

Abstract
Perturbations in the foetal environment predispose individuals to diseases that become apparent in adulthood. These findings prompted researchers to hypothesize that foetal exposure to environmental oestrogens may play a role in the increased incidence of breast cancer observed in European and US populations over the last 50 years. There is widespread human exposure to bisphenol A, an oestrogenic compound that leaches from dental materials and consumer products. In CD-1 mice, perinatal exposure to environmentally relevant bisphenol A levels induced alterations of the mammary gland architecture. Bisphenol A increased the number of terminal end buds at puberty and terminal ends at 6 months of age and increased ductal lateral branching at 4 months of age. Exposed mice also showed an enhanced sensitivity to oestradiol when ovariectomized prior to puberty. All these parameters are associated in human beings with an increased risk for developing breast cancer. To assess whether bisphenol A induces mammary gland neoplasia, we chose a rat model because it more closely mimics the human disease than mouse models. Examination of the mammary glands of Wistar/Furth rats during early adulthood revealed that gestational exposure to bisphenol A induced the development of pre-neoplastic lesions and carcinoma in situ in the absence of any additional treatment aimed at increasing tumour development. Emerging epidemiological data reveal an increased incidence of breast cancer in women exposed to diethylstilboestrol during gestation. Hence, both animal experiments and epidemiological data strengthen the hypothesis that foetal exposure to xenooestrogens may be an underlying cause of the increased incidence of breast cancer observed over the last 50 years.
The hypothesis that prenatal exposure to endocrine disruptors might cause cancer arose when two ingrained notions began to be contested: (i) embryonic development is merely the unfolding of a genetic programme, and (ii) only mutagenic agents can cause cancer. The realization that a reductionist programme has failed to bring about a more robust understanding of complex phenomena has resulted in an ongoing re-appraisal of old research traditions in embryology and cancer research.
Despite studies from the end of the 19th century illustrating phenomenon such as environmentally triggered polyphenism, the dominant view in developmental biology at the end of the 20th century was that development is the unravelling of a genetic programme where the environment plays virtually no relevant role. Two main factors contributed to the dominance of the genetic programme view. One was the advent of developmental mechanics, which concentrated on the inner workings of the embryo rather than on the ecological determination of phenotype. Another was the dominance of a genocentric view originating from the molecular biology revolution.
For most of the 20th century, the dominant stance regarding cancer was the somatic mutation theory [1]. The premises of this theory are: (i) cancer is derived from a single somatic cell that has accumulated multiple DNA mutations, (ii) the default state in metazoan cells is proliferative quiescence, and (iii) cancer is a disease of cell proliferation caused by mutations in genes that control the cell cycle. The research programmes and policies emanating from this theory have fallen short both in explaining the causes of cancer and in providing effective treatments. Due to these shortcomings, an older tradition, centred at the tissue level of organization, has been updated as the tissue organization field theory (TOFT) of carcinogenesis and neoplasia and is gaining momentum [2]. This tradition originated in the late 19th century when pathologists began describing the histological pattern of tumours using merely the light microscope and suggesting that altered tissue organization was at the core of neoplasia, thus linking carcinogenesis to embryonic development. In contrast to the somatic mutation theory, the TOFT postulates that: (i) carcinogenesis represents a problem of tissue organization, (ii) proliferation is the default state of all cells, and (iii) carcinogenesis is a reversible phenomenon [2]. Carcinogens, as well as teratogens, would disrupt the normal dynamic interaction of neighbouring cells and tissues during early development and throughout adulthood [3]. According to this theory, carcinogenesis is comparable to organogenesis gone awry.
We are now witnessing a resurfacing of old theories of development and carcinogenesis in the context provided by the advancements made in genetics, cell biology and molecular biology. The environment is again accepted as a main player in phenotype determination. There are at least three potential ways that environmental cues could dictate the building of a phenotype: (i) a neuroendocrine route, whereby the nervous system monitors the environment and is stimulated to send signals to the endocrine system; responding hormones would then alter gene expression, (ii) the epigenetic route, whereby environmental agents alter transcriptional capabilities through histone modification and DNA methylation, and (iii) direct induction, whereby environmental agents act directly as hormones or disrupt the metabolism or synthesis of endogenous hormones [4]. Ecological developmental biology and the implications of the TOFT provide the appropriate context to explore the following questions: (i) does foetal exposure to endocrine disruptors cause neoplastic development? and, more pointedly, (ii) does breast cancer start in the womb?
The complex field of endocrine disruptors
Following the publication of Rachel Carson’s Silent Spring, researchers continued to make connections between chemical exposures and adverse outcome in animals and human beings. Soon after the massive introduction of the pesticide Dichloro-Diphenyl-Trichloroethane (DDT) and other synthetic chemicals into the environment, evidence emerged linking environmental exposure with a variety of developmental and reproductive abnormalities, while laboratory studies revealed that some of these compounds had oestrogenic activity. This led to the enunciation of the endocrine disruptor hypothesis at the Wingspread Conference, held in Racine, Wisconsin, in 1991. The term endocrine disruptor was coined because the participants proposed that developmental alterations observed in diverse animal species were due to exposure to multiple chemicals that, through different modes of action, disrupted the endocrine systems of metazoan organisms during organogenesis and development at large [5].
Environmental endocrine disruptors are now defined by the US Environmental Protection Agency as ‘exogenous agents that interfere with the synthesis, secretion, transport, binding, action, or elimination of natural hormones in the body that are responsible for the maintenance of homeostasis, reproduction, development, and/or behaviour’. This definition evokes a diversity of targets and pathways that may be affected by exogenous chemicals, implying that endocrine disruptors are heterogeneous agents encompassing diverse structures.
While exposures during adulthood were considered potentially deleterious, the main concern of the conferees was the exposure of developing organisms, because some effects documented in the genital tracts of animals were comparable to those seen in the daughters and sons born to women treated with diethylstilboestrol during pregnancy. The conference participants recognized that the human diethylstilboestrol syndrome was an extreme expression of the plasticity of the foetus in response to environmental cues, providing a template for the potential effects that other hormonally active chemicals could have on human health.
