

Abstract
In order to discover non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs) that are effective against both wild-type (WT) virus and variants that encode the clinically troublesome Tyr181Cys (Y181C) RT mutation, virtual screening by docking was carried out using three RT structures and more than 2 million commercially available compounds. Two of the structures are for WT-virus with different conformations of Tyr181, while the third structure incorporates the Y181C modification. Eventually nine compounds were purchased and assayed. Three of the compounds show low-micromolar anti-viral activity towards either or both the wild-type and Y181C HIV-1 strains. The study illustrates a viable protocol to seek anti-HIV agents with enhanced resistance profiles.
Keywords: HIV reverse transcriptase, virtual screening, consensus docking, Glide, Tyrosine 181
Introduction
Since the recognition of AIDS in 1981 the associated pandemic has claimed more than 25 million lives, and ca. 33 million people are currently infected with the causative agent, human immunodeficiency virus (HIV).1 AZT, which was introduced in 1987, was the first drug to show some success in combating HIV infection, though significant chemotherapeutic progress was not achieved until 1996 with the advent of triple combination therapy, HAART. Though further improvements have made the dosing more tolerable, HAART is only palliative, it is not globally available, serious side-effects are common, and, owing to the HIV’s high mutation rate, resistance to any drug regimen can be expected in time. Coupled with the lack of success in vaccine development,1 it is essential to seek new anti-HIV agents that feature efficacy against a broad spectrum of HIV variants, low cost, easy storage and administration, and reduced side effects.
Though inhibition of multiple HIV proteins is being pursued for suppression of viral replication, HIV reverse transcriptase (RT) has been the key target.2 The enzyme converts single-stranded viral RNA into double-stranded DNA, which is incorporated into the host cell’s genome by HIV integrase. The nucleoside class of RT inhibitors (NRTIs) including AZT are faulty substrates whose incorporation into the product DNA causes premature strand termination, while the non-nucleoside RT inhibitors (NNRTIs) are true inhibitors that bind to an allosteric site ca. 10-Å away from the polymerase active site.2 Notably, the widely prescribed Atripla is a once-a-day coformulation of two NRTIs, tenofovir and emtricitabine, and the NNRTI efavirenz.2
Our own anti-HIV efforts have been directed at discovery of new NNRTIs.3,4 Numerous compounds in multiple series have been found that are both potent against the wild-type (WT) virus and that have auspicious computed pharmacological properties. However, improvement in the performance against some common HIV strains that encode RT mutations is needed. In particular, though some of the NNRTIs are effective against the troublesome Lys103Asn (K103N) variant, the Tyr181Cys (Y181C) mutation has been consistently problematic.3,4 This RT modification arises early upon administration of most NNRTIs and confers resistance to many of them including nevirapine, the first approved drug in the class.2 Consequently, as reported here, virtual screening has been performed to seek leads that show activity against both wild-type HIV as well as a variant that encodes the Y181C mutation. More than two million compounds were screened in the docking calculations using three crystal structures of RT, a conventional WT structure, one with an alternative conformation for Y181, and a structure of Y181C RT. Though only nine compounds were ultimately purchased and assayed, one compound shows low-micromolar activity against both viral strains, while two others show similar activity against either the WT or Y181C strain. The study illustrates a viable protocol to seek leads that are active against viral mutants, and it has provided specific lead series that are being optimized with expectations for enhanced resistance profiles.
Structural Background
In apo HIV-RT structures such as 1rtj, shown in white in Figure 1, the side chains of both Tyr181 and Tyr188 are pointed “down” toward the assumed entrance of the NNRTI binding site.5 However, in the more than seventy reported crystal structures of HIV-RT in complex with NNRTIs, almost all have Tyr181 and Tyr188 rotated “up”, lining the π-rich binding pocket along with Trp229 and Phe227. Typically, the aromatic side chains of the two tyrosines and tryptophan engage in aryl-aryl interactions with the NNRTIs. At the time of this study there were also six crystal structures of RT-NNRTI complexes (1fko, 1rt3, 1rti, 1tv6, 2b5j, and 2be2) in the Protein Data Bank showing Tyr181 in the down orientation as for the apo enzyme.6-10 The structures were obtained from three different laboratories and they were co-crystallized with different ligands. The alternative conformations for Tyr181 and Tyr188 are illustrated in Figure 1.
Figure 1.

Superposition of crystal structures illustrating the alternative orientations of Tyr181 and Tyr188 in the NNRTI binding site. 1rt4 in blue represents a conventional structure for a RT complex, while 2be2 in magenta illustrates the rare alternative with Y181 in the “down” conformation, as found in apo structures such as for 1rtj in white. The ligands in the 1rt4 and 2be2 structures are not shown.
