

Abstract

Synthesis and reactivity of N-methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfonyl)fluoroacetamide, a building block for Julia olefination, is reported. This reagent undergoes condensation reactions with aldehydes and cyclic ketones, to give α-fluorovinyl Weinreb amides. Olefination reactions proceed under mild, DBU-mediated conditions, or in the presence of NaH. DBU-mediated condensations proceed with either E or Z-selectivity, depending upon reaction conditions, whereas NaH-mediated reactions are ≥98% Z-stereoselective. Conversion of the Weinreb amide moiety in N-methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfanyl)fluoroacetamide to ketones, followed by oxidation, resulted in another set of olefination reagents, namely (1,3-benzothiazol-2-ylsulfonyl)fluoromethyl phenyl and propyl ketones. In the presence of DBU, these compounds react with aldehydes tested to give α-fluoroenones with high Z-selectivity. The use of N-methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfanyl)fluoroacetamide as a common fluorinated intermediate in the synthesis of α-fluorovinyl Weinreb amides and α-fluoroenones has been demonstrated. Application of the Weinreb amide to α-fluoro allyl amine synthesis is also shown.
INTRODUCTION
Fluorine containing organic molecules are of high interest, due to altered physical, biological and chemical properties caused by fluorine atom introduction.1 One of the approaches for the synthesis of fluorinated compounds is via the use of appropriate fluorinated building blocks.2 Among various fluoroorganics, functionalized fluoroolefins have been the subject of many investigations, either as end products or as synthetic intermediates. A convenient route for olefination is via the modified Julia reaction.3,4 However, this method has not received much attention for the synthesis of vinyl fluorides until recently. Application of this reaction for preparation of fluoroalkylidenes was initially demonstrated using a single olefination precursor5. We were the first to utilize metalation-fluorination as a general approach for the preparation of novel Julia reagents for the synthesis of functionalized fluoroolefins.6–9 Variously functionalized fluoroolefins are now conveniently accessible by the Julia-Kocienski olefination.6–11 In our work, we have developed a series of isolable and stable fluorinated reagents that have yielded facile access to α-fluorostilbene and styrene derivatives,6 α-fluoroacrylates,7 α-fluorovinyl sulfones8 and α- fluoroacrylonitriles.9
On the basis of our previous results, we became interested in the development and use of functionalized fluorinated building blocks that would allow synthesis of various Julia olefination reagents from common precursors. Among the various functional groups, the (N-methoxy-N-methyl) amide moiety can be easily converted to α-fluoro ketones and α-fluoro aldehydes, and is a useful synthetic intermediate in organic synthesis.12 An appropriate heteroaryl derivative, such as N-methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfonyl)fluoroacetamide, or its precursor N-methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfanyl)fluoroacetamide would therefore offer two potential sites for modification (Figure 1). The benzothiazolyl sulfone moiety would provide one point of diversity, namely olefination,3,4 and the amide part would provide the second point of diversity by virtue of its reactivity towards metal alkyls and hydride reducing agents.12 A two-step synthesis of a fluorovinyl Weinreb amide has been reported from tetrafluoroethane (HFC-134a), albeit in a low 30% yield.13 An example of the Weinreb-Wittig Horner reagent (EtO)2P(O)CHFC(O)NMe(OMe) has been reported as well, however no details on its synthesis, characterization and reactivity were provided.14
FIGURE 1.

Fluorinated Julia-Weinreb amide building block.
Recently, Aidhen et al. reported synthesis and NaH-mediated condensation reactions of N-methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfonyl)acetamide with aldehydes, leading to vinyl Weinreb amides via Julia olefination.15 While this work was in progress, synthesis of α,β-unsaturated Weinreb amides16 and the α-fluoro analogs11 were reported using 3,5-bis(trifluoromethyl)phenyl sulfones.
Herein, we present our synthesis of the benzothiazolyl based fluoro Julia-Weinreb amide reagent and study of its reactions at both reactive centers. Specifically, reactions at the amide functionality of N-methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfanyl)fluoroacetamide, followed by oxidation, led to second set of Julia olefination reagents that were also subjected to condensation reactions to give α-fluoroenones. The effect of reaction conditions on olefination selectivity under mild, DBU-mediated conditions as well as with NaH, is presented. Finally, the utility of the fluoro Julia-Weinreb amide building block for the synthesis of fluoroallyl amines, a class of dipeptide isosteres,1 is demonstrated by a concise synthesis of a known dipeptidyl peptidase II inhibitor.17
RESULTS AND DISCUSSION
At the outset, synthesis of a suitable reagent for Julia-Kocienski olefination3,4 was undertaken. Stabilized carbanions derived from 1,3-benzothiazol-2-yl (BT) sulfone derivatives have been successfully used by us in fluoro Julia olefination reactions.7–9 Thus, we sought synthesis of a Weinreb amide derivative based on the benzothiazolyl moiety.
As shown in Scheme 1, the known BT-sulfide 115 was prepared by reaction of the sodium salt of 2-mercapto-1,3-benzothiazole with 2-bromo-N-methoxy-N-methylacetamide18 (prepared from bromoacetyl bromide and (MeO)MeNH·HCl). Oxidation of BT-sulfide 1 with m-CPBA yielded the known sulfone 215 (Scheme 1). Metalation-fluorination of sulfone 2 using LDA and N-fluorodibenzenesulfonimide (NFSI) in toluene resulted in the monofluoro derivative 3 in 85% yield.
SCHEME 1.

Synthesis of Fluoro Julia-Weinreb Reagent
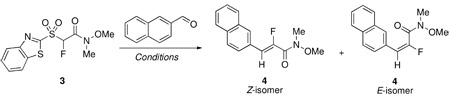
With the desired fluoro Julia-Weinreb reagent 3 in hand, we screened the reactivity of the reagent. Various reaction conditions and additives were tested in condensation reactions of 2-naphthaldehyde (Table 1). All DBU-mediated reactions were performed under Barbier conditions,4 whereas when NaH was used as base, 2-naphthaldehyde was added to a mixture of sulfone and NaH.
TABLE 1.