The vast majority of identified hormonally active agents are oestrogen mimics, also known as xenooestrogens. Endogenous oestrogens are involved in the development and maintenance of the female reproductive tract and secondary sexual characteristics as well as in the regulation of the menstrual/oestrous cycle, pregnancy and lactation. At the cellular level, these endogenous hormones mediate cell proliferation and the synthesis and secretion of cell type-specific proteins in reproductive tissues such as the ovary, oviduct, uterus, vagina, hypothalamus, pituitary and mammary gland. These effects are mediated, for the most part, by oestrogen receptors (α and β). Additionally, the expression of oestrogen receptors in the male reproductive tract and in non-reproductive organs such as the thyroid, cardiovascular system and bone indicates the potentially vast reach of synthetic oestrogenic chemicals.
Until recently, the issue of whether hormonally active agents at low, environmentally relevant doses could alter development was highly controversial. Although xenooestrogens are usually less potent than oestradiol regarding their binding affinity for classical nuclear oestrogen receptors, it is now clear that they act additively with endogenous oestrogens. This may explain how low seemingly insignificant levels of xenooestrogens have an impact when added to the already significant levels of endogenous steroidal hormones [6]. In addition, xenooestrogens bind to plasma-carrier proteins with significantly lower affinities than those of natural oestrogens, and thus are more readily available to target cells than their endogenous counterparts [7]. The same xenooestrogens that seem to be weak agonists for the nuclear oestrogen receptors are strong agonists when acting via membrane oestrogen receptors; this may also explain their ability to produce biological effects at low doses [8].
Toxicologists typically assume that after a chemical exposure, an organism will respond in a monotonic way (i.e. the higher the dose the greater the effect). However, hormones display diverse types of response curves, including those that have a U or inverted U shape, which are known as non-monotonic curves. For example, at low physiological levels, androgens increase the proliferation rate of prostate cell lines, while at high physiological levels they induce proliferative quiescence [9]. Similarly, oestrogens invoke a non-monotonic (inverted U shaped) response in a variety of morphometric parameters in the mammary gland of ovariectomized prepubescent mice [10] (fig. 1). Dose–response curves can also be non-monotonic for some effects observed after prenatal exposure to xenooestrogens. For example, prenatal exposure to methoxychlor alters the response of the adult uterus to oestradiol; low doses increase the response and higher doses reduce it [11]. The frequent occurrence of non-monotonic dose–response curves in biological phenomena indicates the importance of understanding how these complex biological phenomena are regulated [12]. These patterns ultimately highlight the inadequacy of using the response to high doses of a natural or environmental hormone or other toxicant to extrapolate or predict the effects of exposure to low doses of the compound.
Fig. 1.
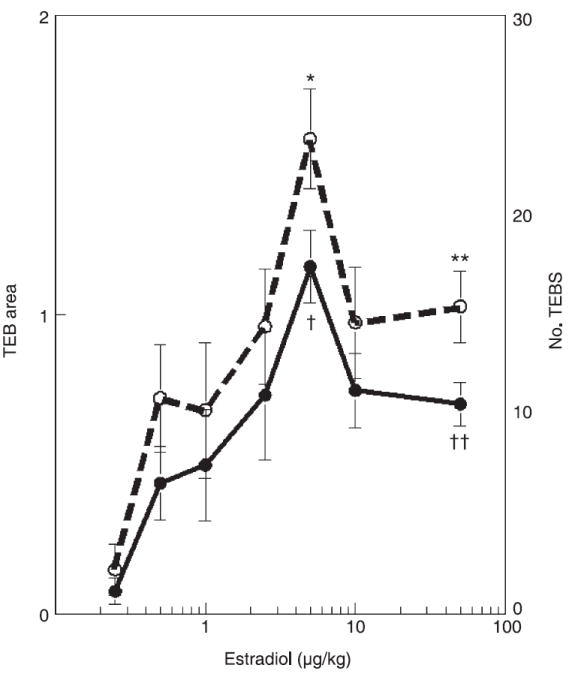
Morphometric parameters in the mammary gland of ovariectomized pubertal mice demonstrate non-monotonic dose–response curves in response to oestradiol. The dashed line denotes the number of terminal end buds (TEB) and the solid line denotes TEB area. * or † indicates the dose where the maximal response was observed, which is statistically different from both its ovariectomized control and the highest administered dose (marked with the ** or ††).
Foetal origins of adult diseases
Epidemiological studies revealed that perturbations in the foetal environment such as maternal malnutrition caused diseases and/or organ dysfunctions, including heart disease, stroke, diabetes and hypertension, that became apparent in adulthood [13]. Recently, scientists have hypothesized that exposure to hormonally active agents may also lead to alterations in the development of a foetus. The high sensitivity to both endogenous and exogenous hormones by the developing organism led Dr. Howard Bern to coin the term the ‘fragile foetus’ [14].
Oestrogen exposure throughout a woman’s life is a major risk factor for the development of breast cancer, as has been demonstrated by the increased risk associated with early age of menarche and late age of menopause [15]. The positive correlation between increased intrauterine levels of oestrogens (a phenomenon observed in twin births) and breast cancer in daughters born from such pregnancies also supports this link [16]. Additional evidence comes from women exposed to high doses of the synthetic oestrogen diethylstilboestrol, a drug administered to pregnant women between the years 1948 and 1971 to prevent miscarriages. Women who were exposed to diethylstilboestrol while pregnant now show a higher incidence of breast cancer [17]. Their daughters, that is, ‘diethylstilboestrol daughters’, are now reaching the age at which breast cancer is diagnosed. The rate ratio for the incidence of breast cancer in diethylstilboestrol-exposed versus unexposed women aged 40 and older was 2.5, indicating a statistically significant increase in the in utero diethylstilboestrol-exposed women [18].