It was originally thought that the Y181-down orientation was associated with weak inhibitors;11 however, our interest in this binding mode was reignited in 2005 by the report of the 2be2 structure and its ligand, R221239, which is a very potent NNRTI against both WT HIV (IC50 = 2 nM) and a Tyr181Cys variant (IC50 = 5 nM).9 A possible complication here was that the Y181-down orientation can also arise for complexes when the crystallization is performed at low pH, as in the 1fko case.12 However, examination of the experimental details indicated that this was not an issue for at least the 1tv6, 2b5j and 2be2 structures, which involved crystallizations near pH 7. Thus, the 2be2 RT structure emerged as a desirable one for virtual screening. In particular, Tyr181 does not appear to be interacting significantly with the ligand in the 2be2 structure (Figure 2); the aryl-aryl contacts are between the 3,5-dimethylphenoxy group of the ligand and Tyr188 and Trp229. Consequently, mutation to Cys181 is expected to be relatively benign. So, the idea was that other ligands that are well-accommodated in the 2be2 structure and that avoid contact with Tyr181 might also be effective towards Y181C variants.
Figure 2.

Superimposed NNRTI binding sites and ligands from the three crystal structures. 1rt4 is displayed in blue, 2be2 in magenta, and 1jla in green. Ligand structures are also shown in Scheme 1.
In addition, to enhance the possibility of finding robust leads that could adapt to alternative RT structures, a conventional RT structure and one for a Y181C mutant were included in the virtual screening. The conventional 1rt4 structure,13 which was used in a prior docking exercise,14 and the high-resolution (2.5-Å) 1jla structure15 containing the Y181C mutation were chosen. The NNRTI binding sites for the three structures are overlaid in Figure 2. The three ligands, UC-781 (1rt4), TNK-651 (1jla), and R221239 (2be2), which were removed for the docking calculations, are illustrated in Scheme 1 in orientations similar to the manner in which they are overlaid in the binding site.
Scheme 1.

Ligands in the 1rt4, 1jla, and 2be2 crystal structures.
Virtual Screening
Our previous, reported effort in the virtual screening of NNRTIs used the 1rt4 structure and covered ca. 70,000 compounds from the Maybridge catalog.14 After an initial similarity filter and docking using Glide 3.5 with standard precision (SP),16,17 the top 500 compounds were re-docked in extra-precision (XP) mode.18 The top 100 of these were post-scored with an MM-GB/SA method that provided high correlation between predicted and observed activities.14 Though assaying of ca. 20 high-scoring library compounds failed to yield any actives, a viable core was identified that did lead to anti-HIV agents, which were potent against WT virus, but inactive towards the Y181C variant.4 In the present case, the latter problem has been approached by use of the three crystal structures and the screening has been expanded to cover the more than 2 million compounds in the ‘drug-like’ subset of the ZINC database, which was downloaded from zinc.docking.org.19 The ZINC collection has been compiled from catalogs of commercially available compounds.
For the three RT structures (1rt4, 1jla, 2be2),9,13,15 the crystallographic water molecules and ligands were removed, and the proteins were prepared using Schrödinger’s Maestro and Glide 3.5 software.16-18 The receptors were subjected to the standard protein preparation workflow, which involves a restrained, partial energy-minimization using the OPLS-AA force field.20 The three structures have less than 2.5-Å Cα root-mean-square deviation from each other in all three pair-wise cases. The atoms of the 17 residues that comprise the binding-site deviate by, at most, 1.2 Å across the three structures.
In our previous study,14 it was shown that Glide did well predicting the structures of the complexes of six NNRTIs with the wild-type 1rt4 structure. In order to investigate the ligand binding modes generated for the two other receptors, the ligands for 1jla and 2be2 were self-docked back into their protein structures using Glide 3.5 in SP mode. The results were essentially perfect, as illustrated in Figure 3. The average all-atom RMSD for comparing the crystal structures and Glide poses for the three ligands is 0.81 Å, with a maximum deviation of 1.03 Å for the 2be2 case.
Figure 3.

Comparison of Glide SP poses (in white) and the crystal structures for self-docking the 1jla (left) and 2be2 (right) ligands. RMSD for 1jla is 0.76 Å, and for 2be2 it is 1.03 Å.
All of the ZINC compounds were then docked into each of the three RT structures using Glide SP.16,17 The value in using multiple protein structures for virtual screening has been noted, though optimal ways to proceed and combine results are unclear.21,22 Presently, two approaches were taken, as summarized in Figure 4. First, the top 5,000 structures from each SP screen were submitted to extra precision (XP) scoring18 in a ‘parallel’ fashion. The XP processing takes about 15 minutes per compound on a 3-GHz Pentium processor, so it is typical to apply it only to compounds that score well using the SP mode. Among the 15,000 structures that passed the SP filter, there were only 17 structures in common, i.e., that were among the top-5000 SP scores for all three receptors.
Figure 4.

The two hierarchical screening protocols used with the three receptors. In the parallel mode, XP scoring was performed on the top 5000 compounds from each SP analysis. In the serial mode, XP scoring was applied to the common 4,684 structures that scored in the top 100,000 in all SP runs.
The second approach started with the common compounds among the top 100,000 in each SP screen. These 4,684 compounds were then processed in XP mode. This ‘serial’ approach relies less on the rank produced from the SP scoring and more on the commonality of ranking reasonably well for all three receptors. The goal is to retain compounds that might end up with very favorable XP scores. In the end, XP scores were obtained for a total of nearly 20,000 compounds. Much additional filtering was still needed, as the intention was to purchase and assay roughly 10-20 compounds. This limit was set by cost considerations for materials and labor.
