

Abstract
The DNA repair enzyme NEIL1 is a DNA glycosylase that is involved in the first step of base excision repair (BER) of oxidatively induced DNA damage. NEIL1 exhibits a strong preference for excision of 4,6-diamino-5-formamidopyrimidine (FapyAde) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) from DNA with no specificity for 8-hydroxyguanine (8-OH-Gua). In this study, we report on the significant accumulation of (5′R)-8,5′-cyclo-2′-deoxyadenosine (R-cdA) and (5′S)-8,5′-cyclo-2′-deoxyadenosine (S-cdA) in liver DNA of neil1−/− mice that were not exposed to exogenous oxidative stress, while no accumulation of these lesions was observed in liver DNA from control or ogg1−/− mice. Significant accumulation of FapyGua was detected in liver DNA of both neil1−/− and ogg1−/− mice, while 8-OH-Gua accumulated in ogg1−/− only. Since R-cdA and S-cdA contain an 8,5′-covalent bond between the base and sugar moieties, they cannot be repaired by BER. There is evidence that these lesions are repaired by nucleotide excision repair (NER). Since the accumulation of R-cdA and S-cdA in neil1−/− mice strongly points to the failure of their repair, these data suggest that NEIL1 is involved in NER of R-cdA and S-cdA. Further studies aimed at elucidating the mechanism of action of NEIL1 in NER are warranted.
Oxidatively induced DNA damage occurs in living cells due to endogenous and exogenous sources that cause formation of oxygen-derived species, including free radicals (1). The hydroxyl radical is one of the most reactive free radicals and causes the formation of numerous lesions in DNA (2). Among these lesions, 8,5′-cyclopurine 2′-deoxynucleosides contain a C8−C5′ covalent bond, causing distortion of the DNA helix. Both (5′R)- and (5′S)-diastereomers of these compounds were identified in DNA in vitro and in vivo under various conditions (3).
Most oxidatively modified DNA bases are repaired by base excision repair (BER),1 a multistep process that is initiated by DNA glycosylases (4). In contrast, 8,5′-cyclopurine 2′-deoxynucleosides cannot be repaired by BER and are subject to nucleotide excision repair (NER) (5,6). In prokaryotic and eukaryotic cells, there are numerous DNA glycosylases that possess varying and sometimes overlapping substrate specificities toward oxidatively modified DNA bases (7). Escherichia coli Nei is similar in amino acid sequence to E. coli Fpg; however, its substrate specificity resembles that of E. coli Nth (endonuclease III), albeit with no sequence homology (8). Recently, homologues of Nei were discovered in mammals and called Nei-like or NEIL proteins (9−12).
Both human and mouse NEIL1 proteins were found to exhibit a strong preference for excision of 4,6-diamino-5-formamidopyrimidine (FapyAde) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) with no significant specificity for 8-hydroxyguanine (8-OH-Gua) (9,13,14). This substrate specificity is distinct from that of OGG1 that efficiently excises FapyGua and 8-OH-Gua, but not FapyAde, from DNA (15,16). Recently, significant accumulations of FapyAde and FapyGua were observed in several organs of neil1−/− mice, unequivocally proving that these lesions are in vivo substrates of NEIL1 (17). Moreover, it was reported that the Cockayne syndrome complementation group B protein (CSB) stimulates the repair of FapyAde and FapyGua by NEIL1 and that CSB and NEIL1 co-immunoprecipitate and colocalize in HeLa cells (18). These results demonstrated that CSB and NEIL1 cooperate in the repair of formamidopyrimidines and that a functional and physical interaction between these proteins exists. In contrast, no direct functional and physical interactions were identified between CSB and OGG1 (19). Recently, the involvement of NEIL1 in the removal of psoralen-induced interstrand DNA cross-links was reported (20). The proposed mechanism involved the excision by NEIL1 of the unhooked interstrand cross-link fragment via hydrolysis of the glycosidic bond of the cross-linked base.
