 57 are not. We propose that the 16 Da increment reveals proline hydroxylation, a post-translational modification known to be common in plant proteins.10 Peptide [61–70] from P1 was also mapped [Fig. 4 (a)] via three b ions and five y ions. The masses of fragment y5 and y6 ions indicate that proline 65 is also hydroxylated in the heavy isoform. The same peptides [42–59] and [61–70] were identified in P3 isoform and the fragmentation patterns of those peptides are essentially the same as that of P1 isoform with minor variation in the abundance of fragment ions (data not shown). Thus, we propose that proline 46, 53, and 65 are hydroxylated in the P3 isoform. In the CID spectrum of peptide [42–58] from light isoform P2, presented in Figure 4 (c), proline residues 46 and 53 are seen to be hydroxylated, while prolines at positions 43, 50 and 55 in P2 are not modified. Similarly, a CID spectrum of peptide [42–58] from the P4 isoform was almost the same as that from the P2 isoform with minor differences in relative peak intensities (data not shown). Consequently, proline residues 46 and 53 in the P4 isoform are hydroxylated. It is worth noting that this modification occurs at analogous sites in both the heavy isoforms and the light isoforms (Fig. 2).
Figure 4.
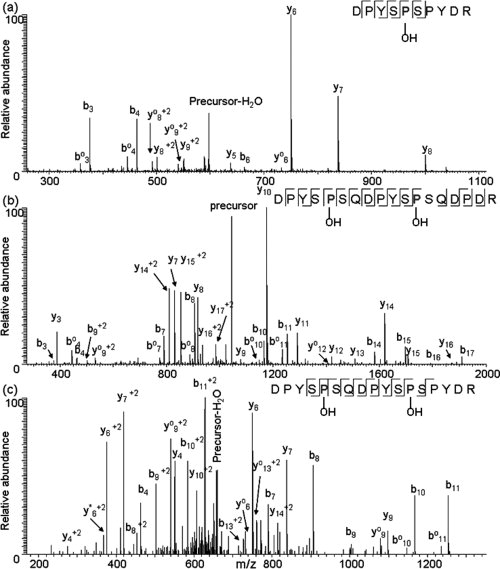
MS/MS spectra of peptides acquired on a LTQ mass spectrometer that were identified with proline hydroxylated. (a) CID spectrum of peptide [61–70] from heavy isoform P1 of Ara h 2, doubly charged ion at m/z 607 was selected as the precursor ion; (b) CID spectrum of peptide [42–59] from heavy isoform P1 of Ara h 2, doubly charged ion at m/z 1042 was selected as the precursor ion; (c) CID spectrum of peptide [42–58] from light isoform P2 of Ara h 2, triply charged ion at m/z 669 was selected as the precursor ion.
The de novo sequencing covered the whole protein sequence of each isoform, respectively. To confirm this manual analysis, a MASCOT search was performed using the spectra of tryptic peptides from P1 acquired on a Q-TOF mass spectrometer. It returned a result with CONG7_ARAHY (P1) identified. Using an E value cut off ≤0.05, the peptides identified in an automated manner covered 47% of the protein sequence excluding the signal peptides. Among those peptides, [35–60], [42–60], and [61–70] were identified with 2, 2, and 1 hydroxyprolines. The E values were 0.00061, 0.0045, and 0.0071, respectively, indicating highly reliable identifications. The localized hydroxylation sites are identical to those described above for the heavy isoform P1. This supports the conclusion from de novo sequencing that there are hydroxyprolines at positions 46, 53, and 65 in the P1 isoform.
The site-specific hydroxylation of proline residues occurs only on the second proline of repeated peptide motifs PYSPS in the middle of the Ara h 2 isoforms, as is the case for amino acids [43–47] and [50–54] in the P2 isoform (Fig. 2). Previous studies have identified the immunodominant IgE binding linear epitopes in Ara h 2 as being overlapped regions at amino acids [39–48] and [47–56].5 Thus, both of these peptides contain the PYSPS motif. However, hydroxylation of prolines was not known to occur when these epitopes were identified, and it is not clear how the presence of hydroxyprolines affects the allergenicity of Ara h 2, Moreover, there is an additional PYSPS motif in isoforms P1 and P3 and it is not known if this additional modified motif can potentially differentiate the allergenicity among these four isoforms.