The three poses for compounds which ranked in the top 500 by XP scoring for each receptor were graphically displayed. Docked conformations from XP Glide frequently showed unreasonably short non-bonded contacts or twisted amides and esters; their rejection eliminated many of the top-ranking structures. Ligands often seem overly large for the binding sites and they end up too compressed. An example is provided in Figure 5 for the ZINC compound which ranked first for both the 2be2 and 1jla structures, and third for 1rt4 using the serial protocol. The poses have favorable interactions such as hydrogen bonding between the oxazinone and the backbone of Lys101 or Lys103, and stacking of the naphthalene moiety with the side chain of Tyr188; however, in each case the ester group is impossibly strained with an O=C-O-C dihedral angle near 90° and with associated overly short 1,6-H,H distances.23
Figure 5.

Many top XP-ranked compounds were not considered for purchase owing to ligand strain stemming from twisted ester and amide linkages or overly short intramolecular contacts. In the serial screen, the illustrated compound ranked first for both the 2be2 and 1jla receptors, and it ranked third for the 1rt4 structure.
Compounds with promising XP poses for multiple receptors as well as interesting and novel binding modes were sought. As further criteria, compounds with functionality that is prone to hydrolysis such as oximes, hydrazones, esters, and tertiary alcohols were avoided along with ones containing electrophilic functionality such as α,β-unsaturated ketones and other Michael acceptors. Such potentially reactive compounds are interesting to contemplate as a source of potentially viable cores, but they are not optimal for purchase given limited resources. With these considerations, compounds 1 - 4 in Figure 6 emerged as top choices. Additionally, after thorough visual inspection of the poses for the 17 molecules ranked in the top 5000 by SP for all three receptors, compound 5 was selected. Finally, top-scoring compounds for the individual receptors were considered, which led to picking compounds 6-9. Several additional compounds were of high interest, but they turned out to be unavailable for purchase. Analogues of some of the compounds also scored well, but to maximize diversity they were not selected except in the case of 1 and 2. Generally, for multiple analogues with similar scores, the smallest ones were given preference in view of the propensity for the docking to favor larger ligands and in order to begin potentially at a lower molecular weight in a lead series. For example, 3 and its 6-benzo-1,3-dioxolyl replacing furanyl analogue both scored well, as was the case for 1, 2, and 1-naphthyl replacing substituted phenyl analogues. Further details on the rankings for 1 - 9 are provided in the results section.
Figure 6.

The purchased compounds. In the virtual screening, compounds 1 - 5 ranked well for all three receptors, while 6 - 9 ranked highly for individual receptors. All compounds also exhibited acceptable predicted properties (Table 1).
As a final check, predictions for pharmacologically important properties were computed using QikProp for the selected compounds.23 When QikProp is run on 1700 known oral drugs, 90% have MW < 470, QP log P < 5.0, QP log S > -5.7, and QP PCaco > 22 nm/s.3c,25 The rms errors for QikProp predictions are 0.5–0.6 log unit. Since the results for 1 – 9 in Table 1 compare well with the above limits and with the results for other NNRTIs, the compounds were purchased. The computed solubilities for 6, 7, and 9 are less than optimal for lead compounds, so this would need to be monitored if their series were pursued.
Table 1.
Predicted Properties for the Purchased Compounds and Known NNRTIs.
| Compound | MWa | QP log Pb | QP log Sc | QP PCacod |
|---|---|---|---|---|
| 1 | 351.9 | 3.0 | -4.2 | 862 |
| 2 | 331.4 | 2.7 | -3.6 | 874 |
| 3 | 333.4 | 3.2 | -5.2 | 1033 |
| 4 | 319.4 | 3.5 | -4.4 | 779 |
| 5 | 336.4 | 3.7 | -4.3 | 827 |
| 6 | 348.8 | 2.5 | -5.8 | 101 |
| 7 | 392.4 | 2.5 | -5.7 | 99 |
| 8 | 329.4 | 1.5 | -3.8 | 175 |
| 9 | 410.9 | 4.0 | -6.0 | 730 |
| nevirapine | 266.3 | 2.5 | -3.2 | 2090 |
| efavirenz | 315.7 | 3.5 | -5.0 | 1585 |
| delavirdine | 456.6 | 2.6 | -5.7 | 218 |
| UC-781 | 335.8 | 5.1 | -5.7 | 6717 |
| rilpivirine | 366.4 | 3.3 | -6.5 | 150 |
| etravirine | 435.3 | 2.7 | -6.7 | 75 |
Molecular weight.
Predicted octanol/water log P from QikProp.
Predicted aqueous solubility from QikProp; S in mol/L.
Predicted Caco-2 cell permeability in nm/s from QikProp.
Experimental Details
Compounds 1 – 9 and three known NNRTIs (Scheme 2), which are approved drugs, were obtained from commercial vendors. 1, 2, 3, and 8 were acquired and assayed as racemic mixtures. The chemical structures and purity of the compounds were verified by 1H NMR (Bruker DRX-400 and DRX-500) and mass spectroscopy (Waters Micromass ZQ) at the Yale Chemical Instrumentation Center. Activities against the wild-type IIIB strain of HIV-126 were determined using MT-2 human T-cells27 at an MOI of 0.1; EC50 values were obtained as the dose required to achieve 50% protection of the infected cells using the MTT colorimetric method. The CC50 for inhibition of MT-2 cell growth by 50% was obtained simultaneously.28 An analogous assay was performed using a variant strain of the virus that encodes the Y181C mutant form of HIV-RT.29
Scheme 2.