Diminution or loss of NEIL1 leads to significant biological consequences. A correlation of inactivating mutations in neil1 with human gastric cancer was demonstrated (21). Knockdown of neil1 was shown to hypersensitize murine cells to killing effects of ionizing radiation (22). Downregulation of NEIL1 increased the level of spontaneous DNA damage and mutations in both Chinese hamster and human bronchial cell lines (23). Moreover, symptoms of metabolic syndrome and several types of cancer were observed in neil1−/− mice (17,24). Collectively, the existing data strongly suggest that NEIL1 with its very distinct substrate specificity and biological properties plays a critical role in the overall maintenance of genomic integrity and in disease prevention.
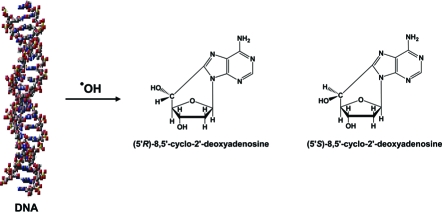
Thus far, the accumulated evidence unequivocally shows that NEIL1 is a DNA glycosylase and may also be preferentially active in transcription- and/or replication-coupled repair of DNA base lesions and interstrand cross-links. No additional role for NEIL1 in DNA repair mechanisms was demonstrated. Recently, neil1−/− mice were shown to exhibit the symptoms of metabolic syndrome and to develop several types of cancer despite the lack of exogenous oxidative stress (17,24). The accumulation of FapyAde and FapyGua, but not 8-OH-Gua, was observed in these mice. In this work, we hypothesize that NEIL1 may be involved in the repair of more complex DNA lesions than those resulting from modifications of DNA bases only. A recent study showed the accumulation of (5′S)-8,5′-cyclo-2′-deoxyadenosine (S-cdA) in several organs of csb−/− mice without exogenous oxidative stress, suggesting a critical role of CSB in the in vivo NER of this lesion (25). The aforementioned cooperation of CSB and NEIL1 in the repair of FapyAde and FapyGua may also be true for other more complex lesions. We tested this hypothesis by analyzing nuclear DNA samples from livers of neil1−/− mice and wild-type mice for R-cdA and S-cdA. We also generated ogg1−/− mice and tested their liver nuclear DNA as well for comparison. The structures of R-cdA and S-cdA are shown in Scheme 1. We utilized a methodology that we had recently developed with the use of liquid chromatography and isotope-dilution tandem mass spectrometry (LC−MS/MS) (26). This technique is capable of the positive identification and accurate quantification of very low endogenous levels of both R-cdA and S-cdA. Mouse liver samples were blinded and shipped on ice. Nuclear DNA was isolated from unfrozen livers immediately upon arrival.
Scheme 1. Structures of R-cdA and S-cdA.

Figure 1 illustrates the ion-current profiles of m/z 250 → m/z 164 (cdA) and m/z 255 → m/z 169 (cdA-15N5) transitions obtained during analysis of a nuclear DNA sample from a neil1−/− mouse liver. The levels of S-cdA in the livers of wild-type, neil1−/−, and ogg1−/− mice are shown in Figure 2. These data unequivocally showed the statistically significant accumulation of S-cdA in neil1−/− mice. In contrast, no accumulation of S-cdA was observed in ogg1−/− mice. R-cdA was quantified in neil1−/− mice only because of its insignificant levels in wild-type and ogg1−/− mice. Its measured level was 0.041 ± 0.013 molecule/106 DNA bases. Thus, we concluded that R-cdA also significantly accumulated in neil1−/− mice. In addition, we measured the levels of FapyGua and 8-OH-Gua using GC−MS.
Figure 1.

Ion-current profiles of the m/z 250 → m/z 164 (cdA) and m/z 255 → m/z 169 (cdA-15N5) transitions.
Figure 2.

Levels of S-cdA in liver DNA of wild-type (1) (n = 4), neil1−/− (2) (n = 7), and ogg1−/− (3) (n = 9) mice. The uncertainties are standard deviations. *p < 0.05.
