

Abstract
Mutations in the preproinsulin protein that affect processing of preproinsulin to proinsulin or lead to misfolding of proinsulin are associated with diabetes. We examined the subcellular localization and secretion of 13 neonatal diabetes-associated human proinsulin proteins (A24D, G32R, G32S, L35P, C43G, G47V, F48C, G84R, R89C, G90C, C96Y, S101C and Y108C) in rat INS-1 insulinoma cells. These mutant proinsulin proteins accumulate in the endoplasmic reticulum (ER) and are poorly secreted except for G84R and in contrast to wild-type and hyperproinsulinemia-associated mutant proteins (H34D and R89H) which were sorted to secretory granules and efficiently secreted. We also examined the effect of C96Y mutant proinsulin on the synthesis and secretion of wild-type insulin and observed a dominant-negative effect of the mutant proinsulin on the synthesis and secretion of wild-type insulin due to induction of the unfolded protein response and resulting attenuation of overall translation.
Keywords: Insulin, Diabetes, Misfolded protein, Unfolded protein response
INTRODUCTION
Recent studies have shown that mutations in the preproinsulin molecule can cause diabetes with onset at birth or shortly thereafter as well as later in life including adulthood [1–8]. They are also the cause of diabetes in the Akita and Munich mouse models of diabetes [9,10]. The diabetes-associated mutations are located in critical regions of the preproinsulin molecule and prevent normal processing of preproinsulin to proinsulin as well as folding and subsequent processing of proinsulin to insulin. They differ from the mutations in insulin and proinsulin associated with familial hyperinsulinemia and familial hyperproinsulinemia which result in the synthesis of an insulin molecule with reduced biological activity or a proinsulin molecule that cannot be processed to insulin [11]. The mutations associated with familial hyperinsulinemia and familial hyperproinsulinemia rarely cause diabetes but rather because of their reduced biological activity lead to accumulation of the mutant proteins in the serum.
Here, we examine the subcellular localization and secretion of 13 neonatal diabetes-associated mutant preproinsulin molecules (A24D, G32R, G32S, L35P, C43G, G47V, F48C, G84R, R89C, G90C, C96Y, S101C and Y108C) and compare the results with wild-type preproinsulin and mutant preproinsulin proteins associated with hyperproinsulinemia (H34D and R89H).
Material and methods
Human insulin cDNA constructs
The human insulin cDNA [12] was cloned in pcDNA3.1(+). Mutations were introduced by overlap extension PCR mutagenesis and the sequence confirmed by DNA sequencing.
Cell culture, measurement of RNA levels and Western blotting
These methods are described in the Supplementary Material.
Subcellular localization of wild-type and mutant human proinsulin
INS-1 (1 × 106), HEK 293 (1 × 105) and AtT20 (1 × 105) cells cultured on glass cover slips in 6 well plates were transfected with 3 µg of pcDNA3.1 (vector), WT or mutant human preproinsulin cDNA together with 0.5 µg of constructs expressing ER-localized monomeric red fluorescent protein (ER-mRFP) [13] or pECFP-Golgi (Clontech, Mountain View, CA), an enhanced cyan fluorescent protein (ECFP) marker for the Golgi apparatus. The cells were cultured for 24 h, fixed with 4% paraformaldehyde for 20 min and then washed with PBS. The fixed cells were incubated with mouse monoclonal anti-human proinsulin C-peptide antibody (Millipore, St. Charles, MO; CBL94) at 1/1,000 dilution in PBS for 1 h at room temperature) or guinea pig polyclonal anti-human insulin antibody (Millipore; 1/1,000 dilution). The antigen was visualized using an anti-mouse secondary antibody conjugated with Alexa Fluor 488 (Invitrogen, Carlsbad, CA; A21202; 1/500 dilution) for human C-peptide or Cy™2-conjugated AffiniPure donkey anti-guinea pig IgG (Jackson ImmunoResearch, West Grove, PA; 1:300 dilution) for human insulin. After immunostaining, the cells were treated with Vectashield® mounting medium with DAPI (Vector, Burlingame, CA) and examined using an Olympus IX80 DSU Spinning Disk confocal microscope with excitation/emission wavelengths of 490/525 for Alexa Fluor 488, 490/528 for Cy2, 565/620 for RFP, 436/470 for ECFP and 350/457 for DAPI. We analyzed overlap of C-peptide or insulin and ER markers using Manders’ overlap coefficient with ImageJ Software (NIH, Bethesda, MD) and the JACoP plug-in [14].
