

Abstract
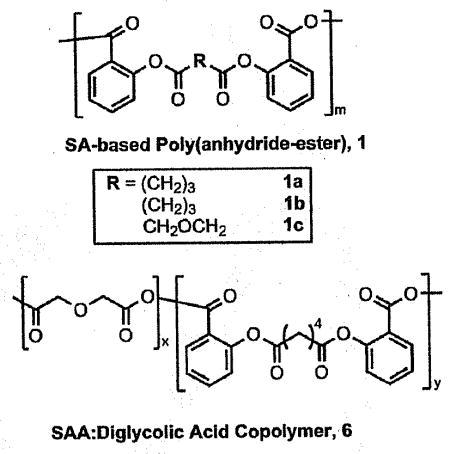
Fast-degrading, salicylate-based poly(anhydride-esters) were designed to degrade and release the active component, salicylic acid (SA), within 1 week. The polymer degradation was enhanced by using shorter or oxygen-containing aliphatic chains. A copolymer of diglycolic acid was also made with a salicylate-based diacid for comparison of polymer properties, including SA release. Both methods resulted in polyanhydrides with molecular weights ranging from 14 500 to 27 800 Da and displayed glass transition temperatures near physiological conditions, namely 33–40°C. the homo- and copolymers completely degraded within one week releasing the chemically incorporated SA.
Keywords: hydrolytic degradation, hydrophilic polymers, poly(anhydride-ester), polyanhydride, polymerization, synthesis
Introduction
Biodegradable polymers have been extensively studied in recent years for biomaterials, packaging materials, agriculture, gene delivery, drug delivery, and tissue engineering[1–4] Polyanhydrides are of. particular interest because of their surface-eroding behavior.[5–7] For most polyanhydrides, bioactive molecules are physically admixed within the matrices and released based upon surface erosion of the polyanhydride matrix.
In contrast, bioactive molecules can be chemically incorporated into poly(anhydride-esters), directly into the backbone of the polymer (e.g., 1a, Figure 1).[8–10] Upon hydrolytic degradation, the labile anhydride and ester bonds are cleaved releasing the bioactive component [e.g., salicylic acid (SA), 2] and the water-soluble biocompatible “linker” molecule (e.g., adipic acid, 3a). The linker molecules chosen are known to be biocompatible or have generally regarded as safe (GRAS) status[11] thus the degradation products are known to be safe. For example, adipic acid is a GRAS and everything added to food in the United States (EAFUS) compound,[11] glutaric acid is naturally occurring in sugar beets[12] and diglycolic acid is used in many biocompatible polymer formulations. [13–15] The polymer degradation rate can be manipulated by altering the chemical structure of the linker or the bioactive[9,10] In this way, the degradation is directly controlled by altering the polymer composition.
Figure 1.
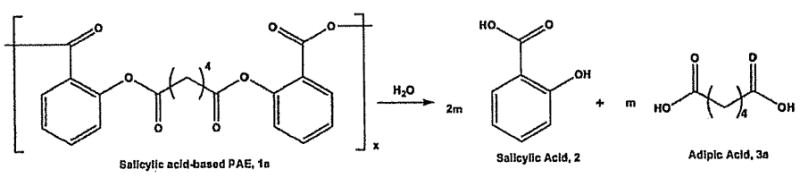
Hydrolysis of SA-based poly(anhydride-ester), 1a, to release the bioactive compound (2) and the biocompatible linker molecule (3a).
An alternate way to control polyanhydride degradation is to modify copolymer compositions. For example, copolymers of salicylate-based poly(anhydride-esters) (e.g., 1a) with poly(1,6-bis(p-carboxyphenoxy)hexane (p-CPH) or poly(1,6-bis(o-carboxyphenoxy)hexane (o-CPH) have been previously synthesized.[16] Increasing the portion of CPH to 1a increased mechanical properties, but decreased the drug loading and release rate of the incorporated SA. Different copolymer compositions can alter the degradation rate and concurrent release of SA from weeks to several months.