We are all exposed involuntarily to a multitude of environmental chemicals with hormonal activities. This is in addition to medically prescribed hormones (hormonal contraceptives or hormone-replacement therapy). Is the increased breast cancer incidence that has been observed during the last 50 years due to this cumulative exposure? A few epidemiological case-control and cohort studies both from the USA and Europe have revealed a positive correlation between breast cancer incidence and serum levels of endocrine disruptors such as dieldrin, DDT and PCBs in women [19]. Controversy abounds on the interpretation of this data, mainly because none of these chemicals are considered to be a marker of a total xenooestrogen exposure [20]. However, assessments of total xenooestrogen exposure in adipose tissue correlated positively with breast cancer incidence [21].
Because a mixture of diverse endocrine disruptors is present in both human and animal tissues, and these exposures occur at all developmental ages, it is expected that human beings and animals will display more florid and diverse syndromes than those laboratory animals exposed to a single chemical at a well-defined developmental stage. However, in an effort to understand the health problems posed by endocrine disruptors, it is useful to examine single agents during specific developmental periods. We have chosen to focus the remainder of this review on the effects of developmental exposure to environmentally relevant levels of the xenooestrogen bisphenol A on mammary gland development and carcinogenesis. Bisphenol A is present ubiquitously in our environment, has high potential for foetal exposure, and the literature already contains multiple examples of developmental effects in experimental models.
Bisphenol A
Bisphenol A (4,4’-isopropylidenediphenol) is widely used in the manufacture of polycarbonate plastics and epoxy resins. Bisphenol A is present in a multitude of products including the interior coating of food cans, wine storage vats, water carboys, milk containers, food storage vessels, baby formula bottles, water pipes, dental materials, automotive lenses, optical lenses, protective coatings, adhesives, protective window glazing, compact discs, thermal paper, paper coatings, and as a developer in dyes [22]. In 2003, the worldwide production of bisphenol A exceeded 6 billion pounds [23]. About 100 tons of bisphenol A are released into the atmosphere each year during production [22]. Studies have shown that incomplete polymerization of bisphenol A during manufacture, and/or depolymerization due to increased temperatures (induced either intentionally for sterilization/heating purposes or unintentionally during storage in warehouses) causes bisphenol A and its derivatives to leach into foods, beverages, infant formula and saliva after application of dental sealants [22]. Bisphenol A has been found in air and dust samples from residential and commercial environments, leachates from waste water treatment plants, river water, and surface and drinking water [22].
Bisphenol A is a diphenyl compound that contains two hydroxyl groups in the ‘para’ position making it remarkably similar to the synthetic oestrogen, diethylstilboestrol (fig. 2). Although bisphenol A has been known to be oestrogenic since 1936 [24], it was in the 1990s that this chemical was serendipitously discovered to leach from polycarbonate plastics in concentrations that were sufficient to up-regulate the expression of progesterone receptor and induce cell proliferation in oestrogen-target, serum-sensitive MCF-7 cells through binding to the oestrogen receptor [25].
Fig. 2.
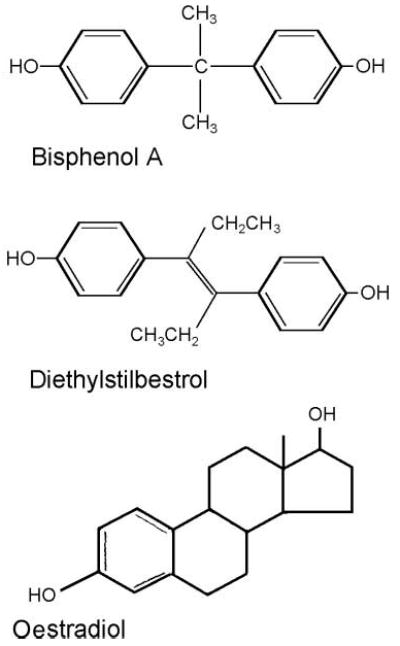
Chemical structures of bisphenol A (BPA), diethylstilbestrol (DES) and oestradiol. The structures of BPA and DES are more similar to one another than they are to the endogenous oestradiol, indicating that chemicals with variable structures are capable of binding to the oestrogen receptor.
The ubiquitous use of bisphenol A provides great potential for exposure of both the developing foetus indirectly through maternal exposure, and the neonate directly through ingestion of infant formula or maternal milk [26]. Indeed, bisphenol A has been measured in maternal and foetal serum and placental tissue at birth in human beings [27]. The range of bisphenol A concentrations in foetal serum ranged from 0.2 to 9.2 ng/ml, indicating that the developing human foetus and neonate are readily exposed to this chemical. A recently published study, the first using a reference adult human population, reported that bisphenol A was found in 95% of 394 urine samples in the USA [28]. From these data, the mean exposure was estimated to be 30–40 ng/kg body weight/day and the 95th percentile was 180–230 ng/kg/day. In a smaller study, Arakawa et al. reported a median daily urinary excretion of bisphenol A of 1.2 μg/day and a maximum daily intake per body weight of 0.23 μg/kg/day [29].
In rodents, bisphenol A has been shown to readily cross the placenta [30] and bind α-foetoprotein but with negligible affinity relative to oestradiol, resulting in enhanced bio-availability during neonatal development [7]. Bisphenol A is present in the mouse foetus and amniotic fluid during maternal exposure in higher concentrations than that of maternal blood. Thus, the pharmacokinetics and pharmaco-dynamics of bisphenol A may exacerbate the impact of this chemical on the developing foetus and neonate [30].