Measurement of C-peptide, proinsulin and insulin in media and cell lysates
Twenty-four hours post-transfection, cells were washed with PBS and then cultured for 4 h in the media described above but with 0.5% bovine serum albumin instead of 10% FBS. The media was collected and the cells were harvested. The cells were washed with PBS and harvested with a cell scraper in lysis buffer at 4° C (50 mM HEPES, 1% Triton X-100 and Complete Protease Inhibitor Cocktail (Roche). The lysate was sonicated for 10 sec (output 2 with microprobe) using a Sonicator W-380 (Heat Systems-Ultrasonics) and then centrifuged at 12,000 rpm for 20 min at 4 °C in an Eppendorf 5415D centrifuge. The supernatant was removed and assayed for human C-peptide (Immulite 2000 C-peptide; Siemens, Los Angeles, CA) and human proinsulin (Human Total Proinsulin ELISA Kit, Millipore, St. Louis, MO; EZHPI-15K).
Statistical analysis
Data are presented as mean ± SEM. Differences between groups were assessed using an unpaired t-test. P < 0.05 was considered significant.
Results
Transient expression of wild-type and mutant human proinsulin in cultured cells
Previous studies indicated that transient expression of mutant proinsulin proteins in INS-1 or MIN6 insulinoma cells can lead to cell death in 48–96 h [5,15,16]. In order to avoid possible confounding effects of ongoing apoptosis, we examined the time-course of expression of WT and mutant (H34D, G84R and C96Y) proinsulin mRNAs in INS-1 cells. The time-course was similar for each with insulin mRNA levels peaking at 24 h and then declining (Supplementary data Fig. 1). Thus, we studied the cells 24 h post-transfection.
Western blotting of INS-1 cell extracts using a human C-peptide antibody showed a band with the mobility of the recombinant human proinsulin standard in cells expressing WT and mutant proinsulin proteins, except for the signal peptide cleavage site mutant proinsulin A24D (Fig. 1). In cells expressing A24D proinsulin, the prominent band was of a larger size consistent with a lack of cleavage of the signal peptide. There was also a second/minor band with the mobility of proinsulin representing less than 5% of the total immunoreactive protein suggesting that INS-1 cells were able to cleave the mutant signal peptide, albeit not efficiently. The site of this cleavage is presently unknown. The human C-peptide antibody used in these studies recognized all the mutant proinsulin proteins except G84R proinsulin. This mutation is located in the C-peptide four residues from the C-terminus and may disrupt the epitope recognized by this particular human C-peptide antibody (Gly does not have a side chain in contrast to Arg and the presence of the side chain of Arg may block binding of the C-peptide antibody by steric hindrance).
Fig. 1.

Expression of human proinsulin in INS-1 cells. Cells were transfected with vector (pcDNA3.1) or WT and mutant human proinsulin cDNAs. Cell lysates were prepared 24 h post-transfection and 15 µg of total protein separated by 15% SDS-PAGE and transferred to a PVDF membrane. The membrane was probed with an anti-human proinsulin C-peptide antibody. The lane labeled “Proinsulin” is the recombinant human proinsulin standard.
Subcellular localization of wild-type and mutant proinsulin
We examined the subcellular localization of the WT and mutant proteins in INS-1 and AtT20 cells by immunohistochemistry using an antibody to human C-peptide to identify human preproinsulin, proinsulin or C-peptide (Fig. 2). This antibody does not cross-react with rodent C-peptideand thus allows us to study the biosynthesis and processing of human proinsulin in rat INS-1 insulinoma cells. The cells were co-transfected with constructs encoding ER-localized RFP that allowed us to visualize the ER and Golgi-localized ECFP that allowed us to visualize the Golgi apparatus.