Structures of known NNRTIs, which were also assayed.
Results and Discussion
The assay results are listed in Table 2. The results for the reference compounds are similar to prior reports using similar assays.4b,30 Among the purchased compounds, 3 shows low-micromolar activity towards both the WT and Y181C-variant HIV-1, while 4 has 7.5-μM activity against the Y181C form, and 5 is a 4.8-μM NNRTI towards the WT virus. In view of the small number of purchased compounds and the prior difficulties in obtaining activity against the Y181C variant, the present virtual screening approach was very successful.
Table 2.
| WT | Y181C | |||
|---|---|---|---|---|
| Compound | EC50 | CC50 | EC50 | CC50 |
| 1 | NA | 60.0 | NA | 53.0 |
| 2 | NA | >100 | NA | >100 |
| 3 | 6.2 | 37.0 | 12.0 | 37.0 |
| 4 | NA | 14.0 | 7.5 | 17.0 |
| 5 | 4.8 | 72.0 | NA | 43.0 |
| 6 | NA | >100 | NA | >100 |
| 7 | NA | >100 | NA | >100 |
| 8 | NA | 2.1 | NA | 1.3 |
| 9 | NA | >100 | NA | >100 |
| nevirapine | 0.11 | >10 | NA | >100 |
| efavirenz | 0.002 | >0.1 | 0.004 | >0.1 |
| etravirine | 0.001 | >0.1 | 0.002 | >0.1 |
For 50% protection in MT2 cells; antiviral curves used triplicate samples at each concentration. NA for EC50 > CC50.
For 50% inhibition of MT-2 cell growth; toxicity curves also used triplicate samples.
It may also be expected that the core structures for some of the remaining compounds in Figure 6 are viable; i.e., modification of substituents could lead to active analogues.4 For example, 1 and 2 bear some resemblance to PNU142721, which has 1-nM potency towards WT HIV-1, though only 1-μM activity towards a Y181C variant.31 The similarity is apparent when the compounds are aligned as in Scheme 3, which reflects the conformation of PNU142721 in the 1ikx crystal structure with K103N HIV-RT.32 This structure is 1rt4-like, having Y181 and Y188 both “up”, and the ligand’s furanopyridine moiety is well-stacked with the phenol of Y181, likely accounting for the loss in activity towards the Y181C mutant. The novel, internal hydrogen bond in the bound conformations of 1 and 2 and facile synthesis of alternative, substituted phenyl-ring derivatives were features that promoted their selections.
Scheme 3.
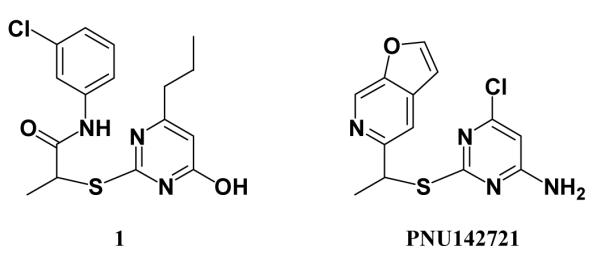
Aligned structures of 1 and a known 1-nM NNRTI.
Glide SP and XP scores and rankings from the different protocol are reported in Table 3 for 1 - 9, while Table 4 contains SP and XP scores for the reference NNRTIs. In order to be in the top 100,000 compounds in each SP screen, a Glide score lower than ca. -9 was needed. Among the known NNRTIs in Table 4, only efavirenz would have reached this threshold for any receptor. Also, the purchased compounds had one or more SP scores at least as favorable as -10.0 with the best score being -11.8 for 6 with the 2be2 structure. The XP scores for the purchased compounds are also generally much better than those for the known NNRTIs with most purchased compounds having one or more XP scores in the -19 to -20 range. The XP results for active compound 3 were particularly auspicious since they were in this range for all three receptors. However, 3 would have been missed using just the parallel protocol (Figure 4) since it was not in the top 5000 compounds for any receptor with the SP scoring (Table 3). Similarly, 4 would have been overlooked in the absence of the serial protocol. 5 is the only active compound that passed the parallel filter, and it did yield extremely low XP scores with the 1jla and 1rt4 receptors. Finally, 6 – 9 were chosen in large part owing to their strong XP showing for one receptor. Though the results here are limited, it appears that this was not a productive strategy. Instead, seeking consensus on good performance for multiple receptors especially via the serial route appears to be the most promising protocol. Again, the serial approach involved performing the XP scoring on the 4,684 compounds that scored in the top 100,000 by SP for all three receptors. One could consider increasing the latter limit, which further deemphasizes the SP ranking, and then skip the parallel approach.
Table 3.