Development of the mouse mammary gland and expression of the oestrogen receptor
In the mouse, at embryonic day (E) 11.5, five placodes appear along each presumptive mammary line as lens-shaped ectodermal structures that later on invaginate into the dermis. The mesenchyme abutting the mammary epithelium becomes denser than the surrounding mesenchyme with several concentric layers of fibroblasts aligning themselves around the epithelial compartment [31]. At E15.5, the epithelial bud elongates to become a cord, and on E16, the primary sprout undergoes a sudden and significant increase in proliferation before it pushes through the closely associated mammary mesenchyme and penetrates the primitive fat pad, a cluster of pre-adipocytes found in the deeper dermal tissue. By E18, branching is apparent and the ductal lumen starts to form [32] (fig. 3).
Fig. 3.
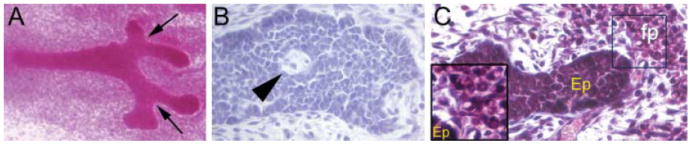
Appearance of the mammary gland at embryonic day 18. (A) Branching points (arrows) are apparent in carmine stained whole mounts. (B) Haematoxylin and eosin-stained paraffin section, demonstrating lumen formation (arrowhead), which is evident in approximately 40% of untreated glands at this age. (C) Lipid droplets are observed in clusters of primitive fat pad (fp) within a short distance of the epithelium (see inset). Ep, epithelium.
Oestrogen receptors α and β are first expressed at E12.5 in the mesenchyme surrounding the bud [33]. Autoradiographic experiments also revealed specific 3H-diethylstilboestrol binding only in the mesenchyme surrounding the epithelial anlagen at E16, suggesting the presence of functional receptors at that time [34]. By E18, oestrogen receptor is detected predominantly in the stroma with punctate expression in the epithelium [32]. This suggests that E18 may be a point of transition because oestrogen receptor α expression is mainly localized to the epithelium at postnatal time-points [35].
During prenatal and neonatal development, the mouse mammary gland grows isometrically with respect to body growth until plasma oestrogen levels rise during the third week of postnatal life. Oestrogens then drive massive peripubertal ductal growth. Bulbous epithelial structures known as terminal end buds (TEB) develop and show both high mitotic and apoptotic activity; the cap cells of the TEB are highly proliferative, permitting rapid ductal elongation as well as the ability of the duct to change direction in the fat pad [36], while death of the body cells in the TEB is essential for the formation of the lumen and the growth of the subtending duct [37]. The ductal tree invades the stroma until it reaches the edge of the fat pad, establishing a network of ducts and a few alveolar buds [36]. This morphology remains relatively quiescent, although minor fluctuations occur with each oestrous cycle. During pregnancy, the entire epithelial compartment undergoes a dramatic proliferation resulting in a plethora of alveolar buds and lobuloalveolar units in preparation for lactation. Once the period of lactation is over, the mammary gland undergoes rapid involution, a process associated with widespread apoptosis and stromal remodelling, to return the gland to its pre-pregnancy state.
Perinatal exposure to bisphenol A alters mammary gland development
Exposure of pregnant mice to either 25 or 250 ng bisphenol A/kg body weight/day, using osmotic mini pumps for 14 days beginning on E8, has been shown to impact certain aspects of development in their female offspring. When examined at E18 (2 days before birth), foetuses of mothers exposed to 250 ng bisphenol A exhibited altered growth of the mammary gland anlagen (fig. 4A). Changes in the appearance of the mammary epithelium were observed, such as decreased cell size and delayed lumen formation, as well as increased ductal area. In the stroma, bisphenol A exposure promoted advanced maturation of the fat pad and altered localization of fibrous collagen [32]. Because maturation of the fat pad is the driving event for ductal growth and branching, it is likely that the increased ductal area in bisphenol A-exposed animals is due to the accelerated formation of their fat pads.
Fig. 4.
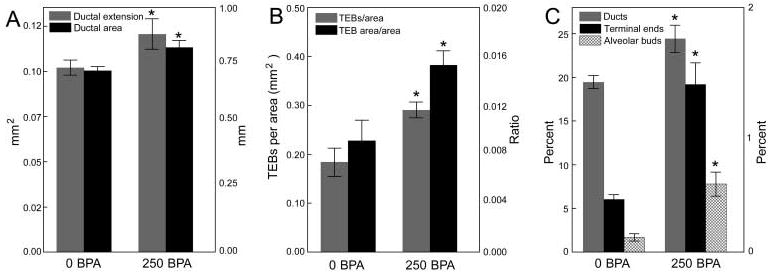
In utero exposure to bisphenol A (BPA) alters tissue organization in the mammary gland at several distinct developmental periods. (A) Ductal area and ductal extension are significantly increased at E18 in BPA-exposed foetuses. (B) Perinatal BPA exposure alters the number of terminal end buds (TEB) and the TEB area relative to total ductal area in the pubertal mammary gland. (C) Perinatally exposed females demonstrate increases in the relative area of ducts, terminal ends and alveolar buds in adulthood (6 months of age). Area of ducts and alvelolar buds: left axis; terminal ends: right axis. * denotes significant differences between animals exposed to 250 ng BPA/kg/day (250 BPA) and the vehicle control (0 BPA).
By postnatal day 10, the percentage of proliferating epithelial cells, measured by the incorporation of bromodeoxyuridine into DNA, was significantly decreased in bisphenol A-exposed mice relative to controls [38]. At 30 days of age, the area and numbers of TEBs relative to the gland ductal area increased (fig. 4B) while apoptotic activity in these structures decreased in bisphenol A-exposed offspring. There was a positive correlation between ductal length and the age at first prooestrus in control females. This correlation was reduced as the bisphenol A dose increased, suggesting that bisphenol A exposure slows down ductal invasion of the stroma. It is likely that the reduced apoptotic index in the TEBs of bisphenol A-exposed females may be the cause of this ductal growth delay, as apoptosis is essential for both the hollowing and the outwards growth of the subtending duct [37]. Collectively, these effects observed at puberty may be attributed to an increased sensitivity to oestradiol that has been observed in bisphenol A-exposed animals [39] (fig. 5).