Effect of Reaction Conditions on Condensation Reactions of 3 with 2-Naphthaldehyde
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | base | solvent | aldehyde:sulfone: base (molar equiv) | additive (molar equiv) | T, rxn time (h) | (%) E/Z ratio;a yieldb | |
| 1 | DBU | CH2Cl2 | 1.0:1.2:4.0 | -- | rt, 2 | 33/67; 90% | |
| 2 | DBU | CH2Cl2 | 1.0:1.2:4.0 | -- | −78 °C, 3.5 | 50/50; 96% | |
| 3 | DBU | THF | 1.0:1.2:4.0 | -- | rt, 2 | 19/81; 84% | |
| 4 | DBU | THF | 1.0:1.2:4.0 | -- | −78 °C, 4 | 4/96; 84% | |
| 5 | DBU | PhMe | 1.0:1.2:4.0 | -- | rt, 48 | 16/84; NAc | |
| 6 | DBU | DMF | 1.0:1.3:2.6 | -- | rt, 20 | 74/26; NAc | |
| 7 | DBU | DMF | 1.3:1.0:2.0 | -- | rt, 15 | 74/26; NAc | |
| 8 | DBU | DMPU | 1.3:1.0:2.0 | -- | rt, 17 | 78/22; 93% | |
| 9 | DBU | DMF-DMPUd | 1.3:1.0:2.0 | -- | −78 °C, 3.5 | 25/75; NAc | |
| 10 | DBU | DMPU | 1.3:1.0:2.0 | -- | 75 °C, 2.5 | 78/22; NAc | |
| 11 | DBU | THF | 1.0:1.3:3.9 | MgBr2 (1.8) | rt, 48 | 10/90; 78% | |
| 12 | DBU | THF | 1.2:1.0:3.0 | MgBr2 (1.4) | rt, 48 | 7/93; 78% | |
| 13 | DBU | THF | 1.2:1.0:3.0 | ZnBr2 (1.4) | rt, 48 | 15/85; 74% | |
| 14 | NaH | THF | 1.0:1.3:2.6 | -- | rt, 20; 70 °C, 2 | Only Z, 84% | |
| 15 | NaH | THF | 1.0:2.0:4.0 | -- | rt, 1 | Only Z, 90% | |
| 16 | NaH | THF | 1.0:2.0:4.0 | 18-Crown-6 (6.0) | rt, 1.5 | 40/60, NAc | |
| 17 | NaH | THF | 1.0:2.0:4.0 | 15-Crown-5 (6.0) | rt, 1.5 | 12/88, NAc | |
| 18e | K2CO3 | DMF | 1.0: 2.0:18.0 | TBAB (0.2) | rt, 18 | 55/45, NAc | |
Relative ratio of diastereomers in the crude reaction mixture determined by 19F NMR prior to isolation. No change in ratio was observed after purification.
Yields of isolated, purified products.
Product not isolated from reaction mixture.
A 1:1 mixture of DMF and DMPU was used to prevent freezing at −78 °C.
Reaction conditions were identical to those described in ref. 11.
Wide changes in stereoselectivity were observed depending upon reaction solvent, reaction temperature and the additive. Good yield and moderate Z-selectivity was observed in the DBU-mediated condensation in CH2Cl2 at room temperature, but there was no selectivity when the reaction was performed at −78 °C (entries 1, 2). Higher Z-selectivity was obtained in THF at room temperature (entry 3) and this increased at −78 °C (entry 4), and products were isolated in good yields in both cases. Room temperature olefination in toluene resulted in a marginal increase in Z-selectivity (entry 5) compared to the room temperature reaction in THF (entry 3), however the reaction was much slower, with approximately 95% conversion after 48 hours. Changing the reaction solvent to DMF resulted in a reversal of selectivity with the E-isomer predominating, but the E/Z ratio was unaffected by a change in the reactant stoichiometry (entries 6, 7). Highest E-selectivity was obtained in DMPU, and the E/Z product mixture was isolated in a high 93% yield (entry 8). Lowering of reaction temperature to −78 °C in a 1:1 mixture of DMF and DMPU (to prevent freezing) again favored formation of Z-isomer (entry 9). Increase in reaction temperature from room temperature to 75 °C did not affect the olefination selectivity in DMPU (compare entries 8 and 10).
Next, reactions were performed in the presence of salt additives. The role of MgBr2 additive on the stereochemical outcome of Julia olefination with benzothiazolyl derived reagents has recently been demonstrated in the synthesis of α-fluoroacrylates.10 In the present case, a room temperature reaction in THF, in the presence of MgBr2 resulted in an increased Z-selectivity, whereas a change in the reactant stoichiometry had little effect on olefination selectivity and yield (entries 11, 12). Addition of ZnBr2 showed only a marginal increase in selectivity (compare entries 3 and 13). Finally, the use of NaH as base in THF resulted in the exclusive formation of the Z-isomer (entries 14, 15). Under these conditions the rate of the reaction and yield increased with higher molar excess of sulfone and NaH (entry 15). In order to assess whether there was any influence of the cation on the stereoselectivity, condensations were performed in the presence of excess of 18-Crown-6 and 15-Crown-5, using NaH. In both cases, a decrease of stereoselectivity was observed, which was more pronounced for 18-Crown-6 (entries 16 and 17). This indicates that the counterion likely also plays a role in the stereoselection. The DBU/MgBr2/THF result (entries 11, 12) is also consistent with this observation. Finally, in order to compare the condensation reactivity of 3 with 2-naphthaldehyde to that of the recently described 3,5-bis(trifluoromethyl)phenyl sulfone based reagent,11 a reaction was performed under the reported conditions11 (entry 18). Under these conditions, no stereoselectivity was obtained in the reaction with 3, likely indicating a link between aryl or heteroarylsulfonyl moiety and the mechanistic path, leading to markedly different stereochemical results.
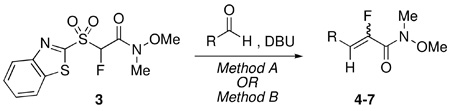
Although we identified reaction conditions that gave exclusive Z-selectivity, we wanted to assess whether the mild DBU-mediated olefinations, which gave condition dependent complementary olefination products with 2-naphthaldehyde, would be applicable to other substrates as well. Table 2 shows condensation reactions of a range of aldehydes using either Method A (DBU, THF, −78 °C, molar ratio of aldehyde:sulfone:DBU = 1:1.4:4, Barbier conditions4), or Method B (DBU, DMPU, room temperature, molar ratio of aldehyde:sulfone:DBU = 1.3:1.0:2, Barbier conditions4).
TABLE 2.
DBU-Mediated Condensation Reactions of 3
 | ||||
|---|---|---|---|---|
| entry | RCHO | conditions, rxn time (h) | products 4–7: (%)E/Z ratio,a yieldb | δ (ppm); mult, J (Hz) |
| 1 | Method A, 4 | 4: 6/94;c 86% c | E-isomer:−110. 5; d, 21.3 | |
| 2 | Method B, 17 | 4: 78/22; 93% | Z-isomer: −120.5; d, 36.6 | |
| 3 |  |
Method A, 2.5 | 5: 3/97; 92% | E-isomer:−104. 8; d, 21.3 |
| 4 | Method B, 16 | 5: 67/33; 83% | Z-isomer: −115.0; d, 33.6 | |
| 5 | Method A, 4 | 6: 74/26; 78% | E-isomer: −115.7; broad s | |
| 6 | Method B, 17 | 6: 86/14; 81% | Z-isomer: −119.7; d, 33.6 | |
| 7 | Method A, 2.5 | 7: 54/46; 74% | E-isomer: −117. 5; broad s | |
| 8 | Method B, 16 | 7: 67/33; 69% | Z-isomer: −125.7; d, 36.6 | |
Relative ratio of diastereomers in the crude reaction mixture determined by 19F NMR prior to isolation. No change in olefin ratio was observed after purification.
Yields of isolated, purified products (reactions were performed under similar conditions using either Method A or Method B, but were not optimized for individual cases).
Practically no change in E/Z ratio and yield was observed upon increasing the sulfone from 1.2 to 1.4 molar equiv (compare entry 4, Table 1).