Fig. 2.
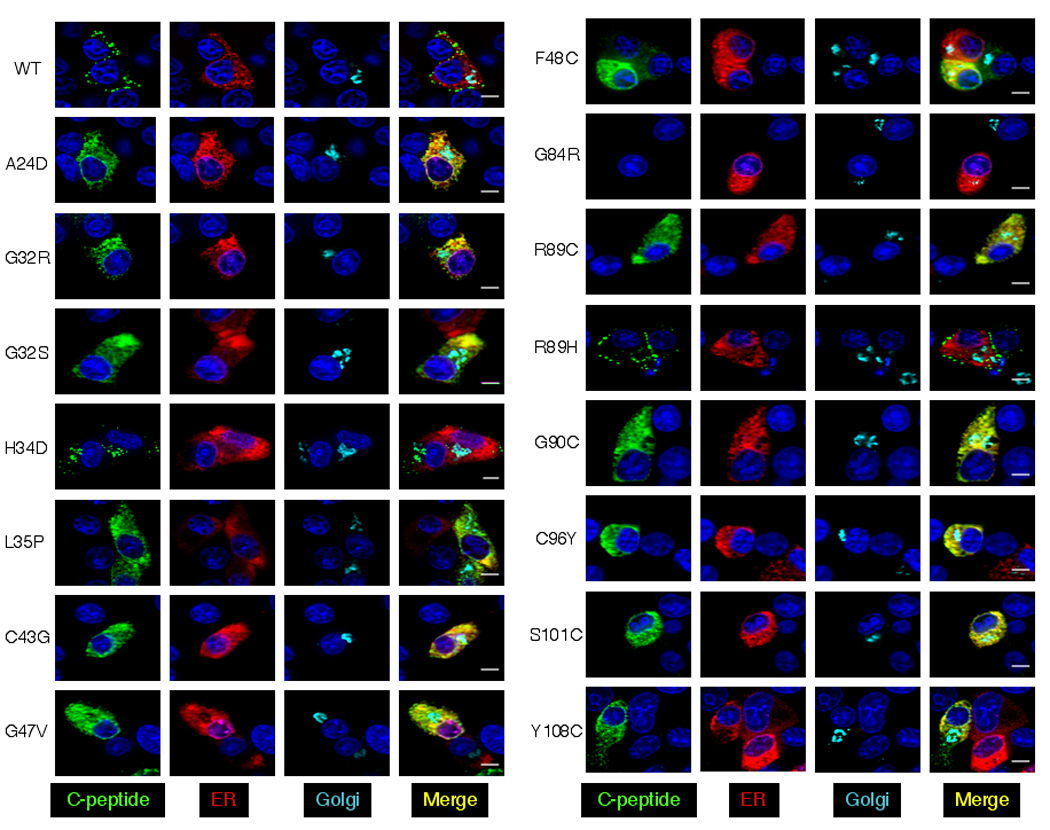

Subcellular localization of wild-type and mutant proinsulin. A. INS-1 cells. B. AtT20 cells. The localization of human C-peptide (INS-1) or human insulin (AtT20) immunoreactive protein is shown in green, ER is shown in red and the Golgi marker in cyan. Nuclei were stained with DAPI (blue). Data shown are representative of three independent experiments. Scale bar: 5 µm.
The staining in cells expressing WT proinsulin and the hyperproinsulinemia mutation R89H showed a punctate pattern at the periphery of the cell suggesting that the protein is localized in secretory granules (Fig. 2A). The hyperproinsulinemia mutation H34D showed a unique pattern with C-peptide staining of secretory granules at the periphery of the cell, as well as perinuclear Golgi staining. The H34D mutation affects hexamer formation and sorting into dense core granules of the regulated secretory pathway [17]. This proinsulin also has 4–5-fold higher receptor binding activity than WT and may bind to insulin receptors in the trans-Golgi and be secreted constitutively or degraded in lysosomes [17,18].