Salicylic acid (SA)-based poly(anhydride-esters) (1) have been proven effective for controlling inflammation[17,18] promoting bone growth,[17,19,20] and preventing biofilm formation[18,21] The polymers can easily be manipulated into different geometries including powders, fibers,[22] microspheres,[23] disks,[9] films, and cardiac stents.[24] Most of these polymers prepared for biomaterials indications have been designed to degrade within weeks to months[17,18,20,24,25]
Salicylic acid (SA) is anti-inflammatory, antiseptic, analgesic, antipyretic and keratolytic, and thus desirable for formulations in many different areas outside of biomaterials[12,26] Relative to sustained drug delivery systems, faster degrading polymers are desirable for wound care, personal care, oral care, cosmetic, and food applications; these applications require rapid but controlled release of the bioactive[27–29] For example, a controlled, high release rate of bioactive is desirable for applications such as wound care in which a high initial release is necessary within 12h-3d[30–33] and for encapsulating flavors and aromas in the food industry or fragrances in the personal care/cosmetics industries, with release in the range of 6–12 h.[30,34] As such, we designed salicylate-based polyanhydrides to release a significant amount of the SA in a short time period (hours to days). This effect was achieved by altering the chemical composition of the linker molecule or introducing a second, more hydrophilic monomer to make poly(anhydride-ester) copolymers.
Experimental part
Materials
Diethyl ether was obtained from Fisher (Fair lawn, NJ, USA). All other chemicals and reagents were purchased from Aldrich (Milwaukee, WI, USA) and used as received.
Chemical Composition
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Varian 500 MHz spectrophotometer. Samples (5–10 mg) were dissolved in an appropriate deuterated solvent (DMSO-d6), which was also the internal reference. Infrared (IR) spectra were measured on a Thermo Nicolet/Avatar 360 FT IR spectrometer, by solvent-casting samples from acetone onto NaCl plate. Negative electrospray ionization mass spectrometry (ESI-MS) was performed using a Finnigan LCQDUO LC/MS/MS spectrometer.
Polymer Properties
A Perkin-Elmer LC system consisting of a Series 200 refractive index detector, a Series 200 LC pump, and an ISS 200 advanced sample processor was used to determine weight-averaged molecular weights(Mw) and polydispersity indices (PDI). For data collection and automation of the system, a Dell OptiPlex GX110 computer running Perkin-Elmer TurboChrom 4 software was utilized. The connection from the LC system to the computer was achieved via a Perkin-Elmer Nelson 900 Series Interface and 600 Series Link. Polymers (1a–c, 6) were dissolved in methylene chloride (10 mg · mL−1) and filtered through 0.45 μm poly(tetrafluoroethylene) (PTFE) syringe filters (Whatman, Clifton, NJ, USA) before elution onto the Jordi divinylbenzene mixed-bed GPC column (7.8 mm × 300 mm) (Alltech Associates, Deerfield, IL, USA). Molecular weights were calculated relative to narrow molecular weight polystyrene standards (Polysciences, Dorval, Canada).
Differential scanning calorimetry was used to determine glass transition (Tg) and melting point (Tm) temperatures using a TA DSC Q200 outfitted with a Dell Dimension 3000 computer running TA Universal Analysis 2000 software was used for data collection and processing. Melting points of diacids (5) were obtained using a heating rate of 10°C · min−1 heating from 25 to 200°C. For glass transition temperatures, samples (~5 mg) were heated under dry nitrogen gas at heating and cooling rates of 10°C · min−1 with a two-cycle minimum. Glass transition temperatures were calculated as half Cp extrapolated. Thermogravimetric analysis for decomposition temperatures (Td) was performed on a Perkin-Elmer TGA7 analyzer with TAC7/DX controller equipped with a Dell OptiPlex Gx110 computer running Perkin-Elmer Pyris software. Samples ~5 mg) were heated under dry nitrogen gas at a heating rate of 10°C · min−1 from 25 to 400°C. Decomposition temperatures were defined as the onset of decomposition.
Static contact angles were measured by dropping deionized water onto polymer films made by solvent-casting polymers in methylene chloride onto glass coverslips using a Rame-Hart Standard Goniometer Model Number 250-00 (Mountain Lakes, NJ, USA) outfitted with a Dell Dimension 3000 computer with DROPimage Advanced software. The measurements taken were in duplicate with an average of five readings recorded within 30 s of depositing the water droplet onto the polymer film.