As can be seen from Table 2, using Method A, high Z-selectivity was observed for aromatic aldehydes (entries 1, 3). The trend reversed in the case of thiophene, where the E isomer predominated (entry 5). No selectivity was observed in the case of n-octanal (entry 7). Condensations using Method B were E-selective in all cases. Selectivity was moderate in the case of electron-poor aromatic and aliphatic aldehydes (entries 4, 8). For the substrates studied, product yields were in the range of 69–93%.
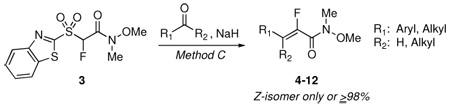
We next explored the scope of the Z-selective olefinations using Method C (NaH, THF, room temperature, molar ratio of carbonyl compound:3:NaH = 1:2:4, aldehyde added to sulfone/NaH mixture). Table 3 shows a series of carbonyl compounds that were subjected to condensation reactions.
TABLE 3.
Condensation Reactions of 3 with Carbonyl Compounds Using NaH
 | ||||
|---|---|---|---|---|
| entry | R1CHO or R1C(O)R2 | conditions, rxn time | products 4–12: (%) E/Z ratio,a yieldb | Z-isomer: δ (ppm); mult, J (Hz) |
| 1 | Method C, 1 h | 4: Z-isomer only, 90% | −120.5; d, 36.6 | |
| 2 | Method C, 1.5 h | 8: 1:99%, 89% | −124.0; d, 36.6 | |
| 3 | 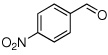 |
Method C, 1.5 h | 5: Z-isomer> 99; 85% | −115.0; d, 33.6 |
| 4 | 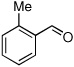 |
Method C, 1.5 h | 9: Z-isomer> 99%; 80% | −122.0; d, 36.6 |
| 5 | 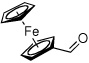 |
Method C, 1.5 h | 10: Z-isomer only; quantitative | −125.1; d, 36.6 |
| 6 | Method C, 1.5 h | 6: 2:98; 99% | −119.7; d, 33.6 | |
| 7 | Method C, 1.5 h | 7: Z-isomer> 99%; 83% | −125.7; d, 36.6 | |
| 8 | Method C, 1.5 h | 11: Z-isomer only; 71% | −124.7; d, 36.6 | |
| 9 | 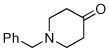 |
Method C,c 45 min | 12: NA; 57% | −126.3 (s) |
| 10 | 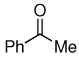 |
Method C, rt–heating, 8 h | Complex rxn mixture -products not identified | -- |
Relative ratio of diastereomers in the crude reaction mixture determined by 19F NMR prior to isolation.
Yields of isolated, purified products.
The ratio of N-benzylpiperidone:3:NaH = 2.5:1:2.
With all aldehydes studied, high Z-stereoselectivity was observed (Z/E ≥99), except in the case of thiophene-2-carboxaldehyde, where 2% of E isomer was formed (Table 3, entry 6). The yields were in the range of 71% to quantitative. The reactivity of ketones with 3 using NaH was tested as well. N-Benzylpiperidone gave a product in 57% yield, but reaction with acetophenone resulted in a complex mixture.
Next, we wanted to test whether the second point of diversity, i.e. the amide in the Julia-Weinreb reagent 3, could be modified prior to olefination reaction. This would allow for the synthesis of second set of Julia reagents from a common precursor. Specifically, we were interested in the synthesis of keto derivatives. Based upon our previous results demonstrating the higher reactivity of aldehydes with fluoro Julia reagents,7–9 we reasoned that keto derivatives of BT-sulfone would undergo selective reactions with aldehydes under mild conditions, without participation of the keto moiety. Since BT-sulfone derivatives are labile under basic conditions, we decided that rather than reaction of 3 with organometallics, the fluorinated sulfide precursor would be a better substrate for this modification. BT-sulfide 1 was therefore subjected to metalation-fluorination using LDA and NFSI in toluene, to give fluoro BT-sulfide derivative 13 in 83% yield.
Reaction of 13 with 2.25 molar equiv of PhLi (−78 °C to room temperature) gave a complex reaction mixture. Upon reaction of 13 with 2.5 molar equiv of PhMgBr at room temperature, some desired product formation was observed in an otherwise complex reaction mixture. Lowering the temperature to −20 °C and use of 6 molar equiv of the Grignard reagent resulted in successful formation of the desired (1,3-benzothiazol-2-ylsulfanyl)fluoromethyl phenyl ketone 14 that was isolated in 82% yield (Scheme 2). Oxidation of 14 using H5IO6 in the presence of catalytic CrO3 in CH3CN19 gave sulfone 16 in 49% isolated yield. In order to test generality, 13 was also reacted with n-propylmagnesium bromide at −20 °C, to give the n-propyl derivative 15 that was isolated in 81% yield. Oxidation of 15 with H5IO6 and catalytic CrO3 in CH3CN19 was fast and smooth at rt to give sulfone 17 in 87% isolated yield (Scheme 2). Since 13 can be potentially converted to 3, we also explored this route (Scheme 2). In fact, 3 can be smoothly prepared from 1 in 68% overall yield, in comparison to the 76% overall yield via Scheme 1. Therefore, 13 serves as a key common intermediate to both types of Julia olefination reagents.
SCHEME 2.

Conversion of 1 to Second Set of Fluoro Julia Olefination Reagents and to 3 via a Common Intermediate
Although α-fluoro-α,β-enones are versatile synthetic intermediates, methods for their synthesis are sparce.13,20 With the two reagents (1,3-benzothiazol-2-ylsulfonyl)fluoromethyl phenyl (16) and n-propyl ketone (17) in hand, we tested their reactivity for the synthesis of α-fluoro-α,β-enones. Reagent 16 was subjected to DBU-mediated olefination with p-methoxybenzaldehyde. A room temperature reaction of 16 in either CH2Cl2 or THF showed very little conversion to product in 24 hours. However, use of refluxing THF resulted in complete conversion to products. In contrast, condensation reactions of 17 led to complex reaction mixtures at higher temperatures. Lowering the reaction temperature to 0 °C resulted in good yields of the α-fluoroenone. Due to lower solubility of 17 in THF, a mixture of CH2Cl2 and THF was used as reaction solvent. In both cases, the reactions were monitored by TLC (after ca 15 min in the case of 16 and after 2 h in the case of 17) and if starting aldehyde was still present, additional sulfone and DBU were added (please see the Experimental Section for details). Condensation reactions of 16 and 17 with some representative aldehydes are displayed in Table 4. In all cases studied, only the Z-isomer was observed. No attempts were made to further optimize the reaction conditions.
TABLE 4.