Cells expressing mutant proinsulin proteins associated with diabetes (A24D, G32R, G32S, L35P, C43G, G47V, F48C, R89C, G90C, C96Y, S101C and Y108C) show C-peptide immunostaining that is localized within an expanded ER. The Golgi in these cells appears very similar in size and shape to that in cells expressing WT proinsulin. The G32R and Y108C mutant proteins show some localization to secretory granules suggesting they can enter the secretory pathway although not efficiently.
The human C-peptide antibody did not recognize the diabetes-associated G84R mutant protein in INS-1 cells (Fig. 2A). Since human insulin antibodies cross-react with rat insulin, we cannot use human insulin antibodies to study the localization of G84R proinsulin/insulin in INS-1 cells. We thus studied the subcellular localization of WT and G84R proinsulin in AtT20 cells using a human insulin antibody (Fig. 2B). The pattern of human insulin staining in G84R proinsulin expressing cells indicates that G84R proinsulin is able to exit the ER and enter the secretory pathway in contrast to the other diabetes-associated mutant proinsulin proteins.
We quantified the overlap between C-peptide (or insulin) and ER-marker staining using Manders’ correlation coefficient to estimate the extent of colocalization of C-peptide and ER [14] (Supplementary data Table 1). There was no significant difference in the overlap between C-peptide and ER between WT and H34D or R89H expressing cells. However, there is increased overlap in cells expressing the diabetes-associated mutations A24D, G32R, G32S, L35P, C43G, G47V, F48C, R89C, G90C, C96Y, S101C and Y108C indicating that these mutant proteins are retained in the ER. We carried out similar analyses comparing the overlap between WT and G84R proinsulin in AtT20 cells but using insulin staining to localize proinsulin/insulin. There was no significant difference in the overlap between insulin and ER in these cells suggesting that the G84R mutant protein can exit the ER.
Secretion of wild-type and mutant proinsulin
We examined the secretion of WT and mutant proteins by static incubation (4 h) using a human C-peptide Immunoassay (Immulite 2000 C-peptide, Siemens) to measure the amount of C-peptide immunoreactive material in the media and cell extracts (intracellular). There were readily detectable levels of C-peptide immunoreactive protein in cell extracts of INS-1 cells expressing WT and mutant proinsulin proteins (Table 1). There were, however, differences in immunoreactive C-peptide levels between the WT and various mutant proteins. It is unknown if these are due to differences in cross-reactivity with the C-peptide antibody or to other mechanism(s) (e.g. the specific effect of the mutant protein on translation or other mechanisms).
Table 1.
Total C-peptide and proinsulin immunoreactive material in cell extracts and media of INS-1 cells expressing wild-type and mutant proinsulin/insulin proteins (4 h static incubation)
| Mutation | Intracellular | Media | % of total in media |
|---|---|---|---|
| pcDNA3 | ND | ND | - |
| WT | 0.87 ± 0.01 | 0.25 ± 0.002 | 22 ± 0.3 |
| A24D | 0.27 ± 0.01 | ND | - |
| G32R | 0.36 ± 0.01 | 0.07 ± 0.002 | 16 ± 0.8 |
| G32S | 0.34 ± 0.004 | ND | - |
| H34D | 0.24 ± 0.01 | 0.17 ± 0.001 | 42 ± 0.6 |
| L35P | 0.24 ± 0.002 | ND | - |
| C43G | 0.07 ± 0.003 | ND | - |
| G47V | 0.44 ± 0.03 | ND | - |
| F48C | 0.15 ± 0.004 | ND | - |
| G84R | 0.85 ± 0.03 | 0.17 ± 0.003 | 17 ± 0.6 |
| R89C | 0.23 ± 0.000 | ND | - |
| R89H | 1.52 ± 0.06 | 0.5 ± 0.05 | 25 ± 1 |
| G90C | 0.32 ± 0.001 | ND | - |
| C96Y | 0.15 ± 0.001 | ND | - |
| S101C | 0.39 ± 0.01 | ND | - |
| Y108C | 0.33 ± 0.004 | 0.09 ± 0.001 | 21 ± 0.01 |
ND: Not-detectable; i.e. C-peptide in sample < 33.1 pmol/L (< 0.0661 pmol of total C-peptide)