Fig. 5.
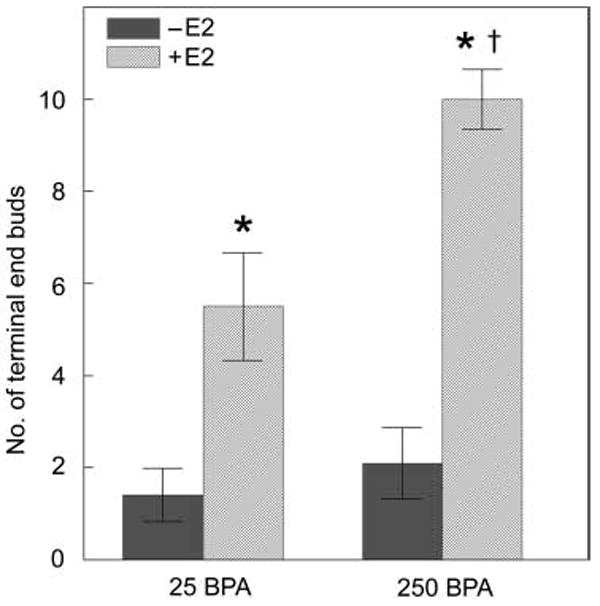
Perinatal bisphenol A (BPA) exposure alters the sensitivity of the mammary gland to oestradiol at puberty. Animals exposed perinatally to vehicle (0 BPA) or 250 ng BPA/kg/day (250 BPA) were ovariectomized and challenged with 0 (−E2) or 0.5 μg oestradiol/kg/day (+E2) at 25 days of age (pre-puberty). BPA-exposed animals had a heightened response in number of terminal end buds (TEB) compared to control animals. * denotes significant differences from −E2 groups. The symbol ‘†’ indicates significant differences from animals exposed perinatally to vehicle and to E2 at 25 days of age.
In animals exposed perinatally to bisphenol A, there was also a significant increase in ductal epithelial cells that were positive for progesterone receptor at puberty. These positive cells were localized in clusters, suggesting future branching points. Indeed, lateral branching was significantly enhanced at 4 months of age in offspring born to mothers exposed to 25 ng bisphenol A/kg body weight/day by osmotic mini pumps [39]. These results are compatible with the notion that increased sensitivity to oestrogens drives the induction of progesterone receptor in epithelial cells, leading to an increase in lateral branching. By 6 months of age, perinatally exposed virgin mice exhibit mammary glands that resemble those of a pregnant mouse. This is reflected by a significant increase in the percentage of ducts, terminal ends and alveolar buds compared to unexposed controls [38] (fig. 4C). In conclusion, these results indicate that perinatal exposure to environmentally relevant doses of bisphenol A results in persistent alterations in mammary gland morphogenesis. Moreover, the altered growth parameters noted in the developing mammary gland at E18 suggest that the foetal gland is a direct target of bisphenol A, and that these alterations cause the phenotypes observed in the mammary gland at puberty and adulthood.
In summary, foetal exposure to low doses of bisphenol A modulates cell proliferation, apoptosis and the timing of development, suggesting that this chemical can predispose the mammary gland to carcinogenesis. Importantly, the observed increased sensitivity to oestrogens may represent a functional equivalent to a known risk factor in human beings, namely, lifetime oestrogen exposure [15]. Moreover, the increased ductal density observed in these mice may be considered equivalent to another acknowledged risk factor in human beings, that is, increased mammographic density [40].
Do bisphenol A studies indicate that breast cancer originates in the womb?
To explore the links between prenatal bisphenol A exposure and mammary gland neoplasia, we used a rat model, because it closely resembles the human disease regarding oestrogen dependency and histopathology [41]. Bisphenol A was administered to pregnant dams at doses of 2.5, 25, 250 and 1000 μg/kg body weight/day using osmotic mini pumps (the estimated lowest-observed effect level was 50 mg/kg body weight/day; the estimated tolerable daily intake was set at 10 μg/kg body weight/day by the European Commission and 50 μg/kg body weight/day by the US Environmental Protection Agency). Foetal exposure to bisphenol A, from E9 to postnatal day 1, resulted in the development of carcinomas in situ in the mammary glands of 33% of the rats exposed to 250 μg/kg body weight/day while none of the unexposed animals developed neoplasias (fig. 6) [42]. These cancers were only observed once the animals had reached young adult age. Foetal exposure to bisphenol A significantly increased the number of pre-neoplastic lesions, namely, intraductal proliferation, by three to four times, an effect also observed in puberty and during adult life (fig. 6). These intraductal hyperplasias are considered the precursors of carcinomas both in rodents and human beings and have been shown to develop into palpable tumours when transplanted into hosts with intact ovaries [43]. The number of intraductal hyperplasias observed at 50 days of age was quantitatively similar at all bisphenol A doses tested. Remarkably, they persisted longer in the animals exposed to the lowest dose. The lesions observed in the bisphenol A-exposed animals were highly proliferative and contained abundant oestrogen receptor-positive cells [42], suggesting that the proliferative activity in these lesions may be oestrogen mediated. As mentioned above, mammary carcinomas in both rats and human beings are predominantly oestrogen dependent, a feature that strengthens the relevance of these findings. Moreover, prenatal exposure to 25 μg bisphenol A/kg body weight/day, followed by treatment at puberty with a ‘subcarcinogenic’ single dose of the chemical carcinogen nitrosomethylurea, resulted in the development of tumours only in the animals exposed in utero to bisphenol A [44].
Fig. 6.
