

Abstract
Wegener's granulomatosis (WG) is an autoimmune condition marked by vasculitis of small and medium sized vessels particularly affecting the upper respiratory tract and kidneys. There is a strong mucosal component similar to other autoimmune conditions such as systemic lupus erythematosus and Behçet's disease. While the pathogenesis of WG is not completely known, auto-antibodies such as IgG ANCAs have been implicated in endovascular damage and modulation of neutrophil / monocyte responses by Fc receptor (FcR) signaling. Due to the substantial mucosal involvement in WG (oral, nasal, and upper respiratory tract involvement), it is probable that IgA antibodies (perhaps IgA ANCAs) play a role in disease. Given discrepancies in associating ANCA levels with disease activity, future work should determine if IgA ANCAs are present in WG patients and examine the biology underlying the ANCAs' signaling partners - the FcRs.
Clinical Vignette
A 57 year old Caucasian gentleman hobbles into the exam room during a Monday morning Internal Medicine clinic at the VA. He is obviously in pain, grimacing and bracing against the counter, as he lowers himself into a chair. His daughter grabs his arm to help him sit, but he shrugs her off. Once comfortable, he grabs a Kleenex from the counter and mops up his nosebleed that has begun trickling onto his shirt. When asked if he wants a wet paper towel, there is no response. His daughter chimes in, “I'm sorry, he doesn't hear so well anymore. It's been getting worse the past year and a half; you just have to speak up sometimes.”
When eliciting his chief complaint, he reports feeling “fine”, but his daughter informs that he has been having sinus infections and nose bleeds intermittently for almost two years. “It might be due to allergies though since his eyes get red from time to time.” As he covers his mouth to cough, several small purpuritic lesions come into view on his elbow. When asked about his pain when sitting down, he responds, “Yea, sometimes my hip or knee hurt. I'm just getting old, but I try not to let it keep me from working.” Upon further questioning, the patient admits that the joint pain, sinus problems, and hearing loss all started about the same time 18 months ago, lasted for several months, improved, and are now returning.
This is his first visit to a physician in thirty years since he had a hernia repaired in the late 1970s. He has a 20 pack year smoking history, drinks alcohol occasionally, is widowed, and works at a quarry. He has a brother with rheumatoid arthritis.
On physical exam, his temperature is 98.9°F, and his vital signs are all stable, except for mild hypertension. Both hearing and visual acuity are reduced with conjunctivitis present. His nasal mucosa appears inflamed with brown crusted lesions on his septum. His oral cavity is moist with multiple half-centimeter ulcers seen on his buccal mucosa. There are no cardiac murmurs, but stridor is present over his trachea with diffuse coarse breath sounds in his lungs. His abdominal exam is normal. There are purpuritic lesions on his right elbow and splinter hemorrhages in his nail beds. Both his right knee and hip appear stiff and painful when moving but are not red or warm to touch.
Routine labwork reveals an elevated creatinine, moderately high blood glucose, mild eosinophilia, and high LDL. His total lymphocyte count, hematocrit, and electrolytes are all normal. Urine analysis reveals glucose, microhematuria, and red cell casts with no indication of a urinary tract infection. A routine nose swab is positive for MRSA.
His constellation of symptoms, including respiratory, upper airway, renal, musculocutaneous, ocular, and auditory involvement, prompts additional labwork and a chest x-ray. The radiograph shows multifocal cavitary lesions. A rheumatological panel reveals an equivocal rheumatoid factor and positive PR3-specific anti-neutrophil cytoplasmic antibodies.
Introduction
Several autoimmune conditions present with mucosal manifestations, such as the oral ulcers, epistaxis, and conjunctivitis mentioned in the clinical vignette. Among others, examples of mucosal autoimmune conditions include systemic lupus erythematosus (SLE) (in which presence of oral ulcers is one of the diagnostic criteria [1]), Behçet's disease (in which oral ulceration is required for diagnosis [2]), pemphigus vulgaris (PV) (in which mucosal ulcers are almost always present [3]), Crohn's disease (in which inflammation can occur in any portion of the gastrointestinal tract), and, the condition described above, Wegener's granulomatosis (WG) [4]. Auto-antibodies, such as anti-nuclear antibodies (ANA) in SLE or anti-desmoglein 3 in PV, are present in several of these conditions providing another immunopathologic similarity between them. Evaluating auto-antibody function in conditions with mucosal manifestations may provide insights for additional research into the pathogenesis of these complex diseases. Given the involvement of upper airway, nasal, and oral mucosa and the established role of auto-antibodies in the disease, WG provides an important model for the role of antibodies in mucosal autoimmunity.
WG, one of the anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitides (AAV), is an autoimmune condition involving small and medium sized vessels. This disease frequently manifests in the respiratory tract (including upper airways) and kidneys [5; 6]. Over 90% of WG patients present with inflammation of their oral or nasal mucosa. In fact, WG is diagnosed by the presence of two out of the 4 criteria established by the American College of Rheumatology (ACR), which includes nasal or oral inflammation, abnormal chest radiograph, excessive urinary sediment, and granulomatous inflammation on biopsy [4]. The Chapel Hill Consensus criteria for systemic vasculitis are another means of classifying disease; it places more emphasis on biopsy results [7]. While not an ACR diagnostic criteria for enrollment in clinical trials, approximately 93% of WG patients have detectable ANCA titers during active disease [8]. The diagnostic emphasis on oral/nasal inflammation and respiratory tract involvement combined with the frequent presence of ANCA support the use of WG as a model of antibody involvement in mucosal autoimmunity.
Although the specific etiology of WG is not known, WG is a complex immunological disease resulting from certain environmental triggers occurring on the background of genetic predisposition [9; 10]. However, what specific factors contribute to a condition that targets primarily the upper respiratory tract and kidney? What elements lead to a condition with systemic vascular and targeted end-organ effects? While WG serves as an example of auto-antibody involvement in mucosal immunity, examining unique features of this disease allows better understanding of the clinical immunology underlying the disorder and of applications for future research and treatment of this vasculitis.
In this short analytical review, we use WG as a model for examining themes of auto-antibodies and mucosal immunity and for evaluating immunological mechanisms unique to this disease.
Wegener's Granulomatosis: Epidemiology and Susceptibility
WG is predominant in Caucasians (97.7% of cases) yet its prevalence is rare (approximately 1 out of 33,000 Caucasian individuals) [11; 12; 13; 14]. Peak onset occurs between ages 45 and 65 with reports showing either a higher prevalence among men [15] or no gender bias [16]. The patient described above is typical demographically for WG – a white male in his late fifties. There also appears to be a differential geographic distribution related to WG and other AAVs. WG is more common in Northern Europe with decreasing prevalence when traveling southward towards the equator [9; 11; 12; 13; 16; 17]. In contrast, microscopic polyangiitis, another AAV, is more prevalent in decreasing latitudes [17]. A similar pattern of prevalence related to relative location to the equator has been observed in the Southern Hemisphere as well [14]. This spread may reflect differing environmental exposures that trigger the respective conditions, may reflect genetic admixture among European individuals, or, likely, may result from some combination of the two.