We observed efficient secretion of WT proinsulin/C-peptide and the hyperproinsulinemia mutant proteins H34D and R89H as well as G84R proinsulin from INS-1 cells (Table 1). The diabetes-associated mutant proinsulins were poorly secreted. We detected low levels of secretion of G32R and Y108C consistent with the localization of some C-peptide immunoreactivity in secretory granule-like structures in these cells. The amounts of C-peptide secreted by the other diabetes-associated mutant proinsulin proteins in INS-1 cells were below the sensitivity of the assay. We also examined secretion by transfected HEK 293 with quantitatively similar results (Supplementary data Table 2). Thus, secretion of the diabetes-associated mutant proteins is impaired whether by the regulated (INS-1 cells) or constitutive pathway (HEK 293 cells).
Effect of C96Y proinsulin on expression and secretion of wild-type proinsulin
We examined the effect of co-expression of increasing amounts of C96Y proinsulin cDNA (0–1.5 µg) with a constant amount of WT proinsulin cDNA (1 µg) on C-peptide immunoreactivity in cell extracts and media. There was a linear increase in human proinsulin mRNA levels with increasing amounts of cDNA input (WT and C96Y) (Fig. 3A).
Fig. 3.

Dominant-negative effect of C96Y proinsulin on wild-type proinsulin biosynthesis. INS-1 cells were transfected with 1 µg of WT insulin cDNA and increasing amounts of C96Y insulin cDNA (0, 0.5, 1.0 and 1.5 µg). Human insulin mRNA and C-peptide levels and rat total and phospho-eIF2a levels were determined 24 h post-transfection by real-time RT-PCR, immunoassay and Western blotting (15 µg of cell lysate), respectively. The results are the mean ± SEM of three independent experiments. A. Relative levels of human mRNA level (normalized to rat TBP mRNA). B. Total human C-peptide levels in cell extracts and media. The C-peptide levels are significantly decreased (P < 0.01) in cells transfected with C96Y insulin cDNA compared to cells transfected with 1 µg of WT insulin cDNA. C. Induction of phospho-eIF2α in cells transfected with pcDNA3.1 (2 µg), WT (2 µg) or C96Y (1 µg) and WT (1 µg) cDNAs. D. Fold induction of phospho-eIF2α.
The levels of C-peptide immunoreactivity in the cell extracts (representing both WT proinsulin/C-peptide and C96Y proinsulin) and media (WT only since as described above C96Y proinsulin is not secreted) were significantly decreased with increasing C96Y proinsulin cDNA input (Fig. 3B). This was accompanied by increased levels of phospho-eIF2α suggesting that the cells co-expressing C96Y proinsulin had activated the unfolded protein response (UPR) leading to attenuation of translation (Fig. 3C and D) [20]. This effect on levels of C-peptide immunoreactivity was not observed when we added increasing amounts of WT proinsulin cDNA (Fig. 3B).
Discussion
In these studies, we have followed the lead of others who have shown that WT and mutant forms of human proinsulin can be studied using in vitro cell culture systems, including cell lines that do not normally synthesize insulin such as HEK 293 and AtT20 cells as well as those that do, such as INS-1 or other insulinoma cell lines [5,15,21–28]. There are advantages to studying heterologous expression of human proinsulin in multiple types of cells as parallel studies allow comparison of secretion via constitutive (HEK 293 cells) and regulated pathways (INS-1 cells). We chose to express “native” human preproinsulin in these systems. Others have used fluorescent protein (GFP, Venus, mCherry)-tagged preproinsulin, either integrated within the C-peptide region of the proinsulin molecule [5,21] or appended to the C-terminus [15,29,30] to monitor proinsulin and insulin biosynthesis and secretion in vitro. We chose not to do so because of the possibility that the presence of the GFP moiety might perturb the folding of the mutant proinsulins or conversely that GFP folding and fluorescence could be negatively affected by proinsulin folding [31]. Comparison of the results of studies using fluorescent protein-tagged preproinsulin [5,21,32] or native preproinsulin showed no apparent differences, at least with respect to the mutations examined to date.