Condensation Reactions of Fluoro Julia Reagents 16 and 17
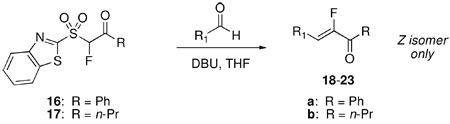 | ||||
|---|---|---|---|---|
| rxn | substrate | sulfone: molar equiv;a DBU: molar equiva | rxn time, T | Products 18–23, yieldb |
| 1 | 16: 3; DBU: 4 | 0.5 h, reflux | R = Ph: 18a, 61% | |
| 2 | 17: 3 ; DBU: 6 | 16 h, 0–5 °C | R = n-Pr. 18b, 89% | |
| 3 | 16: 4; DBU: 6 | 0.5 h, reflux | R = Ph: 19a, 64% | |
| 4 | 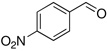 |
16: 2; DBU: 4 | 0.5 h, reflux | R = Ph:20a, 81% |
| 5 | 17: 2; DBU: 6 | 8 h, 0 °C - rt | R = n-Pr: 20b, 86% | |
| 6 | 16: 4; DBU: 3 | 40 min, reflux | R = Ph:21a, 71% | |
| 7 | 17: 3; DBU: 6 | 8 h, 0–5 °C | R = n-Pr: 22b, 90% | |
| 8 | 17: 1.5; DBU: 6 | 3 h, 0–5 °C | R = n-Pr: 23b, 73% | |
Total amount of sulfone and DBU used for complete aldehyde consumption. For good conversions, sequential addition of sulfone and DBU was required (please see the Experimental Section and the Supporting Information for details).
Yields of isolated, purified products.
We were next interested in the application of the methodology to the synthesis of α-fluoro allyl amines. The biological relevance of the α-fluoro allyl amine motif is well documented.1 α-Fluoro-α,β-unsaturated Weinreb amides can be convenient intermediates in α-fluoro allyl amine synthesis (Scheme 3). To explore this application, α-fluoro allyl amine 27 (Scheme 3) was chosen as a target, due to its reported inhibitory activity towards dipeptidyl peptidase II.17 Although we have shown that NaH-mediated condensation of N-benzylpiperidone and 3 led to product 12, we were curious whether the condensation would occur under milder conditions. Successful use of Cs2CO3 as a base has been demonstrated in other Julia-Kocienski olefinations, e.g. in condensations of p-nitrophenyl sulfones21 or in methylenations using tert-butyltetrazolyl sulfones.22 In the present case, Cs2CO3-mediated condensation of 3 with N-benzylpiperidone resulted in a 59% yield of 12 (comparable to the NaH-mediated condensation). In comparison, cyclohexanone gave a 42% yield of condensation product 24 (perhaps due to product volatility). Condensation product 24 was subsequently converted to aldehyde 25 (63%) that was then subjected to imine formation with benzylamine to give 26. Reduction of 26 yielded the desired 27 in 94% yield over two steps.
SCHEME 3.

Synthesis of α-Fluoro Allyl Amine Using Fluoro Julia-Weinreb Reagent 3
CONCLUSIONS
In conclusion, we have synthesized a benzothiazolyl sulfone derived reagent for the synthesis of α-fluorovinyl Weinreb amides via Julia olefination. The reactivity of the reagent was studied under mild, DBU-mediated conditions as well as with NaH. Lower temperatures and relatively nonpolar solvents favor formation of Z-isomer, whereas polar solvents and higher temperatures favor formation of E-isomer. Formation of Z-isomer strongly predominated (>98%) when NaH was used as base. This is to our knowledge the first example showing tunability of reaction condensations leading to the α-fluorovinyl Weinreb amide derivatives. Further, conversion of the amide moiety to a keto functionality in benzothiazolylsulfanyl fluoroacetamide derivative yielded another set of Julia reagents for the synthesis of α-fluoroenones. The methodology presented offers access to two different sets of Julia olefination reagents via a common fluorinated precursor. A series of α-fluoroenones was prepared under mild conditions using DBU as base, with complete Z-stereoselectivity. We have also demonstrated the utility of the reagent for synthesis of α-fluoro allyl amines via Weinreb amides, by preparation of a biologically relevant dipeptidyl peptidase inhibitor.
EXPERIMENTAL SECTION
N-Methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfonyl)fluoroacetamide (3) via Fluorination of 2
A solution of sulfone 2 (1.00 g, 3.33 mmol, 1 molar equiv) in dry toluene (185 mL) was cooled to −78 °C under nitrogen gas. LDA (3.50 mmol, 1.75 mL, 1.05 molar equiv of a 2 M solution in heptane/THF/EtPh) was added to the reaction mixture. After 15 min solid NFSI (1.31 g, 4.17 mmol, 1.25 molar equiv) was added. The mixture was allowed to stir at −78 °C for 50 min, then warmed to rt and stirred for an additional 50 min. Sat aq NH4Cl was added and the mixture was extracted with EtOAc (3 x), and the combined organic layer was washed with sat aq NaHCO3 and brine. The organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude reaction mixture was purified by column chromatography (SiO2, 40% EtOAc in hexanes) to yield 3 (0.905 g, 85%) as a white solid. 1H NMR (500 MHz, CDCl3): δ 8.29 (d, 1H, Ar-H, J = 8.2), 8.04 (d, 1H, Ar-H, J = 7.6), 7.69-7.62 (m, 2H, Ar-H), 6.58 (d, 1H, 2JHF = 47.6), 3.90 (s, 3H), 3.32 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 161.7, 159.7 (d, 2JCF = 22.4), 152.7, 138.0, 128.8, 128.1, 126.1, 122.5, 94.9 (d, 1JCF = 226.1), 62.2, 32.9. 19F NMR (282 MHz, CDCl3): δ−181.5 (d, 2JFH = 48.8). HRMS (ESI) calcd. for C11H11FN2O4S2Na [M + Na]+ 341.0036, found 341.0028.
N-Methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfanyl)fluoroacetamide (13)
A solution of N-methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfanyl)acetamide 1 (2.02 g, 7.51 mmol, 1 molar equiv) in dry toluene (50 mL) was cooled to −78 °C under nitrogen gas. LDA (9.60 mmol, 4.80 mL, 1.28 molar equiv of a 2 M solution in heptane/THF/EtPh) was added to the reaction mixture. After 20 min, solid NFSI (2.84 g, 9.01 mmol, 1.2 molar equiv) was added. The mixture was allowed to stir at −78 °C for 50 min, then warmed to rt and stirred for an additional 50 min. Sat aq NH4Cl (30 mL) was added to the mixture and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 30 mL), and the combined organic layer was washed with sat aq NaHCO3 (30 mL) and brine (30 mL). The organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude reaction mixture was purified by column chromatography (SiO2, 40% EtOAc in hexanes) to yield 13 (1.79 g, 83%) as a pale yellow liquid. 1H NMR (500 MHz, CDCl3): δ 7.98 (d, 1H, Ar-H, J = 8.3), 7.82 (d, 1H, Ar-H, J = 7.8), 7.49-7.36 (m, 3H), 3.81 (s, 3H), 3.28 (s, 3H). 19F NMR (282 MHz, CDCl3): δ −161.0 (d, 2JFH = 51.9). HRMS (ESI) calcd. for C11H11FN2O2S2Na [M + Na]+ 309.0138, found 309.0130.
N-Methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfonyl)fluoroacetamide (3) via Oxidation of 13
H5IO6 (302 mg, 1.32 mmol) was dissolved in CH3CN (20 mL) by vigorous stirring at rt for 30 min. CrO3 (5.0 mg, 0.050 mmol, 15 mol %) was added and the reaction mixture was stirred for an additional 5 min to give an orange colored solution. A solution of fluoro sulfide 13 (95.1 mg, 0.332 mmol) in CH3CN (5 mL) was added dropwise to this mixture, resulting in the formation of a green precipitate. After the addition was complete the reaction mixture was stirred at rt for 3 h at which time TLC (SiO2, 50% EtOAc in hexanes) showed a complete consumption of 13. The reaction mixture was filtered through a Celite pad, the Celite was washed with EtOAc and the solvent was evaporated under reduced pressure. Water was added to the residue (30 mL) and the mixture was extracted with EtOAc (3 × 30 mL), the combined organic layer was washed with sat aq Na2SO3 (30 mL), brine (30 mL) and dried over anhydrous Na2SO4. The organic layer was concentrated under reduced pressure and the crude product was purified by column chromatography (SiO2, 50% EtOAc in hexanes) to afford 3 as a white solid (87.0 mg, 82%).