Genes that have been associated with WG susceptibility encode an array of immunologically relevant molecules, many of which are located in the Major Histocompatibility Complex (MHC) [10]. Alleles of HLA-DPB1 and retinoid X receptor β (RXRB), two neighboring genes in the immune-gene dense MHC class II region, confer one of the strongest associations of any genomic region [18; 19]. Polymorphisms in the genes for a protein tyrosine phosphatase, PTPN22 [20], and a T lymphocyte negative regulator, CTLA4 [21], associate with WG as well as numerous other autoimmune conditions. A genetic study focusing on interleukin 10 (IL10) suggests this cytokine influences auto-antibody production in WG patients [22]. Other immunologically related molecules, including integrin beta 2 (ITGB2, CD18) [23], the SERPIN cluster on Chromosome 14q32.1 [24], and the Fc receptors (FcR) FCGR2A and FCGR3A [25], are implicated by genomic data in WG pathogenesis.
Data have also revealed specific environmental triggers for disease onset or relapse. Chronic nasal carriage of Staphylococcus aureus, as was seen in the clinical vignette, predisposes to disease relapse [26; 27]. This helps explain why adjuvant therapy with trimethoprim-sulfamethoxazole (an antibiotic also known as co-trimexazole, Septra®, or Bactrim®) reduces the rate of active disease relapse in a subgroup of WG patients [28] (although it is also possible that this drug is actually treating bacterial sinusitis mistaken for a WG flare). Inhalation of silica dust in an occupational setting, such as mining, quarrying (the profession of the patient described above), or farming, has been shown to influence emergence of AAVs but not necessarily WG [29]. Various agricultural exposures such as solvents and silica also increase the risk of an ANCA-associated vasculitis [30].
Auto-antibodies in WG Pathogenesis
In WG, vascular damage has been largely attributed to excessive activation of neutrophils and monocytes. Such ill-fated immune responses are derived from a combination of genetic and environmental factors such as those mentioned above [10; 31]. Given the role of neutrophils and monocytes in WG pathogenesis, it is not surprising that the most common immunological marker of disease is the presence of ANCAs [32], as were detected in the patient from the clinical vignette.
ANCAs bind enzymes located in the primary azurophilic granules of neutrophils and in the lysosomes of monocytes. (It is important to note that regardless of the name ANCA targets are expressed by and can activate monocytes.) There are six identified molecules ANCAs recognize as autoantigens; the two most commonly tested in a clinical setting include cytoplasmic and perinuclear ANCAs (cANCA, pANCA). (Their names are derived from their fluorescence staining appearance in neutrophils [32; 33; 34].)
cANCAs target proteinase 3 (PR3, PRTN3 [19p13.3]), a serine protease. PRTN3 contains a polymorphism in a transcription factor binding site of the promoter region that has been genetically associated with WG susceptibility [35]. One report observed cANCAs in more than 90% of patients with active disease and 40% of patients in remission [8].
pANCAs target myeloperoxidase (MPO [17q23.1]), which, like cANCAs, has been genetically associated with WG [36] and are observed in higher levels in patients with active disease [37]. While cANCAs are more common among WG patients, pANCAs are found in higher titers in other vasculitic conditions such as microscopic polyangiitis and Churg-Strauss syndrome [38].
Even though ANCAs are regularly detected among WG patients [39], their presence is not required as a diagnostic criteria from the ACR [4]. However, evidence is compelling as to ANCA involvement in WG pathogenesis: 1.) Neutrophils, which express ANCA targets, have long been observed in the renal lesions of WG patients [5; 6; 40]. 2.) Autoantibodies specific to PR3 (cANCAs) contribute to the inflammatory damage in WG by cross-linking target with antibody receptors or directly initiating a signal cascade to activate an inflammatory response [41; 42; 43]. ANCAs' binding (and subsequent immune reactions) are antigen dependent with cANCA and pANCA inducing differential endpoints in neutrophils activation [39]. In terms of WG, recent work has suggested that cANCAs prime CD14-dependent monocytes and neutrophils [44] to produce cytokines [e.g. tumor necrosis factor (TNF)]. These effects occur by upregulating toll-like receptor (TLR) and nucleotide-binding oligomerization containing molecules (NOD) in response to bacterial antigens [45]. These data also provide a molecular explanation for the relationship of bacterial infection (Staphyloccous aureus) with the incidence of WG [46]. In addition to generating an inflammatory response, TNF-alpha induces an increased surface expression of PR3 on neutrophils [47], thereby accelerating the impact of ANCAs on immune cell activation. 3.) Once bound to neutrophil surface PR3, the Fc portions of ANCAs are recognized by IgG Fc gamma receptors to initiate a signaling cascade [48; 49]. 4.) ANCA-Fc gamma receptor binding triggers an inflammatory reaction such as release of cytokines and chemotactic factors [50; 51] or by causing direct cytotoxicity on endothelial cells such as those in the vasculature [52]. To date, the TH1 cytokine response has been the most examined in WG [51]; however, new evidence also points to a role for the TH17 pathway [53].
As mentioned above, ANCAs may also directly damage endothelial cells in WG. Alveolar cells removed from WG patients after pneumonectomy show a higher level of PR3 expression than alveolar cells removed from non-WG patients. These pneumocytes also showed higher levels of PR3 mRNA expression than alveolar macrophages or neutrophils isolated from the lung tissue [54] suggesting the mechanism of ANCA-mediated inflammation may be direct interaction with target tissues. A similar finding has been reported in the kidney, with glomerular cell expression level of PR3 being associated with crescent formation [55]. It would be interesting to determine PR3 expression level in mucosal tissues impacted by WG.
Another theory suggests that auto-antibodies form specific to the complementary / anti-sense protein of PR3 [56] and that memory T cells from some ANCA-related vasculitic patients recognize the complementary protein [57]. While the specific mechanisms regarding induction of autoimmunity by anti-sense antibodies are not fully understood, the complementary PR3 antibodies can specifically bind plasminogen [58] suggesting a relationship between ANCAs and the coagulation cascade.
Fc Receptors: Signaling Partners of ANCAs
Antibodies, such as ANCAs, signal via Fc receptors (FcRs) to provide immunoregulatory signals including activation and/or inhibition of immune cells [59] such as neutrophils and monocytes [60]. By recognizing the constant portion of antibodies and acute phase molecules such as C-reactive protein [61], FcRs are central to regulating an immune cell's activity in producing an appropriate immune response or an autoimmune reaction. See Figure 1.
Figure 1. Immune regulation by Fc receptors of granuloma formation in Wegner's granulomatosis.