SP and XP Glide Scores and Rankings for Purchased Compounds
| SP Results | ||||||
|---|---|---|---|---|---|---|
| 1jla | 1rt4 | 2be2 | ||||
| Compound | Rank | GScore | Rank | GScore | Rank | GScore |
| 1 | 81323 | -9.9 | 8923 | -9.7 | 9754 | -10.5 |
| 2 | 71029 | -10.0 | 5456 | -9.8 | 56065 | -9.8 |
| 3 | 49321 | -10.1 | 61501 | -9.1 | 16646 | -10.3 |
| 4 | 56728 | -10.0 | 30016 | -9.5 | 22688 | -10.2 |
| 5 | 4955 | -10.9 | 2811 | -10.0 | 4290 | -10.8 |
| 6 | >100000 | -9.0 | >100000 | -8.0 | 159 | -11.8 |
| 7 | 4470 | -10.9 | >100000 | -7.3 | >100000 | -8.9 |
| 8 | >100000 | -9.2 | >100000 | -7.9 | 4750 | -11.2 |
| 9 | 2529 | -11.1 | >100000 | -5.5 | >100000 | -10.4 |
| XP Results | ||||||
|---|---|---|---|---|---|---|
| 1jla | 1rt4 | 2be2 | ||||
| Serial Protocol |
Rank | GScore | Rank | GScore | Rank | GScore |
| 1 | 328 | -18.2 | 66 | -19.9 | 224 | -18.4 |
| 2 | 309 | -18.2 | 94 | -19.4 | 432 | -17.4 |
| 3 | 117 | -19.5 | 81 | -19.5 | 47 | -19.9 |
| 4 | 226 | -18.6 | 496 | -17.0 | 348 | -17.8 |
| Parallel Protocola |
Rank | GScore | Rank | GScore | Rank | GScore |
|---|---|---|---|---|---|---|
| 5 | 45 | -20.4 | 44 | -20.5 | 2925 | -12.4 |
| 6 | NA | NA | 81 | -20.3 | ||
| 7 | 136 | -20.8 | NA | NA | ||
| 8 | NA | NA | 42 | -20.8 | ||
| 9 | 385 | -19.2 | NA | NA | ||
For the parallel protocol compounds 6-9 were only in the top 5000 SP compounds for one receptor, and hence were only rescored with XP for one receptor.
Table 4.
Glide Scores for Docking Reference NNRTIs
| 1jla | 1rt4 | 2be2 | ||||
|---|---|---|---|---|---|---|
| Compound | SP | XP | SP | XP | SP | XP |
| efavirenz | -10.4 | -18.1 | -10.8 | -18.9 | -10.9 | -19.4 |
| nevirapine | -7.7 | -12.5 | -7.8 | -10.3 | -7.5 | -9.3 |
| -etravirine | -8.6 | -13.8 | NAa | -17.5 | -8.8 | -12.6 |
The complex was rejected for being too high in energy.
The Glide XP poses for the active compounds 3 – 5 with the three receptors are shown in Figure 7. 3 adopts a similar conformation and position in the binding site for all three RT structures, while 4 shows a different structure with 2be2 than the other two RT forms, and 5 has a different orientation with the 1rt4 receptor. When one of the binding modes is different, it is usually for the 1rt4 structure with both Tyr181 and Tyr188 “up”. For favorable aryl-aryl interactions between a ligand and Tyr181, the “up” orientation is preferred. This reinforces the sense that targeting the 1jla and 2be2 structures is appropriate for seeking NNRTIs that do not gain binding affinity via interaction with Tyr181 and are, therefore, resilient to the Y181C mutation.
Figure 7.

Complexes for the active compounds 3 (top), 4 (middle), and 5 (bottom) from docking into the three crystal structures for HIV-RT. 3 has a similar binding mode in all cases, while often the binding modes can differ significantly for different RT structures as for 4 and 5.
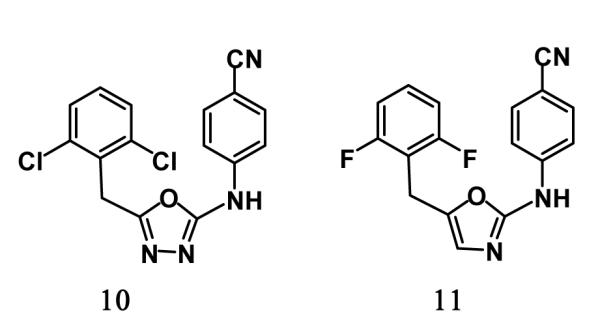
The predicted structures of the complexes for 3 are reasonable based on our prior computations and lead optimization for oxadiazoles and oxazoles such as 10 and 11, which are 130-nM and 13-nM NNRTIs.4,14 The hydrogen bonds between the anilinyl NH and nitrile N of 3 and the backbone oxygen and NH of Lys101 were expected; however, it was not obvious that the larger naphthalene subunit would fit into the Tyr181-Tyr188-Trp229 π-box in place of the dihalobenzyl groups. Subsequent energy minimizations for complexes of the R and S enantiomers of 3 revealed a ca. 2 kcal/mol preference for the S enantiomer based on protein-ligand interaction energies. The results also indicated that the naphthyl group is well accommodated either heading “out” as in the 1jla and 1rt4 structures in Figure 7 or “in” as in the 2be2 structure. This variability could be helpful for maintaining activity against Tyr181 and Tyr188 variants.