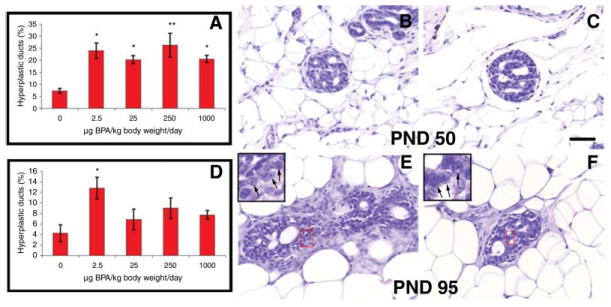
Neoplastic lesions. The percentage of ductal hyperplasias is significantly increased in bisphenol A (BPA)-exposed animals at postnatal day 50 (A) and 95 (D); *P < 0.05; **P < 0.005. Some of the ductal lesions were identified as carcinomas in situ (CIS) and had a cribriform pattern. This can be observed at postnatal day 50 (B and C) and 95 (E and F). The CIS showed not only multiple lumina but also hyperchromatic nuclear staining with visible nucleoli (inset in E and F). Scale bar: 50 μm (figure reproduced with permission, Reproductive Toxicology; 23:383–390, 2007).
All of the morphological and functional alterations described above suggest both direct action of bisphenol A on oestrogen-sensitive organs, as well as bisphenol A-induced alterations in the hypothalamic–pituitary–gonadal axis that would have secondary effects on peripheral organs including the mammary gland. Direct effects of oestrogens on the target reproductive organs and mammary gland are believed to be mediated by regulation of the expression of oestrogen-target genes involved in tissue patterning, histodifferentiation and cytodifferentiation. For example, neonatal exposure to diethylstilboestrol exerts an oestrogenic effect through repression of the Wnt7a signalling pathway in the female reproductive tract [45]. Prenatal exposure to diethylstilboestrol also alters the expression of several Hox genes in the mouse Müllerian duct and uterus [46]. In the words of Block et al., ‘Oestrogens are novel morphogens that directly regulate the expression pattern of posterior Hox genes in a manner analogous to retinoic acid regulation of anterior Hox genes’ [47].
Several members of the Wnt family and Msx2 are expressed during foetal development [48] and the expression of some of these genes is regulated by oestrogens in the adult mammary gland [10]. Hence, it is plausible that foetal xenooestrogen exposure may result in the extemporaneous expression of this set of genes that, in turn, may cause altered morphogenesis and neoplastic development. In addition, foetal bisphenol A exposure may also result in alterations in the methylation patterns of genes involved in the reciprocal tissue interactions that mediate morphogenesis.
Conclusions
The organizational and functional changes reported to date provide important pieces of evidence for the understanding of how xenooestrogen exposure, and bisphenol A exposure in particular, affects foetal development of oestrogen-target organs in human beings and animals. While low-level exposure to bisphenol A or other xenoostrogens during adulthood may not have dramatic effects on females, when exposure occurs in utero or during the perinatal period, it can exert significant and lasting effects on the development of the female reproductive tract, mammary gland, and alter reproductive axis function.
In addition, these results buttress the link between foetal exposure to bisphenol A and the development of neoplasias in the adult mammary gland. These neoplasias may have their origin in the altered morphogenesis that occurs in the foetus during the period of bisphenol A exposure. Furthermore, these results support the hypothesis that exposure to xenooestrogens during foetal life may contribute to the increased incidence of breast cancer observed over the past five decades.
Evidence from the mouse model indicates that bisphenol A alters both the mammary epithelium and stroma during the period of exposure, that is, foetal development, with additional effects that manifest later in life (at puberty and in adulthood). Although oestrogen receptors begin to be expressed in the epithelium at E18, they are present predominantly in the stromal compartment during the period of exposure [33]; thus, it is plausible that bisphenol A is directly altering the development of this tissue compartment, modifying both histogenesis and organogenesis and thus leading to a neoplastic phenotype that manifests in the epithelium in adult life [32]. In the context of the TOFT exposure to bisphenol A would lead to the development of neoplasias in the mammary gland by altering tissue organization.
The findings reviewed above have both practical and theoretical implications. From a practical perspective, it is now evident that animals and human beings are affected by environmental exposure to hormonally active chemicals at levels previously considered to be irrelevant. These data should raise concerns about the potentially deleterious impact of endocrine disrupting chemicals on human development. Extrapolating evidence from animal studies to human beings should be done cautiously, as differences among strains and species have been reported regarding a variety of parameters. However, the mouse and rat have been shown to be excellent surrogate models for the understanding of the diethylstilboestrol syndrome. All of this evidence should encourage regulatory agencies to apply the precautionary principle and thus ban or substitute those chemicals that are likely to be harmful to the normal development of human beings and animals. In fact, the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) Legislation, entered into force in the European Union in June 2007, requires chemical producers to generate safety data for all chemicals produced, manage any risks associated with use of these chemicals, include an analysis of alternatives and a substitution plan where a suitable alternative exists, and communicate this information to consumers.
From a theoretical perspective, the results discussed in this MiniReview suggest that the prevalent view of development as the mere unfolding of a genetically determined programme should be reconsidered. The contamination of our environment with endocrine disrupting chemicals is providing evidence that mammalian development is far more malleable than previously thought, as both natural and synthetic oestrogen exposure during development results in morphological and functional effects that persist into adulthood. The field of environmental endocrine disruption is poised to contribute to the understanding of the mechanisms that underlie the development of hormone-target organs. This quest will require the use of both bottom-up (from genes to organisms) and top-down approaches (from organisms to genes), as well as a new conceptual framework that would take into account the existence of emergent properties, that is, properties of the whole that cannot be explained from the properties of its components. The properties at one level of biological complexity (for instance, tissues) cannot be ascribed directly to their component parts (cells, extracellular matrix, etc.), but arise only because of the interactions among the parts. Developmental biology, guided by this integrative thinking, now has the tools to successfully revisit the old tradition of ecological regulation of development, that is, phenotype plasticity.
Acknowledgments
The authors would like to thank Cheryl Schaeberle for her help with the preparation of this manuscript. This work was supported by National Institutes of Health grants ES012301 and ES08314.