Maximum solubility of diacids (5) was determined by monitoring absorbance of saturated solutions of each diacid (5) in PBS using UV–Vis spectrophotometry (λ = 235 nm for 5a–c; λ = 215 nm for 3c). The absorbance was compared to standard solutions of each diacid (0.001 and 0.01 mg · mL−1).
Synthesis of Salicylic Acid (SA)-Based Diacids (5)
Diacids, 5, were synthesized using previously described techniques.[8,9] The characterization of 5a and b has already been published, however, 5c is a new compound and its properties are outlined below. In brief, SA (2; 32 mmol) was dissolved in 100 mL THF and pyridine (64 mmol). Diglycolyl chloride (4c; 16 mmol) was dissolved in 15/mL THF and added drop-wise to the stirring reaction mixture to give a suspension. The reaction was allowed to stir at room temperature overnight and subsequently quenched by pouring over water (~700 mL) and adding HCl while stirring until pH ~2. The solid formed (5c) was filtered off, washed with water (1L) and allowed to dry under vacuum at room temperature.
Salicylic Acid (SA, Diglycolic) Diacid (5c)
Yield: 70.8% (beige powder). 1H NMR (DMSO-d6): δ(ppm) = 7.93 (d, 2H, Ar–H), 7.65 (t, 2H, Ar–H), 7.40 (t, 2H, Ar–H), 7.24 (d, 2H, Ar–H), 4.55 (s, 4H, CH2). IR (NaCl): 3 600–3 300 (s, OH, COOH), 1780 (vs, C=O, ester), 1757 (vs, C=O, COOH), 1209 cm−1 (s, C–O–C, ether). (C18H14O9)n (374.3)n: ESI-MS m/z 373 z-1; Tm: 182°C.
Synthesis of Salicylic Acid (SA)-based Poly-(anhydride-esters) (1, 6)
Polymer 1a was synthesized by solution polymerization as previously described.[35–36] New polymers (1b, 1c, and 6) were also synthesized using solution polymerization methods.[35–37] In general, the diacid (5; 20 mmol) was dissolved in 20 w/v % anhydrous methylene chloride. Triethylamine (88 mmol) was then added, and the reaction mixture cooled to 0 °C with an ice/water bath. Triphosgene (22 mmol) dissolved in anhydrous methylene chloride was added drop-wise using a syringe pump or addition funnel at a rate of 14 mL · h−1. The reaction was allowed to stir at 0°C for ~2h. Then, the reaction mixture is poured over diethyl ether (~400mL), washed with acidic water ~1L, pH 2 using concentrated HCl). The solid obtained (1 or 6) was dried under vacuum at room temperature.
Salicylic Acid (SA, Diglycolic) Polymer (1c)
Yield: 74.1% (off-white powder). 1H NMR (DMSO-d6): δ(ppm) = 8.18 (b, 2H, Ar–H), 7.81 (b, 2H, Ar–H), 754 (b, 2H, Ar–H), 7.39 (b, 2H, Ar–H), 4.51 (4H,CH2). IR (NaCl): 1786,1727 (vs, C=O, anhydride), 1744 (vs, C=O, ester), 1210 cm−1 (s, C–O–C, ether).
Salicylic (SA) (Adipic)/Diglycolic Copolymer (6)
The molar ratio of 5a to diglycolic acid (3c) incorporated was 60:40. Yield: 50.2% (beige powder). 1H NMR (DMSO-d6): δ(ppm) = 8.08 (b, 2H, Ar–H), 7.78 (b, 2H, Ar–H), 7.47 (b, 2H, Ar–H), 7.31 (b, 2H, Ar–H), 3.32 (b, 4H, CH2), 3.07 (b, 2H, CH2), 1.56 (b, 2H, CH2) IR (NaCl, cm−1): 1790, 1728 (vs, C=O, anhydride), 1763 (vs, C=O, ester), 1208 (s, C–O–C, ether). Composition (by NMR): 61:39.