For 4, the computed structures for the 1jla and 1rt4 complexes are unusual with the phenylthienopyrimidine fragment spanning the binding site diagonally with the phenyl ring near Trp229. The hydrogen bond between the amino group and the oxygen of Lys101 is typical, while the hydrogen bond between the ligand’s hydroxyl group and the backbone oxygen of Tyr188 is novel. The lowest XP score is for 4 with the 1jla structure, which is consistent with its activity towards the Y181C variant (Table 2).
For 5, the complexes with the 1jla and 2be2 models of RT are reminiscent of the actual complexes with TNK-651 and R221239 in these crystal structures (Figures 2 and 3). The benzyl groups of 5 and TNK-651 overlay with the position of the dimethylphenoxy group of R221239 in the π-box. Furthermore, the uracil ring and attached CCO chain are positioned similarly to the COC-phenyl fragment of TNK-651 and CSC-furan unit of R221239, projecting into the channel between Phe227 and Pro236. Though there are also hydrogen bonds between the uracil fragment and the backbone oxygen and NH of Lys103 in the 2be2 pose, the pose received a poor XP score (Table 3). In comparison, the predicted pose for 5 with the 1rt4 structure has little precedent. In this case, the central ring is in the π-box, the uracil ring is exiting the binding site near Glu138, and there is an overly short intramolecular contact between the ether oxygen and an ortho hydrogen of the terminal phenyl ring. In view of the suggested similarities between 5 and R221239, it would be surprising if 5 did not bind to WT HIV-RT in a manner similar to that in the 2be2 structure.
Fundamentally, the present virtual screening exercise provided three compounds that are suitable starting points for lead optimization of NNRTIs that are expected to be active against both WT HIV-1 and Y181C RT variants. It is likely that simple modifications of some of the inactive compounds in Figure 6 can also provide entry into other active series. Since the variety of analogues that can be purchased is limited, our approach to lead optimization is focused synthesis guided by results of free-energy perturbation calculations.3,4 In fact, progress has been made with series based on 1, 3, and 5, as will be described in due course.
Conclusion
Virtual screening was performed by docking ca. 2 million commercially available compounds with HIV reverse transcriptase (RT). Three crystal structures of RT were used in order to find NNRTIs that would be effective against both wild-type HIV-1 and variants possessing the clinically troublesome Y181C RT mutation. Two of the structures, 1rt4 and 2be2, are for WT-virus with different conformations of Tyr181, while the third structure, 1jla, incorporates the Y181C modification. Two protocols were applied for the screening using standard-precision (SP) scoring and the more computational intensive extra-precision (XP) scoring with the Glide program. In the parallel mode, XP scoring was performed on the top 5000 compounds from the SP analysis for each RT structure. In the serial mode, XP scoring was applied to the common 4,684 structures that scored in the top 100,000 in the three SP runs. After much additional filtering by visual inspection of the top-ranked ca. 1500 complexes, the nine compounds in Figure 6 were eventually purchased and assayed. Three of the compounds showed low-micromolar anti-viral activity towards either or both the wild-type and Y181C HIV-1 strains. Though the number of assayed compounds was small, the serial screening strategy appears to be more promising than the parallel one in that it is less biased by the standard-precision scoring. The approach can clearly be expanded to include structures encoding additional resistance mutations. Optimization of the lead compounds and others suggested by the present virtual screening exercise is being pursued.
Acknowledgements
Gratitude is expressed to the National Institutes of Health (AI44616, GM32136, and GM49551) for support of this research. Receipt of the following reagents through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH is also greatly appreciated: MT-2 cells, catalog #237, and nevirapine-resistant HIV-1 (N119), catalog #1392, from Dr. Douglas Richman; HTLV-IIIB/H9, #398, from Dr. Robert Gallo; and HIV-1IIIB (A17 Variant) from Dr. Emilio Emini.