References
- 1.Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–42. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 2.Sonnenschein C, Soto AM. The Society of Cells: Cancer and Control of Cell Proliferation. Springer Verlag; New York: 1999. [Google Scholar]
- 3.Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117:1495–502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert SF. Mechanisms for the environmental regulation of gene expression: ecological aspects of animal development. J Biosci. 2005;30:65–74. doi: 10.1007/BF02705151. [DOI] [PubMed] [Google Scholar]
- 5.Colborn T, Clement C, editors. Chemically Induced Alterations in Sexual and Functional Development: The Wildlife/Human Connection. Princeton Scientific Publishing; Princeton, NJ: 1992. [Google Scholar]
- 6.Silva E, Rajapakse N, Kortenkamp A. Something from ‘nothing’ – eight weak estrogenic chemicals combined at concentrations below NOECs produce significant mixture effects. Environ Sci Technol. 2002;36:1751–6. doi: 10.1021/es0101227. [DOI] [PubMed] [Google Scholar]
- 7.Milligan SR, Khan O, Nash M. Competitive binding of xenobiotic oestrogens to rat α-fetoprotein and to sex steroid binding proteins in human and rainbow trout (Oncorhynchus mykiss) plasma. Gen Comp Endocrinol. 1998;112:89–95. doi: 10.1006/gcen.1998.7146. [DOI] [PubMed] [Google Scholar]
- 8.Wozniak AL, Bulayeva NN, Watson CS. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-α-mediated Ca++ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ Health Perspect. 2005;113:431–9. doi: 10.1289/ehp.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geck P, Maffini MV, Szelei J, Sonnenschein C, Soto AM. Androgen-induced proliferative quiescence in prostate cancer: the role of AS3 as its mediator. Proc Nat Acad Sci USA. 2000;97:10185–90. doi: 10.1073/pnas.97.18.10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vandenberg LN, Wadia PR, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. The mammary gland response to estradiol: monotonic at the cellular level, non-monotonic at the tissue-level of organization? J Steroid Biochem Mol Biol. 2006;101:263–74. doi: 10.1016/j.jsbmb.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Alworth LC, Howdeshell KL, Ruhlen RL, et al. Uterine responsiveness to estradiol and DNA methylation are altered by fetal exposure to diethylstilbestrol and methoxychlor in CD-1 mice: effects of low versus high doses. Toxicol Appl Pharmacol. 2002;183:10–22. doi: 10.1006/taap.2002.9459. [DOI] [PubMed] [Google Scholar]
- 12.Conolly RB, Lutz WK. Nonmonotonic dose-response relationships: mechanistic basis, kinetic modeling, and implications for risk assessment. Toxicol Sci. 2004;77:151–7. doi: 10.1093/toxsci/kfh007. [DOI] [PubMed] [Google Scholar]
- 13.Barker DJP, Hanson MA. Altered regional blood flow in the fetus: the origins of cardiovascular disease? Acta Paediatrica. 2004;93:1559–60. [PubMed] [Google Scholar]
- 14.Bern HA. The fragile fetus. In: Colborn T, Clement C, editors. Chemically-Induced Alterations in Sexual and Functional Development: The Wildlife/Human Connection. Princeton Scientific Publishing Co Inc.; Princeton, NJ: 1992. pp. 9–15. [Google Scholar]
- 15.Pike MC, Spicer DV, Dahmoush L, Press MF. Estrogens, progestogens, normal breast cell proliferation, and breast cancer risk. Epidemiol Rev. 1993;15:17–35. doi: 10.1093/oxfordjournals.epirev.a036102. [DOI] [PubMed] [Google Scholar]
- 16.Ekbom A, Trichopoulos D, Adami HO, Hsieh CC, Lan SJ. Evidence of prenatal influences on breast cancer risk. Lancet. 1992;340:1015–8. doi: 10.1016/0140-6736(92)93019-j. [DOI] [PubMed] [Google Scholar]
- 17.Calle EE, Mervis CA, Thun MJ, Rodriguez C, Wingo PA, Heath CWJ. Diethylstilbestrol and risk of fatal breast cancer in a prospective cohort of US women. Am J Epidemiol. 1996;144:645–52. doi: 10.1093/oxfordjournals.aje.a008976. [DOI] [PubMed] [Google Scholar]
- 18.Palmer JR, Wise LA, Hatch EE, et al. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1509–14. doi: 10.1158/1055-9965.EPI-06-0109. [DOI] [PubMed] [Google Scholar]
- 19.Hoyer AP, Grandjean P, Jorgensen T, Brock JW, Hartvig HB. Organochloride exposure and risk of breast cancer. Lancet. 1998;352:1816–20. doi: 10.1016/S0140-6736(98)04504-8. [DOI] [PubMed] [Google Scholar]
- 20.Soto AM, Fernandez MF, Luizzi MF, Oles Karasko AS, Sonnenschein C. Developing a marker of exposure to xenoestrogen mixtures in human serum. Environ Health Perspect. 1997;105:647–54. doi: 10.1289/ehp.97105s3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ibarluzea JM, Fernández MF, Santa-Marina L, et al. Breast cancer risk and the combined effect of environmental estrogens. Cancer Causes Control. 2004;15:591–600. doi: 10.1023/B:CACO.0000036167.51236.86. [DOI] [PubMed] [Google Scholar]
- 22.Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA) Reproductive Toxicology. 2007;24:139–77. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 23.Burridge E. Bisphenol A: product profile. Eur Chem News. 2003;17:14–20. [Google Scholar]
- 24.Dodds EC, Lawson W. Molecular structure in relation to oestrogenic activity. Compounds without a phenathrene nucleus. Proc Roy Soc Lond B Biol Sci. 1938;125:222–32. [Google Scholar]
- 25.Krishnan AV, Starhis P, Permuth SF, Tokes L, Feldman D. Bisphenol-A: an estrogenic substance is released from polycarbonate flasks during autoclaving. Endocrinology. 1993;132:2279–86. doi: 10.1210/endo.132.6.8504731. [DOI] [PubMed] [Google Scholar]