Peptides identified via de novo sequencing covered 100% of the protein sequence (not all data is shown since it is consistent with the deducted protein sequence from the cDNAs) of each isoform, and accounted for the molecular masses. The primary structures of all the isoforms are shown in Figure 2. Assuming all eight cysteines are involved in disulfide linkages, the calculated average molecular weights of the P1-P4 isoforms, as shown, are 18033, 16661, 17714, and 16341 Da, respectively. The calculated protein masses are essentially identical to the measured protein masses (Fig. 1 inset). The signal peptide is confirmed to be removed, and there is no evidence of carbohydrate side chains. Additionally, no evidence was found in analysis of the tryptic peptides for glycosylation.
Determination of disulfide linkages
As noted above, one of our interests in the study of Ara h 2 is whether and how its structure can be modified by thermal processing. Oxidation of cysteine sidechains is one possible outcome of roasting. Therefore, an understanding of the disulfide bonds in the native protein is important. The general approach for evaluating the cross-linking structure in this protein was through examination of the tryptic digestion products obtained from nonreduced protein, as compared to the products obtained through the standard digestion process where a reducing agent is added. Fractions containing the cross-linked products were collected from the P4 isoform and analyzed. All the tryptic peptides that contain cysteines are given a letter in Table I. Evaluation of the fractions revealed that there were no products corresponding to single crosslink peptides (e.g., A-B, E-F combinations). However, among all the seven tryptic peptides in the P4 isoform that contain cysteine residues (Table I), there are two peptides, C and D, that contain two cysteines. One possibility, that C and D are bridged by two crosslinks (C+D), was also ruled out by the absence of a product with a molecular weight corresponding to that combination. Another possibility, the product, C+D+X+Y (where X and Y are one of the single-cysteine peptides A, B, E, F or G), was again not observed, since no ion was detected with a mass greater than 5000 Da. However, C and D could each be linked to two single-cysteine peptides to create a crosslinked tripeptide (e.g., C+X+Y, where X is A,B, E, F or G). We have considered all the possible disulfide bond linkages for both C and D peptides (Tables II and III). There are eight possible ways to form disulfide bond linkages for the C peptide which include one intrachain linkage and seven interchain linkages (Table II). Similarly, there are eight possible ways for the D peptide (Table III).
Table I.
List of All the Tryptic Peptides that Have Cysteine Residues in P4 Isoform
| Tryptic peptide that contains cysteine residues | Sequence | Residue number | Monoisotopic mass (Da) |
|---|---|---|---|
| A | CQSQLER | [12–18] | 862.4 |
| B | ANLRPCEQHLMQK | [19–31] | 1566.8 |
| C | CCNELNEFENNQR | [70–82] | 1611.6 |
| D | CMCEALQQIMENQSDR | [83–98] | 1897.8 |
| E | NLPQQCGLR | [114–122] | 1027.5 |
| F | CDLDVESGGR | [127–136] | 1049.5 |
| F′ | CDLDVESGGRD | [127–136] | 1164.5 |
Table II.
Possible Disulfide Bond Linkages of C Peptide of P4 Isoform. Mass ≥3000Da Are in Average Mass
| Pattern | Possible linkages of C peptide | Mass (Da) |
|---|---|---|
| 1 | C intrachain linkages | 1609.6 |
| 2 | C+A+B | 4039.5 |
| 3 | C+A+E | 3499.8 |
| 4 | C+A+F | 3521.7 |
| 5 | C+B+E | 4204.8 |
| 6 | C+B+F | 4226.7 |
| 7 | C+E+F | 3687.0 |
| 8 | C+D | 3507.9 |
Table III.
