

Abstract
The ribosomal protein S6 from Thermus thermophilus has served as a model system for the study of protein folding, especially for understanding the effects of circular permutations of secondary structure elements. This study presents the structure of a permutant protein, the 96-residue P54-55, and the structure of its 101-residue parent protein S6wt in solution. The data also characterizes the effects of circular permutation on the backbone dynamics of S6. Consistent with crystallographic data on S6wt, the overall solution structures of both P54-55 and S6wt show a β-sheet of four antiparallel β-strands with two α-helices packed on one side of the sheet. In clear contrast to the crystal data, however, the solution structure of S6wt reveals a disordered loop in the region between β-strands 2 and 3 (Leu43-Phe60) instead of a well-ordered stretch and associated hydrophobic mini-core observed in the crystal structure. Moreover, the data for P54-55 show that the joined wild-type N- and C-terminals form a dynamically robust stretch with a hairpin structure that complies with the in silico design. Taken together, the results explain why the loop region of the S6wt structure is relatively insensitive to mutational perturbations, and why P54-55 is more stable than S6wt: the permutant incision at Lys54-Asp55 is energetically neutral by being located in an already disordered loop whereas the new hairpin between the wild-type N- and C-termini is stabilizing.
Keywords: NMR solution structure, backbone dynamics, S6, permutant, folding
Introduction
The ribosomal protein S6 is one component of the small subunit of the ribosome,1 known to be important for the assembly of the central domain of the small subunit via heterodimerization with S18.2 The crystal structure of the 101 amino acid protein S6 from Thermus thermophilus shows that it folds into a core composed of two β-α-β secondary structure motifs, forming a four-stranded antiparallel β-sheet with the two α-helices assembled on one side of the sheet.3 From this core, a hook-region of conserved residues extends as two antiparallel β-strands, forming a mini-core of hydrophobic residues.2,4–6
S6 has been extensively characterized with respect to its folding properties7,8 and, in particular, how folding is affected by circular permutations, that is how changes in the order of secondary structure elements affect the folding process.9 Detailed φ-value analysis on a number of engineered permutants of S6 reveals that there are two competing folding nuclei, both with analogous structural outline but where the first nucleus (σ1) consists of β1-α1-β3 and the second nucleus (σ2) of β1-α2-β4. As a consequence of the two folding nuclei, the folding of S6 can occur on two parallel pathways with opposite folding order. S6wt is mainly biased towards the σ1 pathway, whereas the permutant P54-55 favors the σ2 pathway.9–11
In this study, we use NMR spectroscopy to characterize the structure and dynamics of S6wt and P54-55 in solution. The results presented here shed new light on how circular permutations affect protein structure, dynamics, and stability.
Results and Discussion
The assigned 15N-HSQC spectra for S6 and P54-55 show well dispersed peaks as expected for folded proteins (Supporting Information Fig. S1). For both proteins, nearly complete backbone assignments and extensive side-chain assignments were obtained. S6wt has a second set of peaks for Ala53 and Leu98. Since these residues are located in the vicinity of Pro51, Pro56, and Pro96, the second set of peaks may be due to proline cis-trans isomerization; the smaller fraction was estimated to ∼20% or less as judged from the relative peak intensities. In addition, the C-terminal residues Asn100 and Ala101 cannot be resolved due to overlapping chemical shifts. P54-55 displays a set of duplicate peaks of approximately equal intensity for Asp2, presumably due to cis-trans interconversion of the subsequent residue, Pro3. The C-terminal residues of P54-55 are also likely to be significantly affected by the presence of a proline at position 93 (Pro93). Ala95 and Lys96 have triplicate or quadruple peaks, where the major conformer has occupancy of 50–60%. Resonances for Tyr92 and Ile94 were not observed in the 15N-HSQC spectrum, probably due to exchange broadening. In addition, the chemical shifts of Leu90 and Ala91 vary with sample preparation.