- 26.Ye X, Kuklenyik Z, Needham J, Calafat AM. Measuring environmental phenols and chlorinated organic chemicals in breast milk using automated on-line column-switching-high performance liquid chromatography-isotope dilution tandem mass spectrometry. J Chromatogr B. 2006;831:110–5. doi: 10.1016/j.jchromb.2005.11.050. [DOI] [PubMed] [Google Scholar]
- 27.Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect. 2002;110:A703–A7. doi: 10.1289/ehp.110-1241091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham JL. Urinary concentrations of bisphenol A and 4-Nonylphenol in a human reference population. Environ Health Perspect. 2005;113:391–5. doi: 10.1289/ehp.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arakawa C, Fujimaki K, Yoshinaga J, Imai H, Serizawa S, Shiraishi H. Daily urinary excretion of bisphenol A. Environ Health Prevent Med. 2004;9:22–6. doi: 10.1265/ehpm.9.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zalko D, Soto AM, Dolo L, et al. Biotransformations of bisphenol A in a mammalian model: answers and new questions raised by low-dose metabolic fate studies in pregnant CD-1 mice. Environ Health Perspect. 2003;111:309–19. doi: 10.1289/ehp.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson GW, Karpf ABC, Kratochwil K. Regulation of mammary gland development by tissue interaction. J Mammary Gland Biol Neoplasia. 1999;4:9–19. doi: 10.1023/a:1018748418447. [DOI] [PubMed] [Google Scholar]
- 32.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to the xenoestrogen bisphenol-A alters development of the fetal mammary gland. Endocrinology. 2007;148:116–27. doi: 10.1210/en.2006-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemmen JG, Broekhof JLM, Kuiper GGJM, Gustafsson JA, Van Der Saag PT, van der Burg B. Expression of estrogen receptor α and β during mouse embryogensis. Mech Dev. 1999;81:163–7. doi: 10.1016/s0925-4773(98)00223-8. [DOI] [PubMed] [Google Scholar]
- 34.Narbaitz R, Stumpf WE, Sar M. Estrogen receptors in mammary gland primordia of fetal mouse. Anat Embryol. 1980;158:161–6. doi: 10.1007/BF00315903. [DOI] [PubMed] [Google Scholar]
- 35.Saji S, Jensen EV, Nilsson S, Rylander T, Warner M, Gustafsson J-A. Estrogen receptors α and β in the rodent mammary gland. Proc Nat Acad Sci USA. 2000;97:337–42. doi: 10.1073/pnas.97.1.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richert MM, Schwertfeger KL, Ryder JW, Anderson SM. An atlas of mouse mammary gland development. J Mammary Gland Biol Neoplasia. 2000;5:227–41. doi: 10.1023/a:1026499523505. [DOI] [PubMed] [Google Scholar]
- 37.Humphreys RC, Krajewska M, Krnacik S, et al. Apoptosis in the terminal end bud of the murine mammary gland: a mechanism of ductal morphogenesis. Development. 1996;122:4013–22. doi: 10.1242/dev.122.12.4013. [DOI] [PubMed] [Google Scholar]
- 38.Markey CM, Luque EH, Munoz de Toro MM, Sonnenschein C, Soto AM. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod. 2001;65:1215–23. doi: 10.1093/biolreprod/65.4.1215. [DOI] [PubMed] [Google Scholar]
- 39.Munoz de Toro MM, Markey CM, Wadia PR, et al. Perinatal exposure to bisphenol A alters peripubertal mammary gland development in mice. Endocrinology. 2005;146:4138–47. doi: 10.1210/en.2005-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCormack VA, Dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:1159–69. doi: 10.1158/1055-9965.EPI-06-0034. [DOI] [PubMed] [Google Scholar]
- 41.Singh M, McGinley JN, Thompson HJ. A comparison of the histopathology of premalignant and malignant mammary gland lesions induced in sexually immature rats with those occurring in the human. Lab Invest. 2000;80:221–31. doi: 10.1038/labinvest.3780025. [DOI] [PubMed] [Google Scholar]
- 42.Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal Bisphenol A exposure. Reprod Toxicol. 2006;23:383–90. doi: 10.1016/j.reprotox.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haslam SZ, Bern HA. Histopathogenesis of 7,12-diemthyl-benz(a)anthracene-induced rat mammary tumors. Proc Natl Acad Sci USA. 1977;74:4020–4. doi: 10.1073/pnas.74.9.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durando M, Kass L, Piva J, et al. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect. 2007;115:80–6. doi: 10.1289/ehp.9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma R, Sassoon DA. PCBs exert an estrogenic effect through repression of the Wnt7a signaling pathway in the female reproductive tract. Environ Health Perspect. 2006;114:898–904. doi: 10.1289/ehp.8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma L, Benson GV, Lim H, Dey SK, Maas RL. Abdominal B (AbdB) Hoxa genes: regulation in adult uterus by estrogen and progesterone and repression in mullerian duct by the synthetic estrogen diethylstilbestrol (DES) Dev Biol. 1998;197:141–54. doi: 10.1006/dbio.1998.8907. [DOI] [PubMed] [Google Scholar]
- 47.Block K, Kardana A, Igarashi P, Taylor HS. In utero diethyl-stilbestrol (DES) exposure alters Hox gene expression in the developing müllerian system. FASEB J. 2000;14:1101–8. doi: 10.1096/fasebj.14.9.1101. [DOI] [PubMed] [Google Scholar]
- 48.Veltmaat JM, Mailleux AA, Thiery JP, Bellusci S. Mouse embryonic mammogenesis as a model for the molecular regulation of pattern formation. Differentiation. 2003;71:1–17. doi: 10.1046/j.1432-0436.2003.700601.x. [DOI] [PubMed] [Google Scholar]