Possible Disulfide Bond Linkages of D Peptide of P4 Isoform. Mass ≥3000Da Are in Average Mass
| Pattern | Possible linkages of D peptide | Mass (Da) |
|---|---|---|
| 1 | D intrachain linkages | 1895.8 |
| 2 | D+A+B | 4326.0 |
| 3 | D+A+E | 3786.3 |
| 4 | D+A+F | 3808.2 |
| 5 | D+B+E | 4491.3 |
| 6 | D+B+F | 4513.2 |
| 7 | D+E+F | 3973.5 |
| 8 | C+D | 3507.9 |
For the P4 isoforms, a peptide with a mass of 4204 Da (Fraction 1) matches the mass of C+B+E (pattern 5 in Table II). In fraction 2, there were two coeluting peptides, with masses of 3807 Da and 3922 Da, respectively. The former mass, 3807 Da, agrees with the of D+A+F (pattern 4 in Table III). The latter mass, 3922 Da, corresponds to D+A+F with an additional aspartic acid at the end of F, resulting from a missed cleavage at the C-terminal peptide. This results in andditional product with a mass 115 Da larger than C+A+F (this product will be denoted as C+A+F′). The 3922 Da peak was dominant so the following analysis of Fraction 2 was based on it.
To confirm these assignments, the fractions containing cross-linked peptides were treated with CNBr (which cleaves on the C-terminal side of methionine) and analyzed by MALDI. In Fraction 2, the 3922 Da peptide (C+A+F′) has two cysteines in D that are at residues 83 and 85, separated by a methionine. The products from CNBr cleavage were calculated for two possibilities of disulfide bond linkages within this disulfide bond linked tripeptide (Table IV). Two peptides, with masses of 1065 Da and 1935 Da, were observed and are in agreement with the values predicted in the left column in Table IV. This indicates that cysteine 12 in peptide A is connected with cysteine 83 in D, and cysteine 85 in D is connected with cysteine 127 in F′.
Table IV.
List of Masses of the Product Peptides from CNBr Cleavage of Disulfide Bond Linked Tri-peptide (Fraction 2) From P4 Isoform
| Disulfide bond linked tri-peptide | D+A+F′ | |
|---|---|---|
| Possible disulfide bond linkages within the cluster | Cys12–Cys83; Cys85–Cys127 | Cys12–Cys85; Cys83–Cys127 |
| Masses of the products from CNBr cleavage (Da) | 1064.4 | 1746.7 |
| 1933.9 | 1251.4 | |
| 747.0 | 747.0 | |
In Fraction 1, the C+B+E tripeptide could not be similarly characterized. Cysteines 70 and 71 in peptide C are adjacent, not separated by a methionine as in D. Thus, we have not found a definitive way to characterize the disulfide linkages in this fraction. Reduction without alkylation generated three peptides with masses of 1625 Da, 1727 Da, and 1086 Da (data not shown here), corresponding to peptides B, C, and E, and further confirming the composition of the C+B+E tripeptide. Thus, we know that Cys25 either links to Cys70 or 71 while Cys119 connects with Cys 71 or 70. The possibility cannot be eliminated that two patterns of adjacent S—S linkages coexist in Fraction 1.
The same analysis was carried out with the peptide fractions from the P3 isoform (data not shown here). Connectivity between Cys12-Cys95 and between Cys97-Cys139 has been determined. Cys 25 is linked either to Cys82 or Cys83, and Cys131 is linked to Cys 83 or Cys82. Although the insertion of 12 amino acids in the P3 isoform shifts the cysteine residue numbers, all crosslinking occurs at comparable positions in isoforms P3 and P4. The disulfide linkages of P1 and P2 isoforms were not studied here, but they are expected to be comparable to P3 and P4, respectively, differing by two more amino acids at the C-termini. These results are consistent with an earlier proposal that the disulfide bonds in Ara h 2 are homologous to those in the bifunctional α-amylase/trypsin inhibitor RBI protein from ragi seed,11 which in turn suggests that there is no disulfide bond scrambling in the process of purifying Ara H2.