References
- (1).Kallings LO. The first postmodern pandemic: 25 years of HIV/AIDS. J. Intern. Med. 2008;263:218–243. doi: 10.1111/j.1365-2796.2007.01910.x. [DOI] [PubMed] [Google Scholar]
- (2) (a).Flexner C. HIV drug development: the next 25 years. Nature Rev. Drug Disc. 2007;6:959–966. doi: 10.1038/nrd2336. [DOI] [PubMed] [Google Scholar]; (c) De Clerq E. The design of drugs for HIV and HCV. Nature Rev. Drug Disc. 2007;6:1001–1018. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- (3) (a).Jorgensen WL, Ruiz-Caro J, Tirado-Rives J, Basavapathruni A, Anderson KS, Hamilton AD. Computer-aided design of non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2006;16:663–667. doi: 10.1016/j.bmcl.2005.10.038. [DOI] [PubMed] [Google Scholar]; (b) Ruiz-Caro J, Basavapathruni A, Kim JT, Wang L, Bailey CM, Anderson KS, Hamilton AD, Jorgensen WL. Optimization of diarylamines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2006;16:668–671. doi: 10.1016/j.bmcl.2005.10.037. [DOI] [PubMed] [Google Scholar]; (c) Thakur VV, Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. Optimization of pyrimidinyl- and triazinyl-amines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 2006;16:5664–5667. doi: 10.1016/j.bmcl.2006.08.037. [DOI] [PubMed] [Google Scholar]; (d) Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. FEP-guided selection of bicyclic heterocycles in lead optimization for non-nucleoside inhibitors of HIV-1 reverse transcriptase. J. Am. Chem. Soc. 2006;128:15372–15373. doi: 10.1021/ja066472g. [DOI] [PubMed] [Google Scholar]
- (4) (a).Barreiro G, Kim JT, Guimaraes CRW, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. From docking false-positive to active anti-HIV agent. J. Med. Chem. 2007;50:5324–5329. doi: 10.1021/jm070683u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zeevaart JG, Wang L, Thakur VV, Leung CS, Tirado-Rives J, Bailey CM, Domaoal RA, Anderson KS, Jorgensen WL. Optimization of Azoles as Anti-HIV Agents Guided by Free-Energy Calculations. J. Am. Chem. Soc. 2008;130:9492–9499. doi: 10.1021/ja8019214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Esnouf R, Ross C, Jones Y, Stammers D, Stuart D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nature Struct. Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- (6).Ren J, Esnouf R, Hopkins A, Jones E, Kirby I, Keeling J, Ross C, Larder B, Stuart D, Stammers D. 3′-Azido-3′-deoxythymidine drug resistance mutations in HIV-1 reverse transcriptase can induce long range conformational changes. P. Natl. Acad. Sci. USA. 1998;95:9518–9523. doi: 10.1073/pnas.95.16.9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ren J, Milton J, Weaver K, Short S, Stuart D, Stammers D. Structural basis for the resilience of efavirenz (DMP-266) to drug resistance mutations in HIV-1 reverse transcriptase. Structure. 2000;8:1089–1094. doi: 10.1016/s0969-2126(00)00513-x. [DOI] [PubMed] [Google Scholar]
- (8).Ren J, Esnouf R, Garman E, Somers D, Ross C, Kirby I, Keeling J, Darby G, Jones Y, Stuart D. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nature Struct. Biol. 1995;2:293–302. doi: 10.1038/nsb0495-293. [DOI] [PubMed] [Google Scholar]
- (9).Himmel DM, Das K, Clark AD, Hughes SH, Benjahad A, Oumouch S, Guillemont J, Coupa S, Poncelet A, Csoka I, Meyer C, Andries K, Nguyen CH, Grierson DS, Arnold E. Crystal structures for HIV-1 reverse transcriptase in complexes with three pyridinone derivatives: A new class of non-nucleoside inhibitors effective against a broad range of drug-resistant strains. J. Med. Chem. 2005;48:7582–7591. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- (10).Pata JD, Stirtan WG, Goldstein SW, Steitz TA. Structure of HIV-1 reverse transcriptase bound to an inhibitor active against mutant reverse transcriptases resistant to other nonnucleoside inhibitors. P. Natl. Acad. Sci. USA. 2004;101:10548–10553. doi: 10.1073/pnas.0404151101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hopkins AL, Ren J, Esnouf RM, Willcox BE, Jones Y, Ross C, Miyasaka T, Walker RT, Tanaka H, Stammers DK, Stuart DI. Complexes of HIV-1 Reverse Transcriptase with Inhibitors of the HEPT Series Reveal Conformational Changes Relevant to the Design of Potent Non-Nucleoside Inhibitors. J. Med. Chem. 1996;39:1589–1600. doi: 10.1021/jm960056x. [DOI] [PubMed] [Google Scholar]
- (12).Udier-Blagović M, Tirado-Rives J, Jorgensen WL. Structural and energetic analyses of the effects of the K103N mutation of HIV-1 reverse transcriptase on efavirenz analogues. J. Med. Chem. 2004;47:2389–2392. doi: 10.1021/jm0303507. [DOI] [PubMed] [Google Scholar]
- (13).Ren J, Esnouf RM, Hopkins AL, Warren J, Balzarini J, Stuart DI, Stammers DK. Crystal structures of HIV-1 reverse transcriptase in complex with carboxanilide derivatives. Biochemistry. 1998;37:14394–14403. doi: 10.1021/bi981309m. [DOI] [PubMed] [Google Scholar]