A total of 745 and 770 nonredundant distance restraints were determined for S6wt and P54-55, respectively (Table I). Dihedral angle restraints were derived, in part, from HNHA experiments using the analysis program Mulder. In addition, the program TALOS provided 65 pairs of backbone (φ,ψ) dihedral angle restraints for both proteins, which was classified as Good by TALOS (Table I). The TALOS-derived restraints were selected only if they were in agreement with the φ-values provided by Mulder. An additional eight (S6wt) and seven (P54-55) φ-angles for nonglycine residues were restrained to negative values,12 based on weak intraresidue NOEs between HN and Hα protons. From the structure calculations, 54 of 300 structures and 53 of 200 structures were accepted for S6wt and P54-55, respectively. The final ensembles consisted of 20 structures [Fig. 1(A and B)] that were selected based on their energies and Ramachandran behavior. Structural statistics are presented in Table I, and ribbon structures of representative conformers of S6wt and P54-55 are presented in Figure 1(C and D).
Table I.
Structural Statistics for the Ensembles of 20 Structures of S6wt and P54-55
| S6wt | P54-55 | |
|---|---|---|
| Number of NOE-derived distance restraints | ||
| Total number | 745 | 770 |
| Intraresidue | 252 | 213 |
| Sequential (|i − j| = 1) | 307 | 303 |
| Medium range (2 ≤ |i − j| ≤ 4) | 97 | 138 |
| Long range (|i − j| ≥ 5) | 89 | 116 |
| Number of dihedral angle restraints | ||
| Backbone φ-angle | 90 | 88 |
| Backbone ψ-angle | 65 | 65 |
| Average number of violations | ||
| Distance restraints >0.5 Å | 0 | 0 |
| Dihedral angle restraints >5° | 0 | 0 |
| Deviations from idealized covalent geometry | ||
| Bonds (Å) | 0.0022 ± 0.0001 | 0.0032 ± 0.0001 |
| Angles (°) | 0.54 ± 0.01 | 0.57 ± 0.01 |
| Impropers (°) | 0.39 ± 0.01 | 0.44 ± 0.01 |
| Ramachandran plot analysis, residues | 3–42, 59–91 | 6–85 |
| Most favored regions (%) | 95.0 | 84.1 |
| Additionally allowed regions (%) | 4.9 | 14.5 |
| Generously allowed regions (%) | 0.1 | 1.3 |
| Disallowed regions (%) | 0.0 | 0.0 |
| Atomic RMSD, residues | 3–42, 59–91 | 6–85 |
| Backbone heavy atoms (Å) | 0.95 | 0.66 |
| All heavy atoms (Å) | 1.60 | 1.44 |
| PDB-ID | 2KJV | 2KJW |
Figure 1.
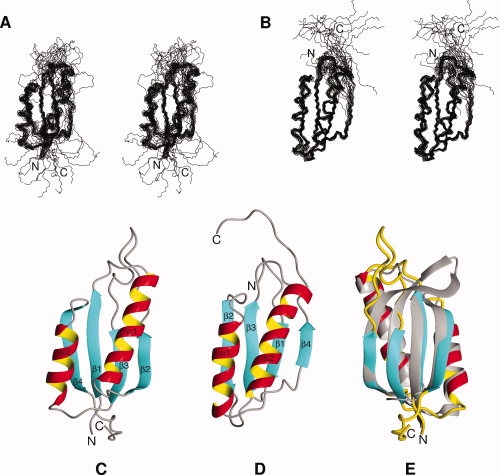
Solution structures of S6wt and P54-55. Stereo views of (A) S6wt and (B) P54-55 solution structures, showing the backbone trace for the final ensembles of 20 calculated structures (PDB-IDs: 2KJV and 2KJW, respectively). The secondary structure regions are superimposed and their N- and C-termini are indicated. Ribbon representations of the lowest-energy structures of (C) S6wt and (D) P54-55, in which the N- and C-terminals as well as secondary structure elements are identified. (E) The solution structure of S6wt (depicted in color) superimposed onto the crystal structure (in grey; PDB-ID: 1RIS). The structures in panel (E) are rotated 180° around a vertical axis compared to the structure in panel (C).