In vitro Hydrolytic Degradation
The release rate of SA was evaluated from hydrolytic degradation, of polymers 1 and 6. Polymers were ground with mortar and pestle to particle sizes ~75–300 μm. Particle size was determined using standard testing sieves (Aldrich, Milwaukee, WI, USA). Ground-up polymer samples (~20 mg) were incubated at 37 °C with agitation at 60 rpm in 10 mL PBS (pH 7.4) in 20 mL Wheaton glass scintillation vials (Fisher, Fair Lawn, NJ) using a controlled environment incubator-shaker (New Brunswick Scientific Co., Edison, NJ, USA). At regular time intervals, the media was analyzed by UV spectrophotometry (Beekman Instruments DU520 UV/Vis spectrophotometer, Fullerton, CA, USA). UV measurements were taken at the wavelength of maximum absorbance for SA (2) (λmax 303 nm), which did not overlap with other polymer degradation products (i.e., diacid, 5). Data were calculated against a calibration curve made from standard solutions of SA in PBS (1 × 10−3, 2.5 × 10−3, 5 × 10−3, 1 × 10−2 mg · mL−1). Degradation was performed in triplicate.
Results and Discussion
Polymer Synthesis
Diacids (5) were synthesized by direct coupling of SA (2) with a diacyl chloride (4) in the presence of base (i.e., pyridine) in THF. Polymers 1 were synthesized as previously described using solution polymerization techniques.[35–37]
The copolymer (6) was synthesized by polymerizing a 60:40 molar ratio of SA-based diacid (5a) with diglycolic acid (3c) using similar solution polymerization techniques as described above and elsewhere (Scheme 1).[35–37] This molar ratio was chosen to have the same weight percent of diglycolic acid in the polymer backbone as in polymer 1c, as shown in Table 1.
Scheme 1.
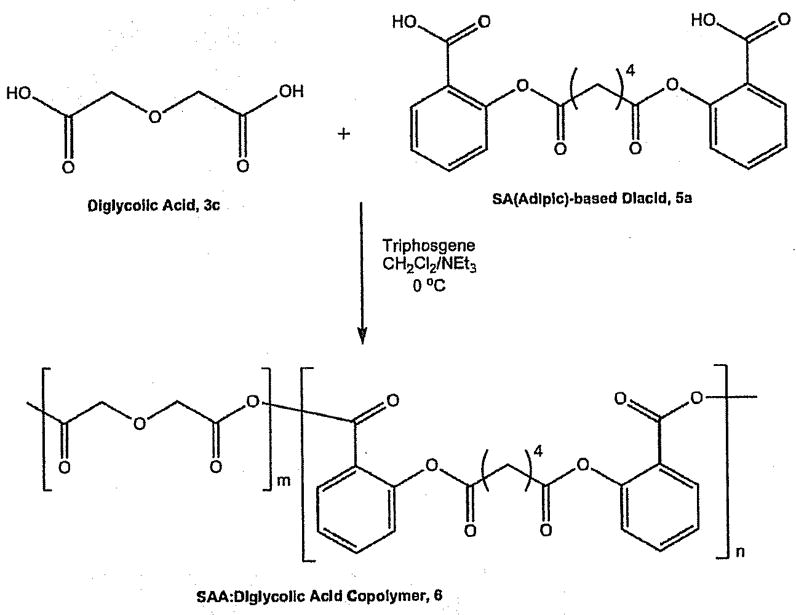
Synthesis of salicylate-based copolymer, 6, from diglycolic acid (3c), and salicylate-based diacid, 5a.
Table 1.
Selected properties of polymers 1 and 6.
| Polymer | wt.-% SA | Mw | PDI |
Tg |
Td |
Contact angle |
|---|---|---|---|---|---|---|
| °C | °C | ° | ||||
| 1a | 71.5 | 27800 | 1.0 | 40 | 283 | 83.1 |
| 1b | 74.2 | 18100 | 1.3 | 38 | 295 | 85.2 |
| 1c | 73.8 | 14500 | 1.5 | 35 | 326 | 49.5 |
| 6 | 61.2 | 21700 | 1.1 | 33 | 269 | 65.4 |
Polymers 1 and 6 were polymerized in solution using triphosgene as the coupling reagent in the presence of triethylamine (Scheme 2). Polymer properties are summarized in Table 1.
Scheme 2.