Figures 3 and 4 illustrate the levels of FapyGua and 8-OH-Gua in liver DNA of wild-type, neil1−/−, and ogg1−/− mice, respectively. A statistically significant accumulation of FapyGua was observed in neil1−/− and ogg1−/− mice. These data confirm our previous findings (17,18) and are on par with the substrate specificities of NEIL1 and OGG1 determined under in vitro conditions (9,13−16). In contrast, 8-OH-Gua accumulated in ogg1−/− mice only (Figure 4), in agreement with the known specificity of OGG1 for 8-OH-Gua. As expected from the substrate specificity of NEIL1, no accumulation of 8-OH-Gua was observed in neil1−/− mice, in agreement with our previous results (17).
Figure 3.

Levels of FapyGua in liver DNA of wild-type (1) (n = 4), neil1−/− (2) (n = 6), and ogg1−/− (3) (n = 7) mice. The uncertainties are standard deviations. *p < 0.05.
Figure 4.

Levels of 8-OH-Gua in liver DNA of wild-type (1) (n = 4), neil1−/− (2) (n = 8), and ogg1−/− (3) (n = 8) mice. The uncertainties are standard deviations. *p < 0.05.
This study strongly suggests that NEIL1 plays a role in the cellular repair of R-cdA and S-cdA. Since these compounds are repaired by NER and not by BER, these results suggest for the first time that NEIL1 is involved in NER in addition to its function as a DNA glycosylase in BER. A previous study demonstrated that CSB stimulates the repair of formamidopyrimidines by NEIL1. Moreover, CSB was shown to play a role in NER of S-cdA. Thus, in turn, NEIL1 may be essential for the involvement of CSB in NER of R-cdA and S-cdA. On the other hand, NEIL1 may also interact with other proteins of the NER complex. Since NEIL1 is not capable of initiating BER at these lesions, it may be possible that it binds to them and accelerates the repair that is normally initiated through NER. Previously, the binding of DNA photolyase to cyclobutane pyrimidine dimers under noncatalytic conditions was shown to facilitate the efficiency of repair via the E. coli UvrABC complex (27,28). These data revealed the principle that DNA lesions can be noncovalently bound by proteins, thereby facilitating their ultimate repair. Whatever the mechanism of action of NEIL1 is in NER, we propose that NEIL1 is involved in NER of R-cdA and S-cdA, and perhaps other complex lesions not subject to BER. NEIL1 may act with CSB and/or any other NER proteins in this process. Further studies to elucidate this mechanism of action of NEIL1 in NER are warranted.
Acknowledgments
Certain commercial equipment or materials are identified in this paper in order to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified is necessarily the best available for the purpose.
Supporting Information Available
Materials and detailed experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported in part by National Institutes of Health Grant DK075974 (R.S.L.).
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: BER, base excision repair; NER, nucleotide excision repair; R-cdA, (5′R)-8,5′-cyclo-2′-deoxyadenosine; S-cdA, (5′S)-8,5′-cyclo-2′-deoxyadenosine; FapyAde, 4,6-diamino-5-formamidopyrimidine; FapyGua, 2,6-diamino-4-hydroxy-5-formamidopyrimidine; 8-OH-Gua, 8-hydroxyguanine; CSB, Cockayne syndrome complementation group B protein; LC−MS/MS, liquid chromatography−tandem mass spectrometry.