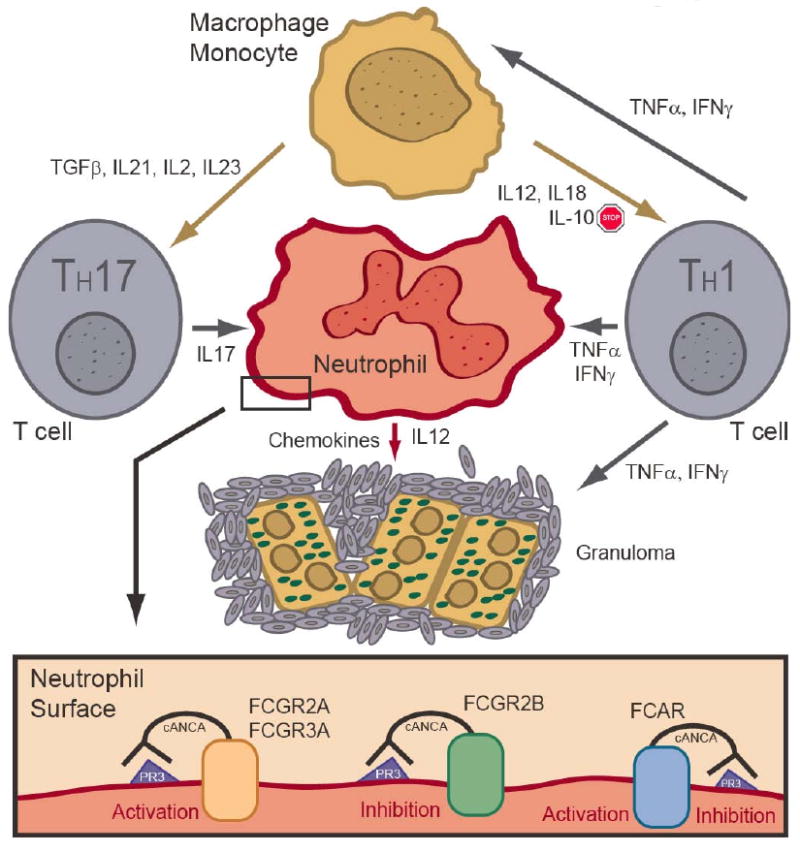
This cartoon depicts some of the cytokines and immune cells involved in granuloma formation. The neutrophil is a central player in WG pathogenesis and is activated and/or inhibited by ANCA signals through the FcRs.
In WG, the downstream effects of cANCA–FcR signaling result in an oxidative burst leading to inflammatory / vasculitic consequences [42; 62]. cANCAs are known to interact with FCGR3B (CD16B) and FCGR2A (CD32A) resulting in neutrophil activation [49; 63]. ANCA binding with FCGR3B also induces cell adhesion thereby promoting a pro-inflammatory state [64]. While the molecular pathway from ANCA-FcR binding to oxidative burst is not completely elucidated, FcR binding by ANCA is required for the Syk phosphorylation involved in neutrophils activation [65] providing evidence of the necessity of the ANCA-FcR interaction.
Phosphatidylinositol 3 kinase (PI3K) activation and subsequent recruitment of protein kinase B (PKB), a serine-threonine kinase, have been observed in immune cell activation by Fc gamma receptors [66; 67]. PI3K activation is also observed in neutrophils stimulated by ANCAs supporting a role for Fc gamma receptors in WG. However, the kinetics of PKB activation differed between conventional Fc gamma receptor binding and ANCA-FcR binding in neutrophils suggesting the involvement of other co-modulating receptors in ANCA stimulation [48].
Even though FCGR2A and FCGR3B have been the focus of research to date, it is possible that other FcRs are involved in ANCA recognition on neutrophils and monocytes especially since other receptors likely participate [48]. Additional FcRs are known to be expressed on neutrophils and monocytes such as the IgA Fc receptor, FCAR (CD89) [68], and the inhibitory Fc gamma receptor, FCGR2B (CD32B) [69].
Genetic Data for Influence of Fc Receptors in WG
Genetic variations within FcR genes, both structural and allelic, are known to influence susceptibility to disease, which emphasizes the importance of these molecules in WG pathogenesis. Polymorphisms in FCGR2A and FCGR3A have been linked to risk of disease relapse among WG patients by allowing chronic nasal carriage of Staphylococcus aureus and altering interactions with ANCAs [25]. Other studies have not found a relationship between WG and FCGR2A variation [70]. However, due to the low prevalence of WG, studies examining genetic associations have analyzed limited numbers of patients with a corresponding limitation in statistical power.
FCGR3B also contains variants, particularly the NA1 allele, that may influence ANCA-associated vasculitis [71; 72]. Neutrophils from individuals with the FCGR3BNA1 allele are more likely to release alpha-defensins, a chemotactic agent, upon stimulation with ANCAs, providing an example of how FcR genetic variation may impact WG [50].
Copy number variation (CNV) is the result of an entire gene or gene segment being duplicated or deleted in some individuals. An altered number of genes may lead to a relative overexpression of protein (in the case of higher copy number) or a deficiency of protein (in the case of gene deletion). Compared to healthy controls, FCGR3B CNV has been associated with both SLE and WG [73]. In SLE, disease susceptibility appears to be associated with a gene deletion; in WG, disease susceptibility is associated with a higher copy number. This suggests a role for FcRs in differentiating between systemic and organ-specific autoimmune reactions [73]. FCGR3B copy number has been correlated with FCGR3B expression level on the cell surface and in soluble form, as well as correlated with neutrophil processing of immune complexes [74].
Additional genotyping of FcR polymorphisms is needed in larger collections to understand more fully the impact of these molecules in WG. Variants among Fc gamma receptors have been associated with other autoimmune conditions characterized by mucosal involvement such as Behçet's disease [75], Crohn's disease [76], and SLE [73; 77]. The breadth of associations highlights the importance of FcR genetics in mucosal autoimmunity. It is also possible that FcR variants will help explain particular disease manifestations within WG. For example, it is likely that variants of FCGR3B are related to renal manifestations given the relationship of FCGR3B CNV to glomerulonephritis in rats [78], lupus nephritis in humans [78; 79], and end stage renal disease in humans [80]. The main barrier in gaining this understanding will be assembling collections with sufficient power to detect associations, especially among clinical subgroups, given the low prevalence of WG.
ANCAs and WG: Is the Mechanism IgG Exclusive?
While it is clear that ANCAs are involved with WG pathogenesis [81], it is less certain if their presence or titer level corresponds with disease activity. Some reports suggest that ANCA titer at the end of aggressive therapy regimen relates to risk of relapse [82; 83; 84]; however, another study reports no relationship between relapse and cANCA level [85]. It would be useful to gauge the risk of relapse and necessary length of cyclophosphamide therapy, which is the first line pharmacologic treatment for WG, based on an immunological marker (i.e cANCA titer) given the adverse effects of the medication such as risk of bladder cancer, leukemia, and lymphoma [86]. Due to the contradictory results of the above studies, perhaps a marker other than the IgG cANCA would be a more appropriate or adjunct marker of disease activity.