- (14).Barreiro G, Guimaraes CRW, Tubert-Brohman I, Lyons TM, Tirado-Rives J, Jorgensen WL. Search for non-nucleoside inhibitors of HIV-1 reverse transcriptase using chemical similarity, molecular docking, and MM-GB/SA scoring. J. Chem. Inf. Model. 2007;47:2416–2428. doi: 10.1021/ci700271z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ren J, Nichols C, Bird L, Chamberlain P, Weaver K, Short S, Stuart DI, Stammers DK. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J. Mol. Biol. 2001;312:795–805. doi: 10.1006/jmbi.2001.4988. [DOI] [PubMed] [Google Scholar]
- (16).Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- (17).Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- (18).Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- (19).Irwin JJ, Shoichet BK. ZINC - A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS All-Atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- (21).Carlson HA. Protein flexibility and drug design: how to hit a moving target. Curr. Opin. Chem. Biol. 2002;6:447–452. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- (22).Huang S-Y, Zou X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins. 2007;66:399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- (23).Price MLP, Ostrovsky D, Jorgensen WL. Gas-Phase and Liquid-State Properties of Esters, Nitriles, and Nitro Compounds with the OPLS-AA Force Field. J. Comput. Chem. 2001;22:1340–1352. [Google Scholar]
- (24).Jorgensen WL. QikProp. v 3.0 Schrödinger LLC; New York: 2006. [Google Scholar]
- (25).Proudfoot JR. The evolution of synthetic oral drug properties. Bioorg. Med. Chem. Lett. 2005;15:1087–1090. doi: 10.1016/j.bmcl.2004.12.024. [DOI] [PubMed] [Google Scholar]
- (26) (a).Popovic M, Read-Connole E, Gallo RC. T4 positive human neoplastic cell lines susceptible to and permissive for HTLV-III. Lancet. 1984;2:1472–1473. doi: 10.1016/s0140-6736(84)91666-0. [DOI] [PubMed] [Google Scholar]; (b) Popovic M, Sarngadharan MG, Read E, Gallo RC. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science. 1984;224:497–500. doi: 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]; (c) Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, Ivanoff L, Petteway SR, Jr., Pearson ML, Lautenberger JA, Papas TS, Ghrayeb J, Chang NT, Gallo RC, Wong-Staal F. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature. 1985;313:277–284. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- (27) (a).Harada S, Koyanagi Y, Yamamoto N. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science. 1985;229:563–566. doi: 10.1126/science.2992081. [DOI] [PubMed] [Google Scholar]; (b) Haertle T, Carrera CJ, Wasson DB, Sowers LC, Richmann DD, Carson DA. Metabolism and anti-human immunodeficiency virus-1 activity of 2-halo-2′,3′-dideoxyadenosine derivatives. J. Biol. Chem. 1988;263:5870–5875. [PubMed] [Google Scholar]
- (28) (a).Lin TS, Luo MZ, Liu MC, Pai SB, Dutschman GE, Cheng YC. Antiviral activity of 2′,3′-dideoxy-β-L-5-fluorocytidine (β-L-FddC) and 2′,3′-dideoxy-beta-L-cytidine (β-L-ddC) against hepatitis B virus and human immunodeficiency virus type 1 in vitro. Biochem. Pharmacol. 1994;47:171–174. doi: 10.1016/0006-2952(94)90002-7. [DOI] [PubMed] [Google Scholar]; (b) Ray AS, Yang Z, Chu CK, Anderson KS. Novel use of a guanosine prodrug approach to convert 2′,3′-didehydro-2′,3′-dideoxyguanosine into a viable antiviral agent. Antimicrob. Agents Chemother. 2002;46:887–891. doi: 10.1128/AAC.46.3.887-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Richman D, Shih C-K, Lowy I, Rose J, Prodanovic P, Goff S, Griffin J. Human immunodeficiency virus type 1 mutants resistant to nonnucleoside inhibitors of reverse transcriptase arise in tissue culture. Proc. Natl. Acad. Sci. USA. 1991;88:11241–11245. doi: 10.1073/pnas.88.24.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ludovici DW, De Corte BL, Kukla MJ, Ye H, Ho CY, Lichtenstein MA, Kavash RW, Andries K, de Béthune M-P, Azijn H, Pauwels R, Lewi PJ, Heeres J, Koymans LMH, de Jonge MR, Van Aken KJA, Daeyaert FFD, Das K, Arnold E, Janssen PAJ. Evolution of Anti-HIV Drug Candidates. Part 3: Diarylpyrimidine (DAPY) Analogues. Bioorg. Med. Chem. Lett. 2001;11:2235–2239. doi: 10.1016/s0960-894x(01)00412-7. [DOI] [PubMed] [Google Scholar]
- (31).Wishka DG, Graber DR, Kopta LA, Olmsted RA, Friis JM, Hosley JD, Adams WJ, Seest EP, Castle TM, Dolak LA, Keiser BJ, Yagi Y, Jeganathan A, Schlachter ST, Murphy MJ, Cleek GJ, Nugent RA, Poppe SM, Swaney SM, Han F, Watt W, White WL, Poel T-J, Thomas RC, Voorman RL, Stefanski KJ, Stehle RG, Tarpley WG, Morris J. (-)-6-Chloro-2-[(1-furo[2,3-c]pyridin-5-ylethyl)thio]-4-pyrimidinamine, PNU-142721, a New Broad Spectrum HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitor. J. Med. Chem. 1998;41:1357–1360. doi: 10.1021/jm9801049. [DOI] [PubMed] [Google Scholar]
- (32).Lindberg J, Sigurosson S, Lowgren S, Andersson HO, Sahlberg C, Noreen R, Fridborg K, Zhang H, Unge T. Structural Basis for the Inhibitory Efficacy of Efavirenz (DMP-266), MSC194, and PNU142721 Towards the HIV-1 RT K103N Mutant. Eur. J. Biochem. 2002;269:1670–1677. doi: 10.1046/j.1432-1327.2002.02811.x. [DOI] [PubMed] [Google Scholar]