Mutations in the preproinsulin protein that affect processing to proinsulin or normal folding of proinsulin result in retention of the mutant protein in the ER and impaired secretion in agreement with studies of others [5,9,21,32]. The severity of the effect of individual mutations on folding is reflected in the amount of C-peptide reactive material found within the cells (see Table 1); e.g. the C43G mutant which cannot form the B19-A20 disulfide bond which is the first to form and may serve as a template for further folding shows the greatest depression in intracellular levels [33]. The mutant protein also leads to attenuation of synthesis and secretion of the WT protein (and presumably other cellular proteins) (Fig. 3). Thus, the misfolded proinsulin protein compromises beta-cell function, decreases insulin secretion and sets in place a pathological process whereby the functionally beta-cells are stimulated to produce more insulin which ultimately leads to beta-cell failure and diabetes.
The results presented here suggest we will be able to classify diabetes-associated mutations based on their effect on proinsulin biosynthesis. For example, the proinsulin mutations (G32R and Y108C) result in the synthesis of a protein that can be targeted to the secretory pathway and secreted although inefficiently. If the efficiency of this process could be improved, it may be possible to delay or prevent the development of diabetes in patients with these mutations. The preproinsulin mutant protein A24D may constitute another class and it seems likely that if the efficiency of cleavage of the signal peptide from this protein could be increased that it would enter the secretory pathway and be secreted. Further studies of other mutant insulin proteins may reveal yet other classes.
The results of the studies of the G84R mutant protein suggest that it is not retained in the ER as are the other diabetes-associated mutations and that it can exit the ER and be sorted to secretory granules. This mutation was identified in a Vietnamese patient diagnosed with diabetes on the first day of life [2]. In addition to diabetes, he has developmental delay and anal atresia. The non-diabetic mother does not carry the mutation and the father is not available for testing. The functional studies presented here suggest that the G84R mutation may not affect proinsulin biosynthesis and thus may not be the cause of this patient’s diabetes.
Continued studies of mutant insulin proteins that cause diabetes could identify the specific proteins and pathways that allow the beta-cell to maintain ER homeostasis and lead to new approaches for preserving beta-cell function in patients expressing a mutant insulin as well as in those that have type 2 diabetes.
Supplementary Material
Acknowledgments
We thank Dr. Vytas Bindokas for his kind advice and assistance with the colocalization studies. These studies were supported in part by U.S. Public Health Service Grants DK-13914 and DK-20595 (The University of Chicago Diabetes Research and Training Center; Diabetes Centers R24 Seeding Collaborative Interdisciplinary Team Science in Diabetes, Endocrinology and Metabolic Diseases) and a gift from the Kovler Family Foundation.