(1,3-Benzothiazol-2-ylsulfanyl)fluoromethyl Phenyl Ketone (14)
To a solution of fluoro sulfide 13 (1.01 g, 3.51 mmol) in dry THF (10.0 mL) was added PhMgBr (21.0 mL, 1M solution in THF, 21.0 mmol) dropwise at −20 °C. The reaction mixture was stirred at −20 °C and after 1 h TLC (SiO2, 20% EtOAc in hexanes) showed disappearance of 13. The reaction was quenched with sat aq NH4Cl (30 mL), the aqueous layer was extracted with EtOAc (3 × 30 mL) and the combined organic layers were washed with NaHCO3 (30 mL), brine (30 mL) and then dried over Na2SO4. The organic layer was concentrated in vacuo and the crude product was purified by column chromatography (SiO2, 10% EtOAc in hexanes) to afford 14 as a colorless liquid (872 mg, 82%). 1H NMR (500 MHz, CDCl3): δ 8.10 (d, 1H, Ar-H, J = 7.8), 8.03 (d, 1H, Ar-H, J = 7.8), 7.95 (d, 1H, 2JHF = 51.6), 7.83 (d, 1H, Ar-H, J = 7.8), 7.65 (t, 1H, Ar-H, J = 7.8), 7.52 (t, 2H, Ar-H, J = 7.4), 7.41 (t, 1H, Ar-H, J = 7.4), 7.31-7.28 (m, 1H, Ar-H), 7.18-7.17 (m, 1H, Ar-H). 19F NMR (CDCl3): δ −162.1 (d, 2JFH = 51.9). HRMS (ESI) calcd. for C15H10FNOS2Na [M + Na]+ 326.0080, observed 326.0076.
(1,3-Benzothiazol-2-ylsulfanyl)fluoromethyl n-Propyl Ketone (15)
To a stirring solution of n-propylmagnesium bromide, prepared from n-propyl bromide (3.40 mL, 37.1 mmol) and Mg (891 mg, 37.1 mmol) in THF (60 mL), at −20 °C was added a solution of fluoro sulfide 13 (1.77 g, 6.19 mmol) in THF (30 mL) dropwise. After complete addition, the reaction mixture was stirred at −20 °C for 1 h, then warmed to 0 °C and quenched with sat NH4Cl. The reaction mixture was diluted with water and extracted with EtOAc (2 × 30 mL). The combined organic layer was thoroughly washed with water and then with brine, dried over anhydrous Na2SO4 and the solvent was evaporated in vacuo. The crude product was purified by column chromatography (SiO2, 5% EtOAc in hexanes) to afford ketone 15 as a yellow oil (1.35 g, 81%). 1H NMR (500 MHz, CDCl3): δ 7.94 (d, 1H, Ar-H, J = 7.9), 7.81 (d, 1H, Ar-H, J = 7.9), 7.47 (t, 1H, Ar-H, J = 7.6), 7.37 (t, 1H, Ar-H, J = 7.6), 6.83 (d, 1H, 2JHF = 51.0), 2.77 (t, 2H, J = 7.2), 1.71 (sext, 2H, J = 7.3), 0.97 (t, 3H, J = 7.4). 19F NMR (282 MHz, CDCl3): δ −163.6 (d, 2JFH = 51.9). HRMS (ESI) calcd. for C12H12FNOS2Na [M + Na]+ 292. 0237, found 292.0230.
(1,3-Benzothiazol-2-ylsulfonyl)fluoromethyl Phenyl Ketone (16)
H5IO6 (2.62 g, 11.5 mmol) was dissolved in CH3CN (50 mL) by vigorous stirring at rt for 30 min. CrO3 (11.0 mg, 0.110 mmol, 4 mol %) was added and the reaction mixture was stirred for an additional 5 min to give an orange colored solution. A solution of fluoro sulfide 14 (872 mg, 2.87 mmol) in CH3CN (10 mL) was added dropwise to this mixture, resulting in an exothermic reaction and formation of a yellowish precipitate. After the addition was complete the reaction mixture was stirred overnight at which time TLC (SiO2, 25% EtOAc in hexanes) showed a complete consumption of 14. The reaction mixture was filtered through a Celite pad, the Celite was washed with CH3CN and the filtrate was concentrated under reduced pressure. Water was added to the residue and the mixture was extracted with EtOAc (3 × 50 mL), the combined organic layer was washed with sat aq Na2SO3 (30 mL), brine (30 mL) and dried over anhydrous Na2SO4. The organic layer was concentrated under reduced pressure and the crude product was purified by column chromatography (SiO2, 40% EtOAc in hexanes) to afford 16 as a colorless solid (472 mg, 49%). 1H NMR (500 MHz, CDCl3): δ 8.29 (d, 1H, Ar-H, J = 7.8), 8.12 (d, 2H, Ar-H, J = 8.3), 8.05 (d, 1H, Ar-H, J = 7.4), 7.71-7.64 (m, 3H, Ar-H), 7.55 (t, 2H, Ar-H, J = 7.8), 6.81 (d, 1H, 2JHF = 47.9). 13C NMR (125 MHz, CDCl3): δ 185.3 (d, 2JCF = 17.5), 161.6, 152.7, 137.8, 135.5, 134.0, 130.1, 129.1, 129.0, 128.2, 126.1, 122.6, 99.5 (d, 1JCF = 233.7). 19F NMR (282 MHz, CDCl3): δ −179.3 (d, 2JFH = 48.8). HRMS (ESI) calcd. for C15H10FNO3S2Na [M + Na]+ 357.9978, observed 357.9974.
(1,3-Benzothiazol-2-ylsulfonyl)fluoromethyl n-Propyl Ketone (17)
H5IO6 (2.51 g, 11.0 mmol) was dissolved in CH3CN (44 mL) by vigorous stirring at rt for 30 min. CrO3 (5.5 mg, 0.055 mmol, 2 mol %) was added and the reaction mixture was stirred for an additional 5 min to give an orange colored solution. A solution of fluoro sulfide 15 (739 mg, 2.75 mmol) in CH3CN (11 mL) was added dropwise to this mixture, resulting in an exothermic reaction and formation of a precipitate. The reaction mixture was stirred at rt and TLC (SiO2, 20% EtOAc in hexanes) showed a complete consumption of 15 after 30 min. The reaction mixture was filtered through a sintered glass funnel and then through a Celite pad, and the Celite was washed with CH3CN (10 mL). The filtrate was concentrated under reduced pressure at rt, water was added to the residue and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic layer was washed with sat aq Na2SO3 and brine, dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography (SiO2, 15% EtOAc in hexanes) to afford sulfone 17 as a white solid (720 mg, 87%). 1H NMR (500 MHz, CDCl3): δ 8.25 (d, 1H, Ar-H, J = 7.9), 8.04 (d, 1H, Ar-H, J = 7.6), 7.69-7.63 (m, 2H, Ar-H), 5.97 (d, 1H, 2JHF = 48.2), 2.88 (dtd, 1H, J = 18.7, 7.2, 2.7), 2.76 (dt, 1H, J = 18.9, 7.1), 1.71 (sext, 2H, J = 7.3), 0.96 (t, 3H, J = 7.3). 19F NMR (282 MHz, CDCl3): δ −182.3 (d, 2JFH = 48.8). 13C NMR (125 MHz, CDCl3): δ 196.6 (d, 2JCF = 19.7), 161.3, 152.4, 137.4, 128.8, 128.1,125.7, 122.4, 100.6 (d, 1JCF = 237.1), 42.1, 16.0, 13.3. HRMS (ESI) calcd. for C12H12FNO3S2Na [M + Na]+ 324. 0135, found 324. 0124.