The solution structure of S6wt has a well-defined core, containing a β-sheet of four antiparallel β-strands and two α-helices packed on one side of the sheet, as well as a flexible loop and C-terminal, see Figure 1(A and C). The connectivity scheme of the secondary structure elements is β1-α1-β2-β3-α2-β4. The N-terminal residues (Met1-Arg3) are followed by the first β-strand (β1, Tyr4-Leu10), a turn (Asn11-Asp15), the first α-helix (α1, Gln16-Asn32), a turn (Tyr33-Arg36), the second β-strand (β2, Val37-Glu42), an 18-residue loop (Leu43-Phe60), the third β-strand (β3, Leu61-Val65) packed antiparallel in between strand β1 and β2, a turn (Glu66-Asp70), the second α-helix (α2, Arg71-Ile81), a turn (Arg82-Asn84), the fourth β-strand (β4, Val85-Val91) packed antiparallel to β1, followed by nine disordered C-terminal residues (Lys92-Ala101). The atomic RMSD:s for the backbone and heavy atoms of residues 3–42 and 59–91 in the ensemble of S6wt structures are 0.95 and 1.6 Å, respectively (Table I).
Similar to its parent structure, the solution structure of the designed permutant protein P54-55 has a well-defined core structure containing a β-sheet of four antiparallel β-strands and two α-helices packed on one side of the sheet [Fig. 1(B and D)]. As intended by the design of the permutant, the secondary structure connectivity is β1-α1-β2-β3-α2-β4 (or with the nomenclature of S6wt: β3-α2-β4-β1-α1-β2). The N-terminal residues (Met1-Phe7), which are disordered, are followed by the first β-strand (β2, Leu8-Tyr10), a turn (Gln11-Pro15), the first α-helix (α1, Glu16-Ile28), a turn (Arg29-Arg33), the second β-strand (β2, Arg34-Ala39), a turn (Ser40-Arg45), the third β-strand (β3, Tyr46-Val51) packed antiparallel in between strand β1 and β2, a turn (Leu52-Asp57), the second α-helix (α2, Gln58-Tyr75), a turn (Gly76-Lys81), the fourth β-strand (β4, Val82-Glu84) packed antiparallel with β1, followed by less well-defined C-terminal residues (Leu85-Lys96). The atomic RMSD:s for the backbone and heavy atoms of residues 6–85 in the ensemble of P54-55 structures are 0.66 and 1.44 Å, respectively (Table I). The core of the P54-55 solution structure is fairly similar to the solution structure of S6wt; their lowest-energy structures have a RMSD of 2.57 Å for the backbone atoms of residues Tyr6-Val38 and Arg45-Glu84 in P54-55 compared with the corresponding residues Tyr59-Val91 and Arg3-Glu42 in S6wt [Fig. 1(C and D)].
The backbone dynamics of S6wt were characterized by measurements of 15N R1, R2 and {1H}-15N NOE [Fig. 2 (A–C)]. In particular the R2 and {1H}-15N NOE data demonstrate the dynamic properties of the loop (Leu43-Phe60) and the C-terminal (Ser93-Ala101). From the model-free analysis of the relaxation data, a rotational correlation time of 7.3 ns and a diffusion tensor ratio of 0.76 were derived. Many residues (25 of 75) were fitted by model i, 15 by model ii, 9 by model iii, 23 by model iv, and 3 by model v. Overall, residues in secondary structure elements display high 1H-15N order parameter (S2), with an average of 0.91 [Fig. 2(D)]. In addition, a number of residues exhibit substantial dynamics on the picosecond-nanosecond timescale [Fig. 2(E)] and chemical exchange on the millisecond timescale [Fig. 2(F)].
Figure 2.