Experimental
Materials
Solvents for HPLC gradients (acetonitrile, water) were purchased from Burdick & Jackson (Morristown, NJ). Formic acid, cyanogen bromide (CNBr), and dithiothreitol (DTT) were purchased from Sigma (St. Louis, MO). Ara h 2 protein, prepared from the Runner variety without using any acid or protease, was purchased from TNO (Zeist, the Netherlands). Trypsin was purchased from Promega (Madison, WI). Trifluoro acetic acid (TFA) was purchased from J.T. Baker (Phillipsburg, NJ). All chemicals and proteins were used without further purification.
Separation of Ara h 2 isoforms
Ara h 2 isoforms were separated using an AKTA purifier FPLC (GE Healthcare Bio-Sciences Corp., Piscataway, NJ). One hundred micrograms of Ara h 2 raw protein suspended in 1mL distilled water was loaded onto a BioSuite DEAE 2.5 μm NP anion exchange column with an ID of 2.6mm and a length of 3.5mm (Waters, Milford, MA). The isoforms were eluted by increasing solvent B from 1%-7% in 40 column volumes (solvent A: 0.1 M Tris buffer, pH 7.4; solvent B: HPLC grade water) using a flow rate of 1mL/min. The chromatogram is shown in Figure 1. Each fraction was collected via a fraction collector that would initiate/stop the collection when the UV absorption level exceeded/dropped below a specified level. Each fraction was desalted and concentrated in a 5000 Da cut off spin column (Millipore, Temecula, California). The protein concentration of the P1 isoform was measured at about 0.05mg/ml by RC DC protein assay (Biorad, Hercules, CA). The concentration of the other three isoforms were not determined experimentally, but they were at comparable concentration to P1 based on their comparable respective chromatographic peak areas in Figure 1. One hundred micrograms of each isoform was loaded onto a reversed-phase C18 column (1.0 mm I.D. ×100 mm) with 1.7 μm packing material on an ACQUITY LC system (Waters, Milford, MA). The gradient was linearly ramped from 0 to 100% solvent D (solvent C: 0.1% Formic acid and 1% acetonitrile in water, solvent D: 0.1% formic acid and 1% water in acetonitrile) in 40 min. The flow rate was set at 100 μL/min to allow the molecular mass of each isoform to be measured using a Q-TOF premier mass spectrometer (Waters, Milford, MA). Each molecular weight was determined by deconvoluting the MS spectrum using the MassEnt1 of the MassLynx software (Waters, Milford, MA).
Proteolytic digestion of Ara h 2 isoforms
A Discover Benchmate microwave system (CEM Corp, Matthews, NC) was utilized to perform the protein digestion. It allows precise control of the temperature, power, and time. Fifty microliters of protein solution (0.05 mg/mL) was incubated with 10 mM DTT at 60°C for 20 min and then heated in the microwave with 2 μL trypsin (0.4 μg/μL) for 30 min at 37°C. Formic acid was added to reach a final concentration of 0.1% to stop the reaction. In experiments designed to minimize missed cleavages, trypsin digestion was carried out in the microwave system for 60 min. The other experimental conditions were the same as described earlier.
LC-MS/MS
The peptide mixtures that were obtained from 30 and 60 min microwave accelerated trypsin digestions were separated on a Waters nanoACQUITY HPLC interfaced to an LTQ-XL ion trap mass spectrometer (Thermo Electron, San Jose CA). Ten microliters product was loaded onto a reverse-phase C18 column 1.0 mm I.D. ×100 mm) with 1.7 μm bridged ethyl hybrid (BEH) particle packing (Waters, Milford, MA). The gradient was ramped from 0 to 10% B in 5 min and then ramped to 30% B in 25 min (solvent A: 0.1% formic acid and 1% acetonitrile in water, solvent B: 0.1% formic acid and 1% water in acetonitrile). CID and ETD were both used when necessary. For CID, 35–40% collision energy was applied to precursor ions depending on the mass of the precursor peptide. For ETD, the reaction time was 80 milliseconds. All spectra were analyzed manually. In all cases, targeted CID was employed, as most of the precursor peptide masses and charge states can be predicted based on the deduced protein sequence.