Representative Procedures for Condensations of Aldehydes with N-Methoxy-N-methyl-(1,3-benzothiazol-2-ylsulfonyl)fluoroacetamide (3) Using Methods A, B and C
Method A. Synthesis of (E/Z)-2-Fluoro-N-methoxy-N-methyl-3-(4-nitrophenyl)propenamide (5)
To a stirred solution of the p-nitrobenzaldehyde (151 mg, 1.00 mmol, 1 molar equiv) and sulfone 3 (445 mg, 1.4 molar equiv) in dry THF (7.8 mL) at −78 °C was added a cooled (−75 °C) solution of DBU ( 609 mg, 4.00 mmol, 4.0 molar equiv) in dry THF (7.8 mL). The reaction mixture was allowed to stir at −78 °C until complete consumption of aldehyde was observed by TLC (3 h), sat aq NH4Cl (15 mL) was added, the reaction mixture was brought to rt and extracted with Et2O (3 × 50 mL). The combined organic layer was washed with 1N NaOH (40 mL), water and brine, dried over Na2SO4, and the solvent was evaporated under reduced pressure. Analysis of the crude reaction mixture by 19F NMR showed the E/Z product ratio of 3:97. The crude product was purified by column chromatography (20% EtOAc in hexanes) to yield 233 mg (92%) of (E/Z)-5 as pale yellow solid. Although 3% of (E)-5 was detected, NMR data are reported only for the major isomer. Major isomer (Z)-5: 1H NMR (500 MHz, CDCl3): δ 8.23 (d, 2H, Ar-H, J = 8.6), 7.75 (d, 2H, Ar-H, J = 8.5), 6.73 (d, 1H, 3JHF = 35.7), 3.81 (s, 3H), 3.30 (s, 3H). 19F NMR (282 MHz, CDCl3): δ −115.0 (d, 3JFH = 33.6). HRMS (ESI) calcd. for C11H11FN2O4Na [M + Na]+ 277.0595, found 277.0593.
Method B. Synthesis of (E/Z)-2-Fluoro-N-methoxy-N-methyl-3-(2-thienyl)propenamide (6)
To a stirred solution of thiophene-2-carboxaldehyde (45.8 mg, 0.409 mmol, 1.3 molar equiv) and 3 (100 mg, 0.314 mmol, 1.0 molar equiv) in DMPU (2.4 mL) at rt was added solution of DBU (95.7 mg, 0.629 mmol, 2 molar equiv) in DMPU (2.4 mL) dropwise. The reaction mixture was allowed to stir for 18 h, at which time complete consumption of 3 was observed by TLC (SiO2, 30% EtOAc in hexanes). The reaction was quenched with sat aq NH4Cl (5 mL), the aqueous layer was extracted with Et2O (3 × 20 mL), the combined organic layer was washed with 1N NaOH (20 mL), water and brine, dried over Na2SO4, and the solvent was evaporated under reduced pressure. Analysis of the crude reaction mixture by 19F NMR showed the E/Z product ratio of 86:14. The crude product was purified by column chromatography (SiO2, 20% EtOAc in hexanes), to afford 55 mg (81%) of (E/Z)-6 as a pale yellow viscous liquid. For the 1H NMR data of (Z)-6 (minor isomer in the present case), please see the Supporting Information. Major isomer (E)-6: 1H NMR (500 MHz, CDCl3): δ 7.29 (d, 1H, Ar-H, J = 4.3), 7.13 (d, 1H, Ar-H, J = 3.1), 6.98 (app t, 1H, Ar-H, J ~ 4.3), 6.71 (d, 1H, 3JHF = 22.0), 3.75 (s, 3H), 3.30 (s, 3H). (E/Z)-6 HRMS (ESI) calcd. for C9H10FNO2SNa [M + Na]+ 238.0308, observed 238.0301.
Method C. Synthesis of (Z)-2-Fluoro-N-methoxy-N-methyl-3-(4-methoxyphenyl)propenamide (8).11
A suspension of NaH (70.5 mg, 2.94 mmol, 4 molar equiv) and 3 (467 mg, 1.47 mmol, 2 molar equiv) in dry THF (8.4 mL) was stirred at rt under a nitrogen atmosphere for 2 min. A solution of p-methoxybenzaldehyde (100 mg, 0.734 mmol, 1 molar equiv) in dry THF (3.4 mL) was added dropwise. The reaction mixture was allowed to stir at rt for 1.5 h and then quenched with sat aq NH4Cl (10 mL). The mixture was extracted with Et2O (3 × 40 mL), the combined organic layers were washed with 1N NaOH (30 mL), water and brine, and dried over Na2SO4. Analysis of crude reaction mixture by 19F NMR showed the E/Z product ratio of 1:99. The crude product was purified by column chromatography (SiO2, 20% EtOAc in hexanes) to yield 156 mg (89%) of 8 as a light brown semisolid. (Z)-8: 1H NMR (500 MHz, CDCl3): δ 7.57 (d, 2H, Ar-H, J = 8.5), 6.90 (d, 2H, Ar-H, J = 8.8), 6.69 (d, 1H, 3JHF = 37.5), 3.82 (s, 3H), 3.78 (s, 3H), 3.27 (s, 3H). 19F NMR (282 MHz, CDCl3): δ −124.0 (d, 3JFH = 36.6). HRMS (ESI) calcd. for C12H14FNO3Na [M + Na]+ 262.0849, found 262.0844.