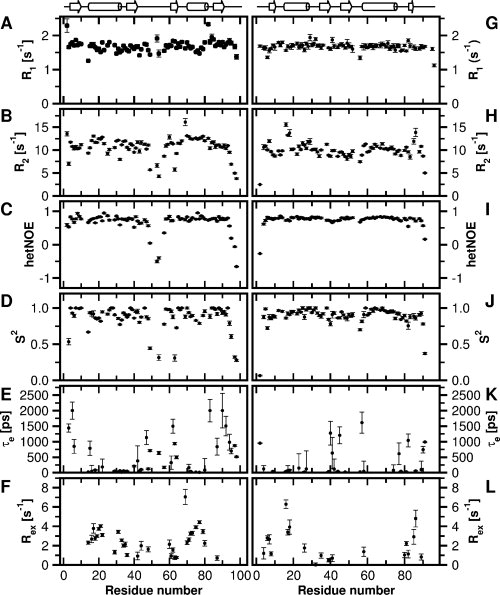
Backbone dynamics of S6wt and P54-55. Backbone 15N NMR relaxation data and motional parameters for (A–F) S6wt and (G–L) P54-55 at 25°C and at 14.1 T, plotted as a function of residue number. Longitudinal relaxation rate constants are shown in (A and G), transverse relaxation rate constants in (B and H), and {1H}-15N NOEs in (C and I). Order parameters (S2), effective internal correlation times (τe) and exchange rates (Rex) are shown in (D and J), (E and K), and (F and L), respectively.
Measurement of backbone dynamics for P54-55 demonstrates that the N- and C-terminal residues are disordered [Fig. 2(G–I)]. Model-free analysis of the relaxation data yielded a rotational correlation time of 6.9 ns and a diffusion tensor ratio of 0.80. Relaxation data for a majority of the residues (42 of 79) were described by model i, 9 by model ii, 7 by model iii, 12 by model iv, and 9 by model v. The H—N order parameters for P54-55 are high overall, with an average of 0.92 for residues in secondary structure elements [Fig. 2(J)]. In comparison to S6wt, the permutant has less number of residues that display dynamics on the picosecond timescale [Fig. 2(K)] and chemical exchange on the millisecond timescale [Fig. 2(L)].
The core of the solution structure of S6wt is very similar to that of the crystal structure (PDB-ID: 1RIS),3 with a RMSD of 1.60 Å for the backbone atoms of residues Arg3-Glu42 and Tyr59-Val91, using the lowest-energy solution structure for comparison [Fig. 1(E)]. The main differences are observed in the end of β-strand β2 and in the beginning of strand β3, which are flexible in solution [Fig. 1(A)]. The flexible properties of the loop-region (Leu43-Phe60) are clearly revealed by the relaxation measurements [Fig. 2(A–F)], where these residues exhibit lower than average R2 values and order parameters, and negative heteronuclear NOEs. The lack of high crystallographic B-factors in this region further confirms this difference (Supporting Information Fig. S2). The crystal structures of S6wt alone or within the ribosome are very similar and show that this loop-region (or so-called hook-region3) forms two well-defined antiparallel β-strands, creating a mini-core of hydrophobic residues. The mini-core appears to be important for protein–protein interactions, either for the crystal packing of S6wt itself or for the interaction with S18 within the ribosome. In a previous folding study,8 two point-mutations, I48A and L52A, were introduced within the assumed mini-core [Fig. 3 (A)], without an expected destabilization of the protein [see the chevron plots in Fig. 3(B)]. The solution structure shows that these sites of mutation are located in a loop-region that is flexible instead of an ordered mini-core. As a result, these mutations have negligible effects on protein stability. In contrast, the chevron plot in Figure 3(B) further reveals a clear difference in stability between S6wt and P54-55. The overall more rigid core structure for P54-55 and the additional hairpin structure between the original N- and C-termini may account for the 0.49 kcal/mol higher stability of the permutant compared to S6wt.
Figure 3.