For purposes of confirmation, a peptide mixture from a 60 min digest of P1 isoform was introduced to a Q-TOF mass spectrometer (Waters, Milford, MA) using the same LC system and the same gradient as described above. CID was performed with a default collision energy profile that provides collision energy adjustment according to the precursor ion mass. The data was submitted for database search as described later to confirm the result from manual analysis of the protein sequence and PTM.
Mascot database searches
The raw data from the 60 min digestion of P1 acquired on the Q-TOF mass spectrometer was processed using the default Q-TOF settings via MASCOT distiller (Matrix Science, London, UK) to generate the Mascot generic data format that is compatible with Mascot database searching. The search was carried out using an in-house mascot server (http://www.matrixscience.com/). Trypsin was indicated as the proteolytic enzyme. Instrument was specified as ESI-QUAD-TOF. Both the mass tolerances of precursor ions and fragment ions were set at ±0.3 Da. No modification on cysteine was specified. Oxidation of proline was specified as a variable modification. Three missed cleavage sites were allowed. The search was carried out against the Green Plant database in the SwissProt database (http://ca.expasy.org/sprot/).
Fraction collection of peptides containing disulfide bonds
P3 and P4 isoforms were digested with trypsin as described above, except that no reducing reagent was added. The peptide mixtures were separated on the same LC system as described for the LC-MS/MS. The concentration of solvent B was linearly increased from 0 to 50% through 50 min. All the possible combinations of disulfide linkages were considered and the corresponding masses of peptides containing interchain disulfide bonds and intrachain disulfide bonds have been calculated. When these peptides are ionized via ESI, they usually carry several charges. Thus their corresponding series of m/z values can be calculated. The retention time of these peptides can be determined easily via reconstructed ion chromatogram using their m/z values. Eventually the disulfide bond containing peptides were manually collected at their respective eluting times.
CNBr treatment of peptide fractions containing disulfide linkages
The CNBr solution was made by adding 529 mg CNBr to 1ml ACN to reach a final concentration of 5 M. Ten microliters of each peptide fraction was mixed with 15 μL TFA and 5 μL CNBr solution. The mixture was incubated at room temperature in the dark for 15 hr. Then the mixture was mixed with an equal volume of distilled water and dried down in the SpeedVac. This step was repeated once. Two microliters of CHCA solution (10 mg of CHCA in 70% ACN, 0.1% TFA) was added to suspend the sample. The sample was spotted on a MALDI plate and analyzed on a 4800 MALDI TOF/TOF mass spectrometer (Applied Biosystem, Foster City, CA). LC-MS/MS was not used because the collected fraction is a simple mixture, including the target peptides that contain the disulfide bonds.
Conclusions
To summarize, the mass spectrometry studies of Ara h 2 in the commercial sample reported here identify a heavy isoform pair and a light isoform pair that are differentiated by a 12 amino acid insertion in the middle of the sequence, and amino acid variations at residues 40 and 142 in the heavy isoforms and residues 40 and 130 in the light isoforms (Fig. 2). The heavy isoform pair has glycine and glutamate at residues 40 and 142, while the light isoform pair carries glutamate and aspartate at residues 40 and 130, respectively. Within each pair, there is a variation in the C-terminus. The C-termini assigned are consistent with a mass difference 319 Da. The two heavy isoforms both contain three hydroxyproline residues, while the two light isoforms have two hydroxyprolines in homologous positions, as shown in Figure 4. No glycosylation was found, in contrast to past literature reports. There are four disulfide bonds in each isoform of Ara h 2. Two out of the four disulfide bonds have been fully defined and the remaining two are localized to two adjacent Cys residues. The 12 amino acid insertion and amino acid variations at residues 40 and 142 in the heavy isoforms (40 and 130 in the light isoforms) are consistent with previously reported sequences translated from the cDNA sequences.3,4 Determinations of site-specific proline hydroxylation, disulfide linkages and C-terminal variation are reported here for the first time.