General Procedure for Synthesis of 18a23–21a via Condensations of Aldehydes with Fluoro Sulfone 16
To a refluxing solution of aldehyde (1 molar equiv) and DBU (3 molar equiv) in THF (28 mL/mmol of aldehyde) was added sulfone 16 (2 molar equiv, dissolved in THF (4.5 mL/mmol of 16) dropwise. The reaction was stirred at reflux for ca 15 min and the conversion was checked by TLC. If sulfone was consumed and unreacted starting aldehyde was observed, an additional 1 molar equiv of each, DBU in THF (2 mL/mmol of DBU) and solid 16 were added (in the case of o-methoxybenzaldehyde, 3 molar equiv of DBU and 2 molar equiv of 16 were added). If both, sulfone and aldehyde were still present, an additional 1 molar equiv of DBU in THF (2 mL/mmol of DBU) was added. After heating under reflux for an additional 15 min, the conversion was again checked by TLC. If unreacted starting aldehyde was present, an additional 1 molar equiv of solid 16 was added. In all cases, consumption of starting aldehyde was observed after 30–40 min of reflux. The reaction was quenched with sat aq NH4Cl and the mixture was extracted with EtOAc (3 x). The organic layer was washed with sat aq NaHCO3 and brine, dried over anhydrous Na2SO4 and concentrated. The crude product was purified by column chromatography. As representative procedures, condensations of p-nitrobenzaldehyde and thiophene-2-carboxaldehyde with 16 are given in the Supporting Information. For other substrates, see Table 4 for details on reagent stoichiometry, reaction time, temperature and yield. See the Supporting Information for additional details, such as TLC and column chromatographic conditions, spectroscopic data of products, as well as literature reference for known compounds.
General Procedure for Synthesis of 18b,20d 20b, 22b, 23b20d via Condensations of Aldehydes with Fluoro Sulfone 17
A solution of aldehyde (1 molar equiv) and DBU (6 molar equiv) in THF (28.0 mL/mmol of aldehyde) was cooled to 0 °C. Sulfone 17 (2 molar equiv, except for 3-phenylpropanal, where 1.5 molar equiv was used) was dissolved in CH2Cl2 (30.0 mL/mmol of 17) and was added slowly, dropwise to the reaction mixture over 2–3 h (1 h in the case of 3-phenylpropanal). Very slow addition of 17 is critical, since sulfone 17 is unstable under the reaction conditions, thus resulting in higher consumption of 17 upon faster additions. The reaction mixture was allowed to stir at 0 °C until complete disappearance of aldehyde was observed by TLC. If sulfone consumption was observed, an additional 1 molar equiv of 17 was added slowly, dropwise over several hours. Upon disappearance of the aldehyde, sat aq NH4Cl was added to the reaction mixture and the mixture was extracted with EtOAc (2x). The combined organic layer was washed with water, brine, dried over anhydrous Na2SO4 and the solvent was evaporated. Condensations of 17 with p-nitrobenzaldehyde and 3-phenylpropanal are given in the Supporting Information as representative procedures. For other substrates, see Table 4 for details on reagent stoichiometry, reaction time, temperature and yield. See the Supporting Information for additional details, such as TLC and column chromatographic conditions, spectroscopic data of products, as well as literature reference for known compounds.
Synthesis of 24 via Condensation of Cyclohexanone with Fluoro Sulfone 3
To a solution of sulfone 3 (251 mg, 0.788 mmol, 1 molar equiv) in dry DMF (10.0 mL) was added Cs2CO3 (1.03 g, 3.16 mmol, 4 molar equiv) and the color of the reaction mixture turned deep yellowish-orange. The suspension was stirred at rt for 30 min, and a solution of cyclohexanone (154 mg, 1.57 mmol, 2 molar equiv) in dry DMF (2.0 mL) was added. The reaction mixture was stirred at rt for 30 h, sat aq NH4Cl (30 mL) was added and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic layer was washed with sat aq NaHCO3 (30 mL), brine (30 mL) and dried over anhydrous Na2SO4. The solvent was removed in vacuo and the crude product was purified by column chromatography (SiO2, 20% EtOAc in hexanes) to give 24 as a clear liquid (66.6 mg, 42%). 1H NMR (500 MHz, CDCl3): δ 3.73 (s, 3H), 3.23(s, 3H), 2.29-2.23 (m, 4H), 1.64-1.57 (m, 6H). 19F NMR (282 MHz, CDCl3): δ −127.0 (br s). HRMS (ESI) calcd. for C10H16FNO2Na [M + Na]+ 224.1057, observed 224.1055.
Reduction of 24
To a suspension of LiAlH4 (18.0 mg, 0.474 mmol) in dry THF (5.0 mL) at 0 °C was added a solution of 24 (43.0 mg, 0.214 mmol) in dry THF (1.0 mL). The reaction mixture was stirred at 0 °C for 15 min and at rt for 1 h, then cooled to 0 °C and 0.1 N HCl (3 mL) was added to the mixture. The mixture was extracted with Et2O (3 × 30 mL) and the combined organic layer was washed with sat aq NaHCO3 (30 mL), brine (30 mL) and dried over Na2SO4. The organic layer was concentrated under reduced pressure, and the crude product was purified by column chromatography (SiO2, CH2Cl2) to yield 2524 as a colorless liquid (18.0 mg, 63%). 1H NMR (500 MHz, CDCl3): δ 9.79 (d, 1H, 3JHF = 18.0), 2.63-2.61 (m, 2H), 2.44-2.42 (m, 2H), 1.72-1.64 (m, 6H). 19F NMR (282 MHz, CDCl3): δ −137.6 (d, 3JFH = 18.3).
Synthesis of 27 from 25
Step 1: Condensation of 25 with Benzylamine
To a mixture of benzylamine (16.0 mg, 0.149 mmol) and aldehyde 25 (17.0 mg, 0.120 mmol) in dry CH2Cl2 (10.0 mL) molecular sieves (4 Å, 200 mg) were added and the mixture was stirred overnight. After 22 h, TLC (SiO2, 20% EtOAc in hexanes) showed complete consumption of 25. The solvent was evaporated and the crude product 26 (33.0 mg) was subjected to reduction without further purification.
Step 2: Reduction of 26
To a solution of imine 26 (33.0 mg, crude product from step 1) in CH3OH (10.0 mL) was added NaBH4 (11.3 mg) at 0 °C. The reaction mixture was stirred for 1 h and after the starting material was fully consumed, the mixture was evaporated. Sat aq NH4Cl (20 mL) was added to the solid residue and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic layer was washed with NaHCO3 (30 mL), brine (30 mL), and dried over Na2SO4. The organic layer was concentrated and the crude product was purified by silica gel chromatography (20% EtOAc in hexanes) to afford 2717 as a colorless liquid (26.0 mg, 94% yield over two steps). 1H NMR (500 MHz, CDCl3, NH exchanged): δ 7.34-7.31 (m, 3H, Ar-H), 7.27-7.25 (m, 2H, Ar-H), 3.78 (s, 2H), 3.41 (d, 2H, 3JHF = 22.6), 2.24 (br s, 2H), 2.02-1.99 (br m, 2H), 1.64-1.52 (br m, 6H). 19F NMR (282 MHz, CDCl3): δ −121.8 (t, 3JFH = 21.4). HRMS (ESI) calcd. for C15H21FN [M + H]+ 234.1653, observed 234.1652.
Supplementary Material
General procedures using methods A, B and C, representative procedures for condensations of 16 and 17, synthetic details, 1H NMR as well as HRMS data, literature references (where applicable) for 1, 2, (Z)-4, (Z)-6, (Z)-7, (Z)-9-(Z)-11, 12, (Z)-18a-(Z)-21a, (Z)-18b, (Z)-20b, (Z)-22b, (Z)-23b, 1H NMR spectra of 1–25, 27 as well as 13C NMR spectra of 3, 16 and 17. This material is available free of charge via the Internet at http://pubs.acs.org.