To date, both experimental studies and clinical diagnostic tools focus on the IgG isotype of ANCA as functionally important [81; 87; 88]. This is understandable since IgG is the most common isotype in general, since IgG ANCA have been clinically related to WG disease activity [81], and since genetic studies have implicated IgG FcRs with WG susceptibility and relapse [25; 73]. However, there may be a role in WG for other immunoglobulin (Ig) isotypes, and another isotype may serve as a marker for disease activity or risk of relapse.
Incubation with an IgM ANCA derived from a patient with severe vasculitis inhibits neutrophil adhesion and increases phagocytosis of apoptotic neutrophils by macrophages, which differ from the pro-inflammatory effects of IgG ANCA incubation [89]. This study focused on an ANCA specific to MPO, which, while present at times, is not the most common ANCA target in WG. The IgM ANCA isotype has also been observed in a patient with pulmonary hemorrhage and acute renal failure [90], symptoms commonly reported in WG. IgM ANCAs may be present and modulating disease severity in WG.
IgE ANCAs have not been reported in the experimental setting or in a case study isolated from a patient. While unlikely to impact WG pathogenesis, IgE ANCAs could play a role in another vasculitis such as Churg-Strauss syndrome that is known to have positive ANCA titers and high IgE levels. It is not clear if IgE ANCAs have been sought and not found or simply not investigated.
The most likely Ig isotype, other than IgG, to influence WG is IgA. As discussed above (and seen in the clinical vignette), mucosal manifestations are common among WG patients with nasal / oral inflammation and respiratory tract involvement being diagnostic markers of disease [4]. Since IgA is the most prevalent antibody isotype at mucosal surfaces [91] and since ANCAs are the most prevalent auto-antibodies involved in WG, it is probable that IgA ANCAs are present and impacting disease in WG patients. WG patients frequently manifest glomerular damage [4], and IgA deposition is the most common cause of glomerulonephritis [92] providing more circumstantial evidence for IgA ANCA involvement in WG.
IgA ANCAs have been observed in erythema elevatum diutinum [93], which is a neutrophil-associated vasculitis of the skin. This identification establishes a precedent for IgA ANCA involvement in neutrophil-related and vasculitic diseases. Other conditions with observed IgA ANCAs include cutaneous vasculitis [94], ulcerative colitis [95], and Henoch-Schönlein purpura [96]. It needs to be determined if IgA ANCAs can be detected in WG patients and if their presence mediates the disease process.
One possible avenue of investigation is the role of the FcR for IgA (FCAR, CD89) in WG. If IgA ANCAs are present, this molecule would be central to any downstream effects. FCAR is a particularly interesting FcR since it has recently been shown to direct both pro-inflammatory and anti-inflammatory effects [97; 98] in an allele specific manner [99]. The activating or inhibitory effects of IgA are due, at least in part, to differential stoichiometric binding of IgA with FCAR. When two FCAR molecules are complexed together on the cell surface, their close proximity creates a different Fc binding site for IgA that signals activation when engaged. When FCAR remains as a monomer on the cell suface, engagement of the traditional Fc binding site signals the cell to downregulate an immune response [97]. In fact, the binding site of IgA to FCAR impacts immune evasion by Staphyloccus aureus to allow chronic nasal carriage [100], a risk factor for WG susceptibility [26]. Supposing that IgA ANCAs are present in WG patients, such paradoxical regulation by FCAR could explain a role for ANCAs in the range of severity observed among WG patients [33; 101]. It needs to be determined if these other ANCA isotypes are present in WG patients and what, if any, impact they have on pathogenesis.
Summary
WG is a rare autoimmune vasculitic condition characterized in part by mucosal involvement and induced by auto-antibody mechanisms. While the pathogenesis of WG is not completely understood, IgG ANCAs have been implicated in causing direct damage to endovasculature and in modulating neutrophil and monocyte responses via FcR signaling, as supported by both functional and genetic studies. Given the prominent mucosal component in WG (oral, nasal, and upper respiratory tract involvement), it is probable that some form of IgA antibodies are present and play a role in disease. IgA ANCAs are a likely candidate to meet that description. Due to discrepancies in relating ANCA titer with disease activity, future work should focus on identifying alternative ANCAs, such as of the IgA isotype, and evaluating the biology and genetics of the auto-antibody signaling partners, the Fc receptors. Additional research into understanding the role of ANCAs in the mucosal manifestations of WG may provide insight into diseases involving the mucosal immune system as a whole and help explain the oral ulcers and epistaxis observed in the patient above.
Acknowledgments
The authors received salary support from NIH R01-AR33062, R01-AR42476, P01-AR49084, and T32 AR07450 (NIAMS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 2.Criteria for diagnosis of Behcet's disease. International Study Group for Behcet's Disease. Lancet. 1990;335:1078–80. [PubMed] [Google Scholar]
- 3.Black M, Mignogna MD, Scully C. Number II. Pemphigus vulgaris. Oral Dis. 2005;11:119–30. doi: 10.1111/j.1601-0825.2005.01139.x. [DOI] [PubMed] [Google Scholar]
- 4.Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, Calabrese LH, Fries JF, Lie JT, Lightfoot RW, Jr, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990;33:1101–7. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 5.Wegener F. About a peculiar rhinogenic granulomatosis with marked involvement of the arterial system and kidneys. Beitr Pathol Anat. 1939;102:533–553. [Google Scholar]
- 6.Wegener F. Über generalisierte, septische efäberkrankungen. Verh Dtsch Pathol Ges. 1936;29:202–210. [Google Scholar]