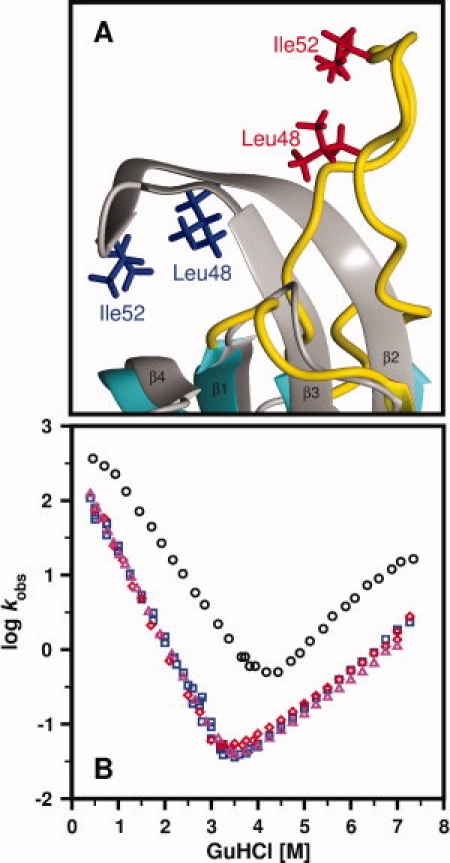
Loop-region of S6wt and chevron plots of S6wt, S6 mutants and P54-55. (A) The loop-region of the S6wt solution structure (depicted in color) superimposed onto the crystal structure (in grey; PDB-ID: 1RIS), where the mutated residues, Leu48 and Ile52, are indicated. (B) Chevron plots of S6wt (blue squares), the mutant L48A (red diamonds), the mutant I52A (magenta triangles) and P54-55 (black rings). The data are adapted from8,11 whereas the chevron plots of the L48A and I52A mutants are not previously published.
In conclusion, the structure and dynamic properties of S6wt in solution explain the insensitivity of mutations in the hook region to overall stability. These results show that this region is more flexible or dynamic in solution than the hydrophobic mini-core defined by the crystal structure. Secondly, the solution structure of P54-55, which to our knowledge represents the first three-dimensional solution structure of a permutant protein, shows that the permutation process itself does not affect the overall three-dimensional structure. Moreover, the conformational dynamics of the permutant protein are reduced overall, which provides new insight into its increased global stability. These results set the basis for further analysis of the dynamic properties of S6wt and engineered permutants of S6 by NMR to accompany the wealth of protein folding and stability data already available for this model system.7,8
Materials and Methods
Sample preparation
Cloning, expression and purification of S6wt and P54-55 were carried out as previously described.7,9–11 Lyophilized proteins were dissolved in a solvent containing 20 mM MES, pH 6.3, 50 mM NaCl and 10% (v/v) D2O, to a concentration of ∼0.5 mM.
NMR spectroscopy
NMR experiments for chemical shift assignments, structure calculations, and backbone dynamics were collected at 25°C on a 600 MHz Bruker AVANCE spectrometer, equipped with a 5 mm triple-resonance, pulsed-field z-gradient warm or cryogenically cooled probe. Recorded datasets were processed using NMRPipe13 or XWINNMR/Topspin (Bruker Biospin) and resonance assignments were determined with Ansig for Windows.14 The sequence specific resonance assignments followed the standard assignment strategy15 and were based on 2D15N-HSQC, 3D 15N-edited NOESY-HSQC, and DIPSI-HSQC, as well as a standard set of 3D triple resonance experiments.16 Backbone and partial side-chain assignments of P54-55 were obtained from CBCANH, CBCA(CO)NH, HNCO, HSQC, HNCA, and HN(CO)CA experiments, while the side-chain assignments were verified and extended using 2D clean-TOCSY and DQF-COSY and 3D 15N-edited DIPSI-HSQC, C(CO)NH, HC(CO)NH, and HCCH-TOCSY experiments. Distance restraints were obtained from 3D 15N-edited NOESY experiments with a mixing time of 100 ms. Dihedral angle restraints were derived from scalar coupling constants measured in HNHA experiments.