ACKNOWLEDGMENT
This work was supported by NSF Grant CHE-0516557, infrastructural support and support for A.K.G. were provided by NIH RCMI Grant 5G12 RR03060. Partial support by PSC CUNY awards 38 and 39 is acknowledged. We thank Dr. Andrew Poss (Honeywell) for a sample of NFSI.
Footnotes
Fluorinated Vinyl Weinreb Amides, Enones and Allyl Amine
REFERENCES
- 1.(a) Welch JT, editor. Selective Fluorination in Organic and Bioorganic Chemistry. Washington, DC: American Chemical Society; 1991. [Google Scholar]; (b) Ojima I, McCarthy JR, Welch JT, editors. Biomedical Frontiers of Fluorine Chemistry. Washington, DC: American Chemical Society; 1996. [Google Scholar]; (c) Bégué J-P, Bonnet-Delpon D. Bioorganic and Medicinal Chemistry of Fluorine. Hoboken, NJ: John Wiley & Sons, Inc; 2008. [Google Scholar]
- 2.(a) Special Issue on Fluorinated Synthons. J. Fluorine Chem. 2004;125:477–645. [Google Scholar]; (b) Soloshonok VA, editor. Fluorine–Containing Synthons. Washington, DC: American Chemical Society; 2005. [Google Scholar]; (c) Soloshonok VA, Mikami K, Yamazaki T, Welch JT, Hoenk JF, editors. Current Fluoroorganic Chemistry: New Synthetic Directions, Technologies, Materials, and Biological Applications. Washington, DC: American Chemical Society; 2007. [Google Scholar]
- 3.Baudin JB, Hareau G, Julia SA, Ruel O. Tetrahedron Lett. 1991;32:1175–1178. [Google Scholar]
- 4.(a) Blakemore PR. J. Chem. Soc., Perkin Trans. I. 2002:2563–2585. [Google Scholar]; (b) Plesniak K, Zarecki A, Wicha J. Top. Curr. Chem. 2007;275:163–250. doi: 10.1007/128_049. [DOI] [PubMed] [Google Scholar]; (c) Aïssa C. Eur. J. Org. Chem. 2009:1831–1844. [Google Scholar]
- 5.Chevrie D, Lequeux T, Demoute JP, Pazenok S. Tetrahedron Lett. 2003;44:8127–8130. [Google Scholar]
- 6.Ghosh AK, Zajc B. Org. Lett. 2006;8:1553–1556. doi: 10.1021/ol060002+. [DOI] [PubMed] [Google Scholar]
- 7.Zajc B, Kake S. Org. Lett. 2006;8:4457–4460. doi: 10.1021/ol0616236. [DOI] [PubMed] [Google Scholar]
- 8.He M, Ghosh AK, Zajc B. Synlett. 2008:999–1004. doi: 10.1055/s-2008-1072513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.del Solar M, Ghosh AK, Zajc B. J. Org. Chem. 2008;73:8206–8211. doi: 10.1021/jo801235x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfund E, Lebargy C, Rouden J, Lequeux T. J. Org. Chem. 2007;72:7871–7877. doi: 10.1021/jo070994c. [DOI] [PubMed] [Google Scholar]
- 11.Alonso DA, Fuensanta M, Gómez-Bengoa E, Nájera C. Adv. Synth. Catal. 2008;350:1823–1829. [Google Scholar]
- 12.(a) Balasubramaniam S, Aidhen IS. Synthesis. 2008:3707–3738. [Google Scholar]; (b) Khlestkin VK, Mazhukin DG. Curr. Org. Chem. 2003;7:967–993. [Google Scholar]; (c) Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- 13.Kanai M, Percy JM. Tetrahedron Lett. 2000;41:2453–2455. [Google Scholar]
- 14.Boumendjel A, Nuzillard J-M, Massiot G. Tetrahedron Lett. 1999;40:9033–9036. [Google Scholar]
- 15.Manjunath BN, Sane NP, Aidhen IS. Eur. J. Org. Chem. 2006:2851–2855. [Google Scholar]
- 16.Alonso DA, Fuensanta M, Gómez-Bengoa E, Nájera C. Eur. J. Org. Chem. 2008:2915–2922. [Google Scholar]
- 17.Van der Veken P, Senten K, Kertesz I, De Meester I, Lambeir A-M, Maes M-B, Scharpe S, Haemers A, Augustyns K. J. Med. Chem. 2005;48:1768–1780. doi: 10.1021/jm0495982. [DOI] [PubMed] [Google Scholar]
- 18.Procedure described for the synthesis of 2-chloro-N-methoxy-N-methylacetamide was used (ref. 14). 2-Bromo-N-methoxy-N-methylacetamide:Hirner S, Panknin O, Edefuhr M, Somfai P. Angew. Chem. Int. Ed. 2008;47:1907–1909. doi: 10.1002/anie.200704689.Mechelke MF, Meyers AI. Tetrahedron Lett. 2000;41:4339–4342.
- 19.Xu L, Cheng J, Trudell ML. J. Org. Chem. 2003;68:5388–5391. doi: 10.1021/jo030031n. [DOI] [PubMed] [Google Scholar]
- 20.(a) Chen C, Wilcoxen K, Zhu Y-F, Kim K-i, McCarthy JR. J. Org. Chem. 1999;64:3476–3482. doi: 10.1021/jo982200n. [DOI] [PubMed] [Google Scholar]; (b) Chen C, Wilcoxen K, Huang CQ, Strack N, McCarthy JR. J. Fluorine Chem. 2000;101:285–290. [Google Scholar]; (c) Bainbridge JM, Corr S, Kanai M, Percy JM. Tetrahedron Lett. 2000;41:971–974. [Google Scholar]; (d) Dutheuil G, Paturel C, Lei X, Couve-Bonnaire S, Pannecoucke X. J. Org. Chem. 2006;71:4316–4319. doi: 10.1021/jo0604787. [DOI] [PubMed] [Google Scholar]; (e) Prakash GKS, Chacko S, Vaghoo H, Shao N, Gurung L, Mathew T, Olah GA. Org. Lett. 2009;11:1127–1130. doi: 10.1021/ol8029627. [DOI] [PubMed] [Google Scholar]
- 21.Mirk D, Grassot J-M, Zhu J. Synlett. 2006:1255–1259. [Google Scholar]
- 22.Aïssa C. J. Org. Chem. 2006;71:360–363. doi: 10.1021/jo051693a. [DOI] [PubMed] [Google Scholar]
- 23.Hata H, Kobayashi T, Amii H, Uneyama K, Welch JT. Tetrahedron Lett. 2002;43:6099–6102. [Google Scholar]
- 24.Sauvetre R, Masure D, Chuit C, Normant JF. Synthesis. 1978:128–130. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General procedures using methods A, B and C, representative procedures for condensations of 16 and 17, synthetic details, 1H NMR as well as HRMS data, literature references (where applicable) for 1, 2, (Z)-4, (Z)-6, (Z)-7, (Z)-9-(Z)-11, 12, (Z)-18a-(Z)-21a, (Z)-18b, (Z)-20b, (Z)-22b, (Z)-23b, 1H NMR spectra of 1–25, 27 as well as 13C NMR spectra of 3, 16 and 17. This material is available free of charge via the Internet at http://pubs.acs.org.