- 7.Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallenberg CG, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 8.Nolle B, Specks U, Ludemann J, Rohrbach MS, DeRemee RA, Gross WL. Anticytoplasmic autoantibodies: their immunodiagnostic value in Wegener granulomatosis. Ann Intern Med. 1989;111:28–40. doi: 10.7326/0003-4819-111-1-28. [DOI] [PubMed] [Google Scholar]
- 9.Mahr AD, Neogi T, Merkel PA. Epidemiology of Wegener's granulomatosis: Lessons from descriptive studies and analyses of genetic and environmental risk determinants. Clin Exp Rheumatol. 2006;24:S82–91. [PubMed] [Google Scholar]
- 10.de Lind van Wijngaarden RA, van Rijn L, Hagen EC, Watts RA, Gregorini G, Tervaert JW, Mahr AD, Niles JL, de Heer E, Bruijn JA, Bajema IM. Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody associated vasculitis: the cause is hidden, but the result is known. Clin J Am Soc Nephrol. 2008;3:237–52. doi: 10.2215/CJN.03550807. [DOI] [PubMed] [Google Scholar]
- 11.Cotch MF, Hoffman GS, Yerg DE, Kaufman GI, Targonski P, Kaslow RA. The epidemiology of Wegener's granulomatosis. Estimates of the five-year period prevalence, annual mortality, and geographic disease distribution from population-based data sources. Arthritis Rheum. 1996;39:87–92. doi: 10.1002/art.1780390112. [DOI] [PubMed] [Google Scholar]
- 12.Koldingsnes W, Nossent H. Epidemiology of Wegener's granulomatosis in northern Norway. Arthritis Rheum. 2000;43:2481–7. doi: 10.1002/1529-0131(200011)43:11<2481::AID-ANR15>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Watts RA, Lane SE, Bentham G, Scott DG. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis Rheum. 2000;43:414–9. doi: 10.1002/1529-0131(200002)43:2<414::AID-ANR23>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 14.Gibson A, Stamp LK, Chapman PT, O'Donnell JL. The epidemiology of Wegener's granulomatosis and microscopic polyangiitis in a Southern Hemisphere region. Rheumatology (Oxford) 2006;45:624–8. doi: 10.1093/rheumatology/kei259. [DOI] [PubMed] [Google Scholar]
- 15.Watts RA, Lane SE, Scott DG, Koldingsnes W, Nossent H, Gonzalez-Gay MA, Garcia-Porrua C, Bentham GA. Epidemiology of vasculitis in Europe. Ann Rheum Dis. 2001;60:1156–7. doi: 10.1136/ard.60.12.1156a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abdou NI, Kullman GJ, Hoffman GS, Sharp GC, Specks U, McDonald T, Garrity J, Goeken JA, Allen NB. Wegener's granulomatosis: survey of 701 patients in North America. Changes in outcome in the 1990s. J Rheumatol. 2002;29:309–16. [PubMed] [Google Scholar]
- 17.Watts RA, Gonzalez-Gay MA, Lane SE, Garcia-Porrua C, Bentham G, Scott DG. Geoepidemiology of systemic vasculitis: comparison of the incidence in two regions of Europe. Ann Rheum Dis. 2001;60:170–2. doi: 10.1136/ard.60.2.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagiello P, Gencik M, Arning L, Wieczorek S, Kunstmann E, Csernok E, Gross WL, Epplen JT. New genomic region for Wegener's granulomatosis as revealed by an extended association screen with 202 apoptosis-related genes. Hum Genet. 2004;114:468–77. doi: 10.1007/s00439-004-1092-z. [DOI] [PubMed] [Google Scholar]
- 19.Szyld P, Jagiello P, Csernok E, Gross WL, Epplen JT. On the Wegener granulomatosis associated region on chromosome 6p21.3. BMC Med Genet. 2006;7:21. doi: 10.1186/1471-2350-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jagiello P, Aries P, Arning L, Wagenleiter SE, Csernok E, Hellmich B, Gross WL, Epplen JT. The PTPN22 620W allele is a risk factor for Wegener's granulomatosis. Arthritis Rheum. 2005;52:4039–43. doi: 10.1002/art.21487. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Huang D, Paris PL, Sauter CS, Prock KA, Hoffman GS. An analysis of CTLA-4 and proinflammatory cytokine genes in Wegener's granulomatosis. Arthritis Rheum. 2004;50:2645–50. doi: 10.1002/art.20385. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Y, Giscombe R, Huang D, Lefvert AK. Novel genetic association of Wegener's granulomatosis with the interleukin 10 gene. J Rheumatol. 2002;29:317–20. [PubMed] [Google Scholar]
- 23.Meller S, Jagiello P, Borgmann S, Fricke H, Epplen JT, Gencik M. Novel SNPs in the CD18 gene validate the association with MPO-ANCA+ vasculitis. Genes Immun. 2001;2:269–72. doi: 10.1038/sj.gene.6363781. [DOI] [PubMed] [Google Scholar]
- 24.Borgmann S, Endisch G, Urban S, Sitter T, Fricke H. A linkage disequilibrium between genes at the serine protease inhibitor gene cluster on chromosome 14q32.1 is associated with Wegener's granulomatosis. Clin Immunol. 2001;98:244–8. doi: 10.1006/clim.2000.4962. [DOI] [PubMed] [Google Scholar]
- 25.Dijstelbloem HM, Scheepers RH, Oost WW, Stegeman CA, van der Pol WL, Sluiter WJ, Kallenberg CG, van de Winkel JG, Tervaert JW. Fcgamma receptor polymorphisms in Wegener's granulomatosis: risk factors for disease relapse. Arthritis Rheum. 1999;42:1823–7. doi: 10.1002/1529-0131(199909)42:9<1823::AID-ANR5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 26.Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. 1994;120:12–7. doi: 10.7326/0003-4819-120-1-199401010-00003. [DOI] [PubMed] [Google Scholar]
- 27.Popa ER, Stegeman CA, Abdulahad WH, van der Meer B, Arends J, Manson WM, Bos NA, Kallenberg CG, Cohen Tervaert JW. Staphylococcal toxic-shock-syndrome-toxin-1 as a risk factor for disease relapse in Wegener's granulomatosis. Rheumatology (Oxford) 2007;46:1029–33. doi: 10.1093/rheumatology/kem022. [DOI] [PubMed] [Google Scholar]
- 28.Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener's granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N Engl J Med. 1996;335:16–20. doi: 10.1056/NEJM199607043350103. [DOI] [PubMed] [Google Scholar]
- 29.Hogan SL, Cooper GS, Savitz DA, Nylander-French LA, Parks CG, Chin H, Jennette CE, Lionaki S, Jennette JC, Falk RJ. Association of silica exposure with anti-neutrophil cytoplasmic autoantibody small-vessel vasculitis: a population-based, case-control study. Clin J Am Soc Nephrol. 2007;2:290–9. doi: 10.2215/CJN.03501006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lane SE, Watts RA, Bentham G, Innes NJ, Scott DG. Are environmental factors important in primary systemic vasculitis? A case-control study. Arthritis Rheum. 2003;48:814–23. doi: 10.1002/art.10830. [DOI] [PubMed] [Google Scholar]