Structural restraints
The analysis of the NOESY spectra resulted in a set of unambiguously assigned NOEs, which were calibrated to known distances found in both α-helical and β-sheet secondary structure elements. The NOE-based restraints were divided into three classes: strong (1.8–2.7 Å), medium (1.8–3.6 Å) and weak (1.8–5.5/6.0 Å). By using an iterative combination of structure calculation and a set of unambiguous distance restraints, many ambiguous NOE cross-peaks were resolved and assigned. The obtained distance restraints were processed by AQUA to remove redundant restraints.17 Dihedral angle restraints were derived from coupling constants using the Mulder program18 and chemical shifts using the TALOS program.19
Structure calculation and analysis
Structures were calculated and analyzed using XPLOR, (v. 3.851). Additional structural analysis and molecular visualization were carried out using MOLMOL20 and Procheck-NMR.17 A total number of 200 (P54-55) or 300 (S6wt) structures were calculated from the structural restraints, using the XPLOR scripts sa.inp and two rounds of refine.inp. The duration of the first high-temperature molecular dynamics step in the sa.inp protocol was increased to 45 ps, while the annealing step was increased to 30 ps. Similarily, the duration of the simulated annealing in the refinement protocol was increased to 30 ps. Distance restraints for nonstereo specifically assigned protons were included using r−6 summation.21 Selection of calculated structures was based on the criteria in the XPLOR script accept.inp (RMSD for bonds and angles less than 0.01 Å and 1°, respectively; no NOE violation larger than 0.5 Å and no dihedral angle restraint violation larger than 5°).
Backbone dynamics
Experiments for measurement of 15N longitudinal and transverse relaxation rate constants and {1H}-15N steady state NOE were recorded as described previously.22 For backbone amide 15N longitudinal relaxation rate constants, R1, two-dimensional correlation spectra were measured for 11 different relaxation delays: 0, 0.010, 0.050, 0.120, 0.220, 0.350, 0.510, 0.700, 0.920, 1.170, and 1.450 s. Transverse relaxation rate constants, R2, were determined correlation spectra measured with 10 different relaxation delays: 0, 0.017, 0.034, 0.050, 0.067, 0.084, 0.101, 0.134, 0.168, 0.202 s. {1H}-15N steady state NOE experiments were recorded in an interleaved manner with and without 1H saturation during the 5 second recycle delay. Relaxation rate constants were determined from the single exponential decay of peak intensities in correlation spectra using NMRView23 and Grace (http://plasma-gate.weizmann.ac.il/Grace), while steady-state NOE values were determined in NMRView from the ratio of peak-heights for spectra recorded with and without 1H saturation.
The inertia and diffusion tensors of representative structures of S6wt and P54-55 were determined using the programs pdbinertia and r2r1_diffusion,24 respectively (A.G. Palmer, Columbia University; http://www.palmer.hs.columbia.edu/software/diffusion. html). 15N relaxation data were analyzed with the Lipari-Szabo model-free formalism, using Modelfree (v4.2)25,26 and the Fast-Modelfree interface27. Each N—H pair was evaluated using the following models for internal motion: (i) S2, (ii) S2 and τe, (iii) S2 and Rex, (iv) S2, τe, and Rex, and, (v) 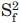 , S2, and τe, where S2 and denote the generalized order parameter and the order parameter for fast internal motion, respectively, τe is the effective internal correlation time, and Rex the exchange of transverse relaxation.
, S2, and τe, where S2 and denote the generalized order parameter and the order parameter for fast internal motion, respectively, τe is the effective internal correlation time, and Rex the exchange of transverse relaxation.
Accession numbers
The sequence specific resonance assignments of S6wt and P54-55 have been deposited in the BioMagResBank (accession numbers: BMRB-16344 and BMRB-16345). Coordinates of the final ensembles of 20 structures of S6wt and P54-55 have been deposited in the Protein Data Bank (PDB ID-codes: 2KJV and 2KJW).
Glossary
Abbreviations:
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- RMSD
root mean square deviation.