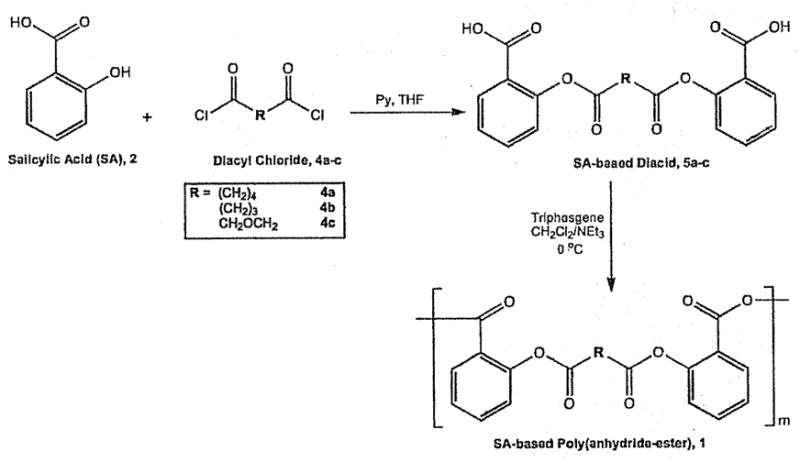
Synthesis SA-based poly(anhydride-esters), 1.
Resulting polymer properties such as molecular weight, ranging from ~15000 to 28000, are typical for most polyanhydrides made by solution polymerization methods.[35–37] Isolating polymers by precipitation removes most small oligomers and residual monomer, resulting in polymers with low polydispersity indices. The high drug loading (by weight) of SA for the polymers varied from 61.2 for polymer 6 up to 74.2% for polymer 1b. A single glass transition temperature was noted for polymer 6, indicating no phase separation. As seen with homopolymers, 1, no melting temperatures were observed for the polymers demonstrating that all polymers are amorphous.
Polymer Degradation
Polymer degradation is influenced by multiple factors, including water permeation into the-polymer matrix and water-solubility of the degradation products.[5–9,22] The contact angles for all polymers were measured (Table 1); by introducing diglycolic acid (3c) into the polymeric backbone either as a “linker” molecule (1c) or as a co-monomer (6), the resulting poly-(anhydride-ester) becomes more hydrophilic. Polymers. 1b and 1a have three and four methylenes, respectively, and display similar contact angles. When diglycolic acid (3c) is incorporated as the linker, the polymer (1c) becomes much more hydrophilic (49.5°). The copolymer (6) comprised of diacid 5a and diglycolic acid (3c) has an intermediate contact angle between polymers 1a and 1c.
Diacid solubility was determined using UV/Vis spectrophotometry. The results are summarized in Table 2. Comparing diacids (5), 5c with the oxygen-containing linker was most soluble in PBS, correlating with contact angle measurements, which indicated the resulting polymer (1c) was most hydrophilic. The co-monomer in polymer 6, diglycolic acid (3c), however, was overall the most soluble in PBS. Copolymer 6 had an intermediate contact angle (65.4°) between that of 1a (83.1°) and 1c (49.5°).
Table 2.
Maximum solubilities of diacids 3c and 5 in PBS.
| Diacid | Maximum solubility |
|---|---|
| mg · mL−1 | |
| 3c | 16.0 |
| 5a | 4.38 |
| 5b | 7.68 |
| 5c | 8.12 |
SA release studies were conducted on ground-up powdered polymer samples in PBS at physiological conditions to determine how much SA (2) is released over a 2 day period. Again, 48 h is the critical time period for wound care and other applications that require a controlled, yet high release rate, and is thus the chosen end point for the degradation study. The results are shown in Figure 2.
Figure 2.
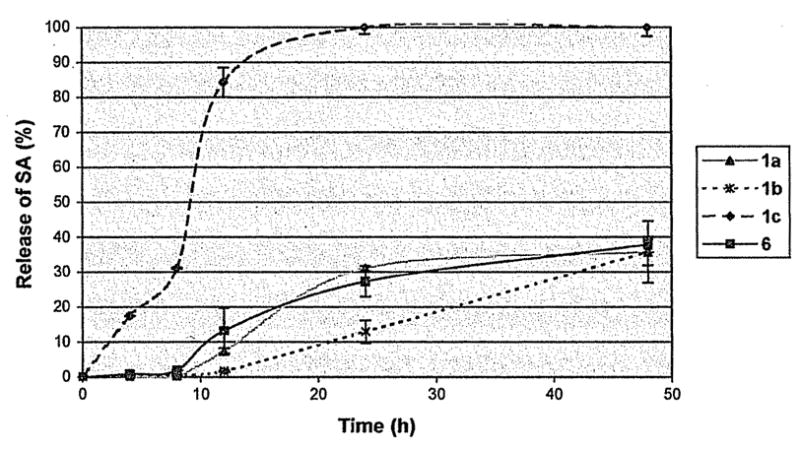
Cumulative release of-SA, 2, from salicylate-based poly(anhydride-esters), 1 and 6.