Knowledge of the full sequence and modifications enables on-going studies of the way in which the protein is modified by thermal processing (such as roasting) and provides information that may be useful in the development of sensitive and automated assays, such as multiple reaction monitoring (MRM) using a triple quadruple mass spectrometer for detecting Ara h 2 in food. The discovery and definition of PTM is expected to facilitate continued studies of factors that may affect the structural determinants of allergenicity in the Ara h 2 family, particularly where processing may produce modification of the protein.
References
- 1.Koppelman S, Vlooswijk R, Knippels L, Hessing M, Knol E, Van Reijsen F, Bruijnzeel-Koomen C. Quantification of major peanut allergens Ara h 1 and Ara h 2 in the peanut varieties Runner, Spanish, Virginia, and Valencia, bred in different parts of the world. Allergy. 2001;56:132–137. doi: 10.1034/j.1398-9995.2001.056002132.x. [DOI] [PubMed] [Google Scholar]
- 2.Carr W. Clinical pearls and pitfalls: peanut allergy. Allergy Asthma Pro. 2005;26:145–147. [PubMed] [Google Scholar]
- 3.Chatel JM, Bernard H, Orson F. Isolation and characterization of two complete Ara h 2 isoforms cDNA. Int Arch Allergy Immunol. 2003;131:14–18. doi: 10.1159/000070429. [DOI] [PubMed] [Google Scholar]
- 4.Yan YS, Lin XD, Zhang YS, Wang L, Wu K, Huang SZ. Isolation of peanut genes encoding arachins and conglutins by expressed sequence tags. Plant Sci. 2005;169:439–445. [Google Scholar]
- 5.Viquez O, Summer C, Dodo H. Isolation and molecular characterization of the first genomic clone of a major peanut allergen, Ara h 2. J Allergy Clin Immunol. 2001;107:713–717. doi: 10.1067/mai.2001.113522. [DOI] [PubMed] [Google Scholar]
- 6.Careri M, Costa A, Elviri L, Lagos J, Mangia A, Terenghi M, Cereti A, Garoffo L. Use of specific peptide biomarkers for quantitative confirmation of hidden allergenic peanut proteins Ara h 2 and Ara h 3/4 for food control by liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem. 2007;389:1901–1907. doi: 10.1007/s00216-007-1595-2. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt H, Gelhaus C, Latendorf T, Nebendahl M, Petersen A, Krause S, Leippe M, Becker WM, Janssen O. 2-D DIGE analysis of the proteome of extracts from peanut variants reveals striking differences in major allergen contents. Proteomics. 2009;9:3507–3521. doi: 10.1002/pmic.200800938. [DOI] [PubMed] [Google Scholar]
- 8.Burks A, Williams L, Connaughton C, Cockrell G, O'brien T, Helm R. Identification and characterization of a second major peanut allergen, Ara h II, with use of the sera of patients with atopic dermatitis and positive peanut challenge. J Allergy Clin Immunol. 1992;90:962–969. doi: 10.1016/0091-6749(92)90469-i. [DOI] [PubMed] [Google Scholar]
- 9.Pramanik B, Mirza U, Ing Y, Liu Y, Bartner P, Weber P, Bose A. Microwave-enhanced enzyme reaction for protein mapping by mass spectrometry: a new approach to protein digestion in minutes. Protein Sci. 2002;11:2676–2687. doi: 10.1110/ps.0213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamport DTA. Hydroxyproline-O-glycosidic linkage of the plant cell wall glycoprotein extensin. Nature. 1967;216:1322–1324. [Google Scholar]
- 11.Maleki S, Viquez O, Jacks T, Dodo H, Champagne E, Chung S, Landry S. The major peanut allergen, Ara h 2, function as a trypsin inhibitor, and roasting enhances this function. J Allergy Clin Immunol. 2003;112:190–195. doi: 10.1067/mai.2003.1551. [DOI] [PubMed] [Google Scholar]