Appendix A. Supplementary Material
Supplementary Material associated with this article can be found, in the online version, at [insert information].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Støy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, Lipton RB, Greeley SA, Patch AM, Ellard S, Steiner DF, Hattersley AT, Philipson LH, Bell GI. Neonatal Diabetes International Collaborative Group, Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. USA. 2007;104:15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, MacDonald MJ, Støy J, Steiner DF, Philipson LH, Bell GI, Neonatal Diabetes International Collaborative Group. Hattersley AT, Ellard S. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57:1034–1042. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polak M, Dechaume A, Cavé H, Nimri R, Crosnier H, Sulmont V, de Kerdanet M, Scharfmann R, Lebenthal Y, Froguel P, Vaxillaire M. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy. A report from the French ND (neonatal diabetes) study group. Diabetes. 2008;57:1115–1119. doi: 10.2337/db07-1358. [DOI] [PubMed] [Google Scholar]
- 4.Molven A, Ringdal M, Nordbø AM, Ræder H, Støy J, Lipkind GM, Steiner DF, Philipson LH, Bergmann I, Aarskog D, Undlien DE, Joner G, Søvik O, the Norwegian Childhood Diabetes Study Group. Bell GI, Njølstad PR. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes. 2008;57:1131–1135. doi: 10.2337/db07-1467. [DOI] [PubMed] [Google Scholar]
- 5.Colombo C, Porzio O, Liu M, Massa O, Vasta M, Salardi S, Beccaria L, Monciotti C, Toni S, Pedersen O, Hansen T, Federici L, Pesavento R, Cadario F, Federici G, Ghirri P, Arvan P, Iafusco D, Barbetti F and the Early Onset Diabetes Study Group of the Italian Society of Pediatric Endocrinology and Diabetes (SIEDP) Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Invest. 2008;118:2148–2156. doi: 10.1172/JCI33777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahamed A, Unnikrishnan AG, Pendsey SS, Nampoothiri S, Bhavani V, Praveen VP, Kumar H, Jayakumar RV, Nair V, Ellard S, Edghill E. Permanent neonatal diabetes mellitus due to a C96Y heterozygous mutation in the insulin gene: a case report. J. Pancreas. 2008;9:715–718. [PubMed] [Google Scholar]
- 7.Bonfanti R, Colombo C, Nocerino V, Massa O, Lampasona V, Iafusco D, Viscardi M, Chiumello G, Meschi F, Barbetti F. Insulin gene mutations as cause of diabetes in children negative for five type 1 diabetes autoantibodies. Diabetes Care. 2009;32:123–125. doi: 10.2337/dc08-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubio-Cabezas O, Edghill E, Argente J, Hattersley AT. Testing for monogenic diabetes among children with antibody-negative clinically defined type 1 diabetes. Diabet. Med. 2009;26:1070–1074. doi: 10.1111/j.1464-5491.2009.02812.x. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Takeuchi T, Tanaka S, Kubo S-K, Kayo T, Lu D, Takata K, Koizumi A, Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herbach N, Rathkolb B, Kemter E, Pichl L, Klaften M, Hrabéde Andelis M, Halban PA, Wolf E, Aigner B, Wanke R. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe beta-cell loss in Munich Ins2C95Smutant mice. Diabetes. 2007;56:1268–1276. doi: 10.2337/db06-0658. [DOI] [PubMed] [Google Scholar]
- 11.Steiner DF, Tager HS, Nanjo K, Chan SJ, Rubenstein AH. Familial syndromes of hyperproinsulinemia and hyperinsulinemia with mild diabetes. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic Basis of Inherited Disease. 7th ed. New York: McGraw-Hill; 1995. pp. 897–904. [Google Scholar]
- 12.Bell GI, Swain WF, Pictet R, Cordell B, Goodman HM, Rutter WJ. Nucleotide sequence of a cDNA clone encoding human preproinsulin. Nature. 1979;282:525–527. doi: 10.1038/282525a0. [DOI] [PubMed] [Google Scholar]
- 13.Snapp EL, Sharma A, Lippincott-Schwartz J, Hegde RS. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. USA. 2006;103:6536–6541. doi: 10.1073/pnas.0510657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J. Microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 15.Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nozaki J, Kubota H, Yoshida H, Naitoh M, Goji J, Yoshinago T, Mori K, Koizumi A, Nagata K. The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/Akita pancreatic beta cells. Genes Cells. 2004;9:261–270. doi: 10.1111/j.1356-9597.2004.00721.x. [DOI] [PubMed] [Google Scholar]