- 31.van Rossum AP, Limburg PC, Kallenberg CG. Activation, apoptosis, and clearance of neutrophils in Wegener's granulomatosis. Ann N Y Acad Sci. 2005;1051:1–11. doi: 10.1196/annals.1361.041. [DOI] [PubMed] [Google Scholar]
- 32.van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, van der Giessen M, van der Hem GK, The TH. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener's granulomatosis. Lancet. 1985;1:425–9. doi: 10.1016/s0140-6736(85)91147-x. [DOI] [PubMed] [Google Scholar]
- 33.Specks U, Wheatley CL, McDonald TJ, Rohrbach MS, DeRemee RA. Anticytoplasmic autoantibodies in the diagnosis and follow-up of Wegener's granulomatosis. Mayo Clin Proc. 1989;64:28–36. doi: 10.1016/s0025-6196(12)65300-2. [DOI] [PubMed] [Google Scholar]
- 34.Tervaert JW, van der Woude FJ, Fauci AS, Ambrus JL, Velosa J, Keane WF, Meijer S, van der Giessen M, van der Hem GK, The TH, et al. Association between active Wegener's granulomatosis and anticytoplasmic antibodies. Arch Intern Med. 1989;149:2461–5. doi: 10.1001/archinte.149.11.2461. [DOI] [PubMed] [Google Scholar]
- 35.Gencik M, Meller S, Borgmann S, Fricke H. Proteinase 3 gene polymorphisms and Wegener's granulomatosis. Kidney Int. 2000;58:2473–7. doi: 10.1046/j.1523-1755.2000.00430.x. [DOI] [PubMed] [Google Scholar]
- 36.Reynolds WF, Stegeman CA, Tervaert JW. -463 G/A myeloperoxidase promoter polymorphism is associated with clinical manifestations and the course of disease in MPO-ANCA-associated vasculitis. Clin Immunol. 2002;103:154–60. doi: 10.1006/clim.2002.5206. [DOI] [PubMed] [Google Scholar]
- 37.Tervaert JW, Goldschmeding R, Elema JD, Limburg PC, van der Giessen M, Huitema MG, Koolen MI, Hene RJ, The TH, van der Hem GK, et al. Association of autoantibodies to myeloperoxidase with different forms of vasculitis. Arthritis Rheum. 1990;33:1264–72. doi: 10.1002/art.1780330829. [DOI] [PubMed] [Google Scholar]
- 38.Seo P, Stone JH. The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med. 2004;117:39–50. doi: 10.1016/j.amjmed.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 39.Bosch X, Guilabert A, Font J. Antineutrophil cytoplasmic antibodies. Lancet. 2006;368:404–18. doi: 10.1016/S0140-6736(06)69114-9. [DOI] [PubMed] [Google Scholar]
- 40.Godman GC, Churg J. Wegener's granulomatosis: pathology and review of the literature. AMA Arch Pathol. 1954;58:533–53. [PubMed] [Google Scholar]
- 41.Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337:1512–23. doi: 10.1056/NEJM199711203372106. [DOI] [PubMed] [Google Scholar]
- 42.Kallenberg CG. Pathogenesis of PR3-ANCA associated vasculitis. J Autoimmun. 2008;30:29–36. doi: 10.1016/j.jaut.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Kettritz R, Jennette JC, Falk RJ. Crosslinking of ANCA-antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol. 1997;8:386–94. doi: 10.1681/ASN.V83386. [DOI] [PubMed] [Google Scholar]
- 44.Hattar K, van Burck S, Bickenbach A, Grandel U, Maus U, Lohmeyer J, Csernok E, Hartung T, Seeger W, Grimminger F, Sibelius U. Anti-proteinase 3 antibodies (c-ANCA) prime CD14-dependent leukocyte activation. J Leukoc Biol. 2005;78:992–1000. doi: 10.1189/jlb.0902442. [DOI] [PubMed] [Google Scholar]
- 45.Uehara A, Iwashiro A, Sato T, Yokota S, Takada H. Antibodies to proteinase 3 prime human monocytic cells via protease-activated receptor-2 and NF-kappaB for Toll-like receptor- and NOD-dependent activation. Mol Immunol. 2007;44:3552–62. doi: 10.1016/j.molimm.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 46.Popa ER, Stegeman CA, Kallenberg CG, Tervaert JW. Staphylococcus aureus and Wegener's granulomatosis. Arthritis Res. 2002;4:77–9. doi: 10.1186/ar392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Rossum AP, Rarok AA, Huitema MG, Fassina G, Limburg PC, Kallenberg CG. Constitutive membrane expression of proteinase 3 (PR3) and neutrophil activation by anti-PR3 antibodies. J Leukoc Biol. 2004;76:1162–70. doi: 10.1189/jlb.0604319. [DOI] [PubMed] [Google Scholar]
- 48.Ben-Smith A, Dove SK, Martin A, Wakelam MJ, Savage CO. Antineutrophil cytoplasm autoantibodies from patients with systemic vasculitis activate neutrophils through distinct signaling cascades: comparison with conventional Fcgamma receptor ligation. Blood. 2001;98:1448–55. doi: 10.1182/blood.v98.5.1448. [DOI] [PubMed] [Google Scholar]
- 49.Porges AJ, Redecha PB, Kimberly WT, Csernok E, Gross WL, Kimberly RP. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via Fc gamma RIIa. J Immunol. 1994;153:1271–80. [PubMed] [Google Scholar]
- 50.Tanaka S, Edberg JC, Chatham W, Fassina G, Kimberly RP. Fc gamma RIIIb allelesensitive release of alpha-defensins: anti-neutrophil cytoplasmic antibody-induced release of chemotaxins. J Immunol. 2003;171:6090–6. doi: 10.4049/jimmunol.171.11.6090. [DOI] [PubMed] [Google Scholar]
- 51.Masutani K, Tokumoto M, Nakashima H, Tsuruya K, Kashiwagi M, Kudoh Y, Fukuda K, Kanai H, Akahoshi M, Otsuka T, Hirakata H, Iida M. Strong polarization toward Th1 immune response in ANCA-associated glomerulonephritis. Clin Nephrol. 2003;59:395–405. doi: 10.5414/cnp59395. [DOI] [PubMed] [Google Scholar]
- 52.Mayet WJ, Schwarting A, Meyer zum Buschenfelde KH. Cytotoxic effects of antibodies to proteinase 3 (C-ANCA) on human endothelial cells. Clin Exp Immunol. 1994;97:458–65. doi: 10.1111/j.1365-2249.1994.tb06110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CG. Skewed distribution of Th17 lymphocytes in patients with Wegener's granulomatosis in remission. Arthritis Rheum. 2008;58:2196–205. doi: 10.1002/art.23557. [DOI] [PubMed] [Google Scholar]
- 54.Brockmann H, Schwarting A, Kriegsmann J, Petrow P, Gaumann A, Muller KM, Galle PR, Mayet W. Proteinase-3 as the major autoantigen of c-ANCA is strongly expressed in lung tissue of patients with Wegener's granulomatosis. Arthritis Res. 2002;4:220–5. doi: 10.1186/ar410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwarting A, Hagen D, Odenthal M, Brockmann H, Dienes HP, Wandel E, Rumpelt HJ, Zum Buschenfelde KH, Galle PR, Mayet W. Proteinase-3 mRNA expressed by glomerular epithelial cells correlates with crescent formation in Wegener's granulomatosis. Kidney Int. 2000;57:2412–22. doi: 10.1046/j.1523-1755.2000.00100.x. [DOI] [PubMed] [Google Scholar]