Supplementary Material
References
- Halliwell B., and Gutteridge J. M. C. (2007) Free Radicals in Biology and Medicine, 4th ed., Oxford University Press, Oxford, U.K. [Google Scholar]
- Evans M. D.; Dizdaroglu M.; Cooke M. S. (2004) Mutat. Res. 567, 1–61. [DOI] [PubMed] [Google Scholar]
- Jaruga P.; Dizdaroglu M. (2008) DNA Repair 7, 1413–1425. [DOI] [PubMed] [Google Scholar]
- Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., and Ellenberger T. (2005) DNA Repair and Mutagenesis, ASM Press, Washington, DC. [Google Scholar]
- Brooks P. J.; Wise D. S.; Berry D. A.; Kosmoski J. V.; Smerdon M. J.; Somers R. L.; Mackie H.; Spoonde A. Y.; Ackerman E. J.; Coleman K.; Tarone R. E.; Robbins J. H. (2000) J. Biol. Chem. 275, 22355–22362. [DOI] [PubMed] [Google Scholar]
- Kuraoka I.; Bender C.; Romieu A.; Cadet J.; Wood R. D.; Lindahl T. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 3832–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M. (2005) Mutat. Res. 591, 45–59. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M.; Burgess S. M.; Jaruga P.; Hazra T. K.; Rodriguez H.; Lloyd R. S. (2001) Biochemistry 40, 12150–12156. [DOI] [PubMed] [Google Scholar]
- Hazra T. K.; Izumi T.; Boldogh I.; Imhoff B.; Kow Y. W.; Jaruga P.; Dizdaroglu M.; Mitra S. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru V.; Sunkara S.; Wallace S. S.; Bond J. P. (2002) DNA Repair 1, 517–529. [DOI] [PubMed] [Google Scholar]
- Morland I.; Rolseth V.; Luna L.; Rognes T.; Bjoras M.; Seeberg E. (2002) Nucleic Acids Res. 30, 4926–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao M.; Kanno S.; Kobayashi K.; Zhang Q. M.; Yonei S.; van der Horst G. T.; Yasui A. (2002) J. Biol. Chem. 277, 42205–42213. [DOI] [PubMed] [Google Scholar]
- Jaruga P.; Birincioglu M.; Rosenquist T. A.; Dizdaroglu M. (2004) Biochemistry 43, 15909–15914. [DOI] [PubMed] [Google Scholar]
- Roy L. M.; Jaruga P.; Wood T. G.; McCullough A. K.; Dizdaroglu M.; Lloyd R. S. (2007) J. Biol. Chem. 282, 15790–15798. [DOI] [PubMed] [Google Scholar]
- Dherin C.; Radicella J. P.; Dizdaroglu M.; Boiteux S. (1999) Nucleic Acids Res. 27, 4001–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audebert M.; Radicella J. P.; Dizdaroglu M. (2000) Nucleic Acids Res. 28, 2672–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan M. K.; Ocampo-Hafalla M. T.; Vartanian V.; Jaruga P.; Kirkali G.; Koenig K. L.; Brown S.; Lloyd R. S.; Dizdaroglu M.; Teebor G. W. (2009) DNA Repair 8, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M.; de Souza-Pinto N. C.; Dogan A.; Aamann M.; Stevnsner T.; Rybanska I.; Kirkali G.; Dizdaroglu M.; Bohr V. A. (2009) J. Biol. Chem. 284, 9270–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo J.; Chen C.; Zeng X.; Christiansen M.; Bohr V. A. (2002) DNA Repair 1, 913–927. [DOI] [PubMed] [Google Scholar]
- Couve S.; Mace-Aime G.; Rosselli F.; Saparbaev M. K. (2009) J. Biol. Chem. 284, 11963–11970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinmura K.; Tao H.; Goto M.; Igarashi H.; Taniguchi T.; Maekawa M.; Takezaki T.; Sugimura H. (2004) Carcinogenesis 25, 2311–2317. [DOI] [PubMed] [Google Scholar]
- Rosenquist T. A.; Zaika E.; Fernandes A. S.; Zharkov D. O.; Miller H.; Grollman A. P. (2003) DNA Repair 2, 581–591. [DOI] [PubMed] [Google Scholar]
- Maiti A. K.; Boldogh I.; Spratt H.; Mitra S.; Hazra T. K. (2008) DNA Repair 7, 1213–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian V.; Lowell B.; Minko I. G.; Wood T. G.; Ceci J. D.; George S.; Ballinger S. W.; Corless C. L.; McCullough A. K.; Lloyd R. S. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkali G.; de Souza-Pinto N. C.; Jaruga P.; Bohr V. A.; Dizdaroglu M. (2009) DNA Repair 8, 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaruga P.; Xiao Y.; Nelson B. C.; Dizdaroglu M. (2009) Biochem. Biophys. Res. Commun. 386, 656–660. [DOI] [PubMed] [Google Scholar]
- Sancar A.; Franklin K. A.; Sancar G. B. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 7397–7401.6390436 [Google Scholar]
- Sancar G. B.; Smith F. W. (1989) Mol. Cell. Biol. 9, 4767–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.