References
- 1.Steitz TA. A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol. 2008;9:242–253. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- 2.Agalarov SC, Sridhar Prasad G, Funke PM, Stout CD, Williamson JR. Structure of the S15,S6,S18-rRNA complex: assembly of the 30S ribosome central domain. Science. 2000;288:107–113. doi: 10.1126/science.288.5463.107. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl M, Svensson LA, Liljas A, Sedelnikova SE, Eliseikina IA, Fomenkova NP, Nevskaya N, Nikonov SV, Garber MB, Muranova TA, Rykanova AI, Amons R. Crystal structure of the ribosomal protein S6 from Thermus thermophilus. Embo J. 1994;13:1249–1254. doi: 10.2210/pdb1ris/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, Yonath A. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 5.Wimberly BT, Brodersen DE, Clemons WM, Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 6.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 7.Otzen DE, Kristensen O, Proctor M, Oliveberg M. Structural changes in the transition state of protein folding: alternative interpretations of curved chevron plots. Biochemistry. 1999;38:6499–6511. doi: 10.1021/bi982819j. [DOI] [PubMed] [Google Scholar]
- 8.Otzen DE, Oliveberg M. Conformational plasticity in folding of the split beta-alpha-beta protein S6: evidence for burst-phase disruption of the native state. J Mol Biol. 2002;317:613–627. doi: 10.1006/jmbi.2002.5423. [DOI] [PubMed] [Google Scholar]
- 9.Lindberg M, Tangrot J, Oliveberg M. Complete change of the protein folding transition state upon circular permutation. Nat Struct Biol. 2002;9:818–822. doi: 10.1038/nsb847. [DOI] [PubMed] [Google Scholar]
- 10.Haglund E, Lindberg MO, Oliveberg M. Changes of protein folding pathways by circular permutation. Overlapping nuclei promote global cooperativity. J Biol Chem. 2008;283:27904–27915. doi: 10.1074/jbc.M801776200. [DOI] [PubMed] [Google Scholar]
- 11.Lindberg MO, Haglund E, Hubner IA, Shakhnovich EI, Oliveberg M. Identification of the minimal protein-folding nucleus through loop-entropy perturbations. Proc Natl Acad Sci USA. 2006;103:4083–4088. doi: 10.1073/pnas.0508863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ludvigsen S, Poulsen FM. Positive theta-angles in proteins by nuclear magnetic resonance spectroscopy. J Biomol NMR. 1992;2:227–233. doi: 10.1007/BF01875318. [DOI] [PubMed] [Google Scholar]
- 13.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 14.Helgstrand M, Kraulis P, Allard P, Härd T. Ansig for Windows: an interactive computer program for semiautomatic assignment of protein NMR spectra. J Biomol NMR. 2000;18:329–336. doi: 10.1023/a:1026729404698. [DOI] [PubMed] [Google Scholar]
- 15.Wüthrich K. NMR of proteins and nucleic acids. New York: John Wiley & Sons; 1986. [Google Scholar]
- 16.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog NMR Spectrosc. 1999;34:66. [Google Scholar]
- 17.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 18.Padrta P, Sklenar V. Program MULDER—a tool for extracting torsion angles from NMR data. J Biomol NMR. 2002;24:339–349. doi: 10.1023/a:1021656808607. [DOI] [PubMed] [Google Scholar]
- 19.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 20.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- 21.Fletcher CM, Jones DNM, Diamond R, Neuhaus D. Treatment of NOE constraints involving equivalent or nonstereoassigned protons in calculations of biomacromolecular structures. J Biomol NMR. 1996;8:19. doi: 10.1007/BF00410328. [DOI] [PubMed] [Google Scholar]
- 22.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 23.Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J Biomolecular NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 24.Lee LK, Rance M, Chazin WJ, Palmer AG., III Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13C alpha nuclear spin relaxation. J Biomol NMR. 1997;9:287–298. doi: 10.1023/a:1018631009583. [DOI] [PubMed] [Google Scholar]
- 25.Mandel AM, Akke M, Palmer AG., III Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 26.Palmer AG, Rance M, Wright PE. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance carbon-13 heteronuclear NMR spectroscopy. JACS. 1991;113:10. [Google Scholar]
- 27.Cole R, Loria JP. FAST-Modelfree: a program for rapid automated analysis of solution NMR spin-relaxation data. J Biomol NMR. 2003;26:203–213. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]