- 56.Pendergraft WF, 3rd, Preston GA, Shah RR, Tropsha A, Carter CW, Jr, Jennette JC, Falk RJ. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004;10:72–9. doi: 10.1038/nm968. [DOI] [PubMed] [Google Scholar]
- 57.Yang J, Bautz DJ, Lionaki S, Hogan SL, Chin H, Tisch RM, Schmitz JL, Pressler BM, Jennette JC, Falk RJ, Preston GA. ANCA patients have T cells responsive to complementary PR-3 antigen. Kidney Int. 2008;74:1159–69. doi: 10.1038/ki.2008.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bautz DJ, Preston GA, Lionaki S, Hewins P, Wolberg AS, Yang JJ, Hogan SL, Chin H, Moll S, Jennette JC, Falk RJ. Antibodies with dual reactivity to plasminogen and complementary PR3 in PR3-ANCA vasculitis. J Am Soc Nephrol. 2008;19:2421–9. doi: 10.1681/ASN.2008030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daeron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–34. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 60.Ralston DR, Marsh CB, Lowe MP, Wewers MD. Antineutrophil cytoplasmic antibodies induce monocyte IL-8 release. Role of surface proteinase-3, alpha1-antitrypsin, and Fcgamma receptors. J Clin Invest. 1997;100:1416–24. doi: 10.1172/JCI119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marnell L, Mold C, Du Clos TW. C-reactive protein: ligands, receptors and role in inflammation. Clin Immunol. 2005;117:104–11. doi: 10.1016/j.clim.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 62.Weidner S, Neupert W, Goppelt-Struebe M, Rupprecht HD. Antineutrophil cytoplasmic antibodies induce human monocytes to produce oxygen radicals in vitro. Arthritis Rheum. 2001;44:1698–706. doi: 10.1002/1529-0131(200107)44:7<1698::AID-ART294>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 63.Kocher M, Edberg JC, Fleit HB, Kimberly RP. Antineutrophil cytoplasmic antibodies preferentially engage Fc gammaRIIIb on human neutrophils. J Immunol. 1998;161:6909–14. [PubMed] [Google Scholar]
- 64.Kocher M, Siegel ME, Edberg JC, Kimberly RP. Cross-linking of Fc gamma receptor IIa and Fc gamma receptor IIIb induces different proadhesive phenotypes on human neutrophils. J Immunol. 1997;159:3940–8. [PubMed] [Google Scholar]
- 65.Hewins P, Williams JM, Wakelam MJ, Savage CO. Activation of Syk in neutrophils by antineutrophil cytoplasm antibodies occurs via Fcgamma receptors and CD18. J Am Soc Nephrol. 2004;15:796–808. doi: 10.1097/01.asn.0000113241.98702.77. [DOI] [PubMed] [Google Scholar]
- 66.Ninomiya N, Hazeki K, Fukui Y, Seya T, Okada T, Hazeki O, Ui M. Involvement of phosphatidylinositol 3-kinase in Fc gamma receptor signaling. J Biol Chem. 1994;269:22732–7. [PubMed] [Google Scholar]
- 67.Tilton B, Andjelkovic M, Didichenko SA, Hemmings BA, Thelen M. G-Protein-coupled receptors and Fcgamma-receptors mediate activation of Akt/protein kinase B in human phagocytes. J Biol Chem. 1997;272:28096–101. doi: 10.1074/jbc.272.44.28096. [DOI] [PubMed] [Google Scholar]
- 68.Lawrence DA, Weigle WO, Spiegelberg HL. Immunoglobulins cytophilic for human lymphocytes, monocytes, and neutrophils. J Clin Invest. 1975;55:368–76. doi: 10.1172/JCI107940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, Zhou T, Edberg JC, Kimberly RP. Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. 2007;178:3272–80. doi: 10.4049/jimmunol.178.5.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tse WY, Abadeh S, McTiernan A, Jefferis R, Savage CO, Adu D. No association between neutrophil FcgammaRIIa allelic polymorphism and anti-neutrophil cytoplasmic antibody (ANCA)-positive systemic vasculitis. Clin Exp Immunol. 1999;117:198–205. doi: 10.1046/j.1365-2249.1999.00960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tse WY, Abadeh S, Jefferis R, Savage CO, Adu D. Neutrophil FcgammaRIIIb allelic polymorphism in anti-neutrophil cytoplasmic antibody (ANCA)-positive systemic vasculitis. Clin Exp Immunol. 2000;119:574–7. doi: 10.1046/j.1365-2249.2000.01182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edberg JC, Wainstein E, Wu J, Csernok E, Sneller MC, Hoffman GS, Keystone EC, Gross WL, Kimberly RP. Analysis of FcgammaRII gene polymorphisms in Wegener's granulomatosis. Exp Clin Immunogenet. 1997;14:183–95. [PubMed] [Google Scholar]
- 73.Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, Heward JM, Gough SC, de Smith A, Blakemore AI, Froguel P, Owen CJ, Pearce SH, Teixeira L, Guillevin L, Graham DS, Pusey CD, Cook HT, Vyse TJ, Aitman TJ. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–3. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Willcocks LC, Lyons PA, Clatworthy MR, Robinson JI, Yang W, Newland SA, Plagnol V, McGovern NN, Condliffe AM, Chilvers ER, Adu D, Jolly EC, Watts R, Lau YL, Morgan AW, Nash G, Smith KG. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med. 2008;205:1573–82. doi: 10.1084/jem.20072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aksu K, Kitapcioglu G, Keser G, Berdeli A, Karabulut G, Kobak S, Ozmen M, Inal V, Kabasakal Y, Oksel F, Kocanaogullari H, Doganavsargil E. FcgammaRIIa, IIIa and IIIb gene polymorphisms in Behcet's disease: do they have any clinical implications? Clin Exp Rheumatol. 2008;26:S77–83. [PubMed] [Google Scholar]
- 76.Latiano A, Palmieri O, Valvano MR, Bossa F, Latiano T, Corritore G, DeSanto E, Andriulli A, Annese V. Evaluating the role of the genetic variations of PTPN22, NFKB1, and FcGRIIIA genes in inflammatory bowel disease: a meta-analysis. Inflamm Bowel Dis. 2007;13:1212–9. doi: 10.1002/ibd.20185. [DOI] [PubMed] [Google Scholar]
- 77.Brown EE, Edberg JC, Kimberly RP. Fc receptor genes and the systemic lupus erythematosus diathesis. Autoimmunity. 2007;40:567–81. doi: 10.1080/08916930701763710. [DOI] [PubMed] [Google Scholar]
- 78.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, Hodges MD, Bhangal G, Patel SG, Sheehan-Rooney K, Duda M, Cook PR, Evans DJ, Domin J, Flint J, Boyle JJ, Pusey CD, Cook HT. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–5. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 79.Jonsen A, Gunnarsson I, Gullstrand B, Svenungsson E, Bengtsson AA, Nived O, Lundberg IE, Truedsson L, Sturfelt G. Association between SLE nephritis and polymorphic variants of the CRP and FcgammaRIIIa genes. Rheumatology (Oxford) 2007;46:1417–21. doi: 10.1093/rheumatology/kem167. [DOI] [PubMed] [Google Scholar]