- 17.Carroll RJ, Hammer RE, Chan SJ, Swift HN, Rubenstein AH, Steiner DF. A mutant human proinsulin is secreted from islets of Langerhans in increased amounts via an unregulated pathway. Proc. Natl. Acad. Sci. USA. 1988;85:8943–8947. doi: 10.1073/pnas.85.23.8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steiner DF, Tager HS, Chan SJ, Nanjo K, Sanke T, Rubenstein AH. Lessons learned from molecular biology of insulin-gene mutations. Diabetes Care. 1990;13:600–609. doi: 10.2337/diacare.13.6.600. [DOI] [PubMed] [Google Scholar]
- 19.Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA, Takeuchi T. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes. 2003;52:409–416. doi: 10.2337/diabetes.52.2.409. [DOI] [PubMed] [Google Scholar]
- 20.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endo. Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci USA. 2007;104:15841–15846. doi: 10.1073/pnas.0702697104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore HP, Walker MD, Lee F, Kelly RB. Expressing a human proinsulin cDNA in a mouse ACTH-secreting cell. Intracellular storage, proteolytic processing, and secretion on stimulation. Cell. 1983;35:531–538. doi: 10.1016/0092-8674(83)90187-3. [DOI] [PubMed] [Google Scholar]
- 23.Gross DJ, Halban PA, Kahn CR, Weir GC, Villa-Komaroff L. Partial diversion of a mutant proinsulin (B10 aspartic acid) from the regulated to the constitutive secretory pathway in transfected AtT-20 cells. Proc. Natl. Acad. Sci. USA. 1989;86:4107–4111. doi: 10.1073/pnas.86.11.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferber S, Gross DJ, Villa-Komaroff L, Danehy F, Vollenweider F, Meyer K, Loeken MR, Kahn CR, Halban PA. Heterogeneity of expression and secretion of native and mutant [AspB10] insulin in AtT20 cells. Mol. Endo. 1991;5:319–326. doi: 10.1210/mend-5-3-319. [DOI] [PubMed] [Google Scholar]
- 25.Zhu YL, Abdo A, Gesmonde JF, Zawalich KC, Zawalich W, Dannies PS. Aggregation and lack of secretion of most newly synthesized proinsulin in non-beta-cell lines. Endocrinology. 2004;145:3840–3849. doi: 10.1210/en.2003-1512. [DOI] [PubMed] [Google Scholar]
- 26.Liu M, Li Y, Cavener D, Arvan P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J. Biol. Chem. 2005;280:13209–13212. doi: 10.1074/jbc.C400475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hua QX, Liu M, Hu SQ, Jia W, Arvan P, Weiss MA. A conserved histidine in insulin is required for the foldability of human proinsulin: structure and function of an ALAB5 analog. J. Biol. Chem. 2006;281:24889–24899. doi: 10.1074/jbc.M602617200. [DOI] [PubMed] [Google Scholar]
- 28.Fan JY, Roth J, Zuber C. Expression of mutant Ins2C96Y results in enhanced tubule formation causing enlargement of pre-Golgi intermediates in CHO cells. Histochem. Cell Biol. 2007;128:161–173. doi: 10.1007/s00418-007-0304-8. [DOI] [PubMed] [Google Scholar]
- 29.Ohara-Imaizumi M, Nakamichi Y, Tanaka T, Ishida H, Nagamatsu S. Imaging exocytosis of single insulin secretory granules with evanescent wave microscopy: distinct behavior of granule motion in biphasic insulin release. J. Biol. Chem. 2002;277:3805–3808. doi: 10.1074/jbc.C100712200. [DOI] [PubMed] [Google Scholar]
- 30.Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by camp. Proc. Natl. Acad. Sci. USA. 2007;104:19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 32.Rajan S, Eames SC, Park SY, Labno C, Bell GI, Prince V, Philipson LH. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am. J. Physiol. Endocrinol Metab. 2009 doi: 10.1152/ajpendo.00592.2009. Dec 1. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weiss MA. Proinsulin and the genetics of diabetes mellitus. J. Biol. Chem. 2009;284:19159–19163. doi: 10.1074/jbc.R109.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.