- 80.Xu G, He Q, Shou Z, Wang H, Wang R, Jiang H, Chen Y, Chen J. Association of Fc gamma receptor IIIB polymorphism with renal-allogrft in Chinese. Transpl Immunol. 2007;18:28–31. doi: 10.1016/j.trim.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 81.Finkielman JD, Lee AS, Hummel AM, Viss MA, Jacob GL, Homburger HA, Peikert T, Hoffman GS, Merkel PA, Spiera R, St Clair EW, Davis JC, Jr, McCune WJ, Tibbs AK, Ytterberg SR, Stone JH, Specks U. ANCA are detectable in nearly all patients with active severe Wegener's granulomatosis. Am J Med. 2007;120643:e9–14. doi: 10.1016/j.amjmed.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 82.Slot MC, Tervaert JW, Boomsma MM, Stegeman CA. Positive classic antineutrophil cytoplasmic antibody (C-ANCA) titer at switch to azathioprine therapy associated with relapse in proteinase 3-related vasculitis. Arthritis Rheum. 2004;51:269–73. doi: 10.1002/art.20234. [DOI] [PubMed] [Google Scholar]
- 83.Sanders JS, Huitma MG, Kallenberg CG, Stegeman CA. Prediction of relapses in PR3-ANCA-associated vasculitis by assessing responses of ANCA titres to treatment. Rheumatology (Oxford) 2006;45:724–9. doi: 10.1093/rheumatology/kei272. [DOI] [PubMed] [Google Scholar]
- 84.Boomsma MM, Stegeman CA, van der Leij MJ, Oost W, Hermans J, Kallenberg CG, Limburg PC, Tervaert JW. Prediction of relapses in Wegener's granulomatosis by measurement of antineutrophil cytoplasmic antibody levels: a prospective study. Arthritis Rheum. 2000;43:2025–33. doi: 10.1002/1529-0131(200009)43:9<2025::AID-ANR13>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 85.Finkielman JD, Merkel PA, Schroeder D, Hoffman GS, Spiera R, St Clair EW, Davis JC, Jr, McCune WJ, Lears AK, Ytterberg SR, Hummel AM, Viss MA, Peikert T, Stone JH, Specks U. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med. 2007;147:611–9. doi: 10.7326/0003-4819-147-9-200711060-00005. [DOI] [PubMed] [Google Scholar]
- 86.Hoffman GS, Stone JH, Langford C. Implications of antineutrophil cytoplasmic antibody status when switching to maintenance therapy. Arthritis Rheum. 2005;53:1–2. doi: 10.1002/art.20921. [DOI] [PubMed] [Google Scholar]
- 87.Holland M, Hewins P, Goodall M, Adu D, Jefferis R, Savage CO. Anti-neutrophil cytoplasm antibody IgG subclasses in Wegener's granulomatosis: a possible pathogenic role for the IgG4 subclass. Clin Exp Immunol. 2004;138:183–92. doi: 10.1111/j.1365-2249.2004.02566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williams JM, Ben-Smith A, Hewins P, Dove SK, Hughes P, McEwan R, Wakelam MJ, Savage CO. Activation of the G(i) heterotrimeric G protein by ANCA IgG F(ab')2 fragments is necessary but not sufficient to stimulate the recruitment of those downstream mediators used by intact ANCA IgG. J Am Soc Nephrol. 2003;14:661–9. doi: 10.1097/01.asn.0000050223.34749.f4. [DOI] [PubMed] [Google Scholar]
- 89.Hayes MJ, Cambridge G. An IgM class anti-neutrophil cytoplasm antibody inhibits neutrophil adhesion and apoptosis via a Syk dependent signaling cascade. Mol Immunol. 2004;41:457–68. doi: 10.1016/j.molimm.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 90.O'Riordan A, Keogh JA, Watson A. Pulmonary renal syndrome due to anti-GBM and IgM C-ANCA disease requiring the use of novel therapeutic agents. Ren Fail. 2005;27:791–4. doi: 10.1080/08860220500244872. [DOI] [PubMed] [Google Scholar]
- 91.Underdown BJ, Schiff JM. Immunoglobulin A: strategic defense initiative at the mucosal surface. Annu Rev Immunol. 1986;4:389–417. doi: 10.1146/annurev.iy.04.040186.002133. [DOI] [PubMed] [Google Scholar]
- 92.Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347:738–48. doi: 10.1056/NEJMra020109. [DOI] [PubMed] [Google Scholar]
- 93.Ayoub N, Charuel JL, Diemert MC, Barete S, Andre M, Fermand JP, Piette JC, Frances C. Antineutrophil cytoplasmic antibodies of IgA class in neutrophilic dermatoses with emphasis on erythema elevatum diutinum. Arch Dermatol. 2004;140:931–6. doi: 10.1001/archderm.140.8.931. [DOI] [PubMed] [Google Scholar]
- 94.Rovel-Guitera P, Diemert MC, Charuel JL, Laporte JL, Musset L, Chosidow O, Piette JC, Frances C. IgA antineutrophil cytoplasmic antibodies in cutaneous vasculitis. Br J Dermatol. 2000;143:99–103. doi: 10.1046/j.1365-2133.2000.03597.x. [DOI] [PubMed] [Google Scholar]
- 95.Gigase P, De Clerck LS, Van Cotthem KA, Bridts CH, Stevens WJ, Van Outryve M, Pelckmans PA. Anti-neutrophil cytoplasmic antibodies in inflammatory bowel disease with special attention for IgA-class antibodies. Dig Dis Sci. 1997;42:2171–4. doi: 10.1023/a:1018803509150. [DOI] [PubMed] [Google Scholar]
- 96.Ozaltin F, Bakkaloglu A, Ozen S, Topaloglu R, Kavak U, Kalyoncu M, Besbas N. The significance of IgA class of antineutrophil cytoplasmic antibodies (ANCA) in childhood Henoch-Schonlein purpura. Clin Rheumatol. 2004;23:426–9. doi: 10.1007/s10067-004-0910-y. [DOI] [PubMed] [Google Scholar]
- 97.Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, Henin D, Benhamou M, Pretolani M, Blank U, Monteiro RC. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. 2005;22:31–42. doi: 10.1016/j.immuni.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 98.Kanamaru Y, Pfirsch S, Aloulou M, Vrtovsnik F, Essig M, Loirat C, Deschenes G, Guerin-Marchand C, Blank U, Monteiro RC. Inhibitory ITAM signaling by Fc{alpha}RI-FcR{gamma} chain controls multiple activating responses and prevents renal inflammation. J Immunol. 2008;180:2669–78. doi: 10.4049/jimmunol.180.4.2669. [DOI] [PubMed] [Google Scholar]
- 99.Wu J, Ji C, Xie F, Langefeld CD, Qian K, Gibson AW, Edberg JC, Kimberly RP. FcalphaRI (CD89) alleles determine the proinflammatory potential of serum IgA. J Immunol. 2007;178:3973–82. doi: 10.4049/jimmunol.178.6.3973. [DOI] [PubMed] [Google Scholar]
- 100.Ramsland PA, Willoughby N, Trist HM, Farrugia W, Hogarth PM, Fraser JD, Wines BD. Structural basis for evasion of IgA immunity by Staphylococcus aureus revealed in the complex of SSL7 with Fc of human IgA1. Proc Natl Acad Sci U S A. 2007;104:15051–6. doi: 10.1073/pnas.0706028104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Etanercept plus standard therapy for Wegener's granulomatosis. N Engl J Med. 2005;352:351–61. doi: 10.1056/NEJMoa041884. [DOI] [PubMed] [Google Scholar]