By 24 h, 100% of the SA is released from polymers 1c. After 48 h, ~35% of SA is released from polymers 1a and 1b, whereas 38% is released from polymer 6. In less than 1 wk, all polymers were completely degraded. When diglycolic acid (3c) was incorporated into the polymer as a linker, the SA release was faster compared with copolymer 6 at the same weight SA percent.
Conclusion
Fast-degrading, bioactive-based polyanhydrides (1) were synthesized by altering the structure of linker molecule to incorporate small aliphatic chains (i.e., adipic and glutaric acid) or more hydrophilic, oxygen-containing aliphatics (i.e., diglycolic acid). For comparison, a copolymer (6) of diglycolic acid was made with a salicylate-based diacid containing a short, aliphatic linker. Both methods rendered polyanhydrides that released a significant amount of the chemically incorporated SA and completely degraded within 1 wk. The controlled, sustained release over the first few days may be beneficial for wound, personal/oral care, cosmetic, and food applications. Furthermore, such poly(anhydride-esters) have good manipulation and are easy to formulate into different formats such as micro-particles, films, and fibers.
Based on results from preliminary experiments using polymers 1 and 6, we recommend these polymers in fast-acting applications. For specific applications such as wound care, which require a significant to complete release of bioactive in the early stages of healing (12–24 h), polymer 1c is most suitable. For other applications that require a slightly longer release, specifically within ~1 week, polymers 1a–b and 6 are more appropriate.
Acknowledgments
The authors thank the National Institutes of Health (DE 13207) for financial support.
References
- 1.Kumar N, Langer R, Domb A. Adv Drug Delivery Rev. 2002;54:889. doi: 10.1016/s0169-409x(02)00050-9. [DOI] [PubMed] [Google Scholar]
- 2.Shikanov A, Kumar N, Domb A. Isr J Chem. 2005;45:393. [Google Scholar]
- 3.Jain J, Modi S, Domb A, Kumar N. J Controlled Release. 2005;103:541. doi: 10.1016/j.jconrel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 4.Gunatillake P, Mayadunne R, Adhikari R. Biotechnol Annu Rev. 2006;12:301. doi: 10.1016/S1387-2656(06)12009-8. [DOI] [PubMed] [Google Scholar]
- 5.Gopferich A. Biomaterials. 1996;17:103. doi: 10.1016/0142-9612(96)85755-3. [DOI] [PubMed] [Google Scholar]
- 6.Tamada J, Langer R. Proc Natl Acad Sci U S A. 1993;90:552. doi: 10.1073/pnas.90.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitaker-Brothers K, Uhrich K. J Biomed Mater Res. 2006;76A:470. doi: 10.1002/jbm.a.30356. [DOI] [PubMed] [Google Scholar]
- 8.Erdmann L, Uhrich K. Biomaterials. 2000;20:1941. doi: 10.1016/s0142-9612(00)00073-9. [DOI] [PubMed] [Google Scholar]
- 9.Prudencio A, Schmeltzer R, Uhrich K. Macromolecules. 2005;38:6895. doi: 10.1021/ma048051u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmeltzer R, Schmalenberg K, Uhrich K. Biomacromolecules. 2005;72A:354. doi: 10.1021/bm049544+. [DOI] [PubMed] [Google Scholar]
- 11.Smith Jim, Honc Lily. Food Additives Data Book. Blackwell Science Ltd; 2003. [Google Scholar]
- 12.The Merck Index. Merck Research Laboratories; Whitehouse Station, NJ: 2001. [Google Scholar]
- 13.Brocchini S, James K, Tangpasuthadol V, Kohn J. J Biomed Mater Res. 1998;42:66. doi: 10.1002/(sici)1097-4636(199810)42:1<66::aid-jbm9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 14.Andjelic S, Yuan J, Jamiolkowski D, Diluccio R, Bezwada R, Zhang H, Mijovic J. Pharm Res. 2006;23:821. doi: 10.1007/s11095-006-9664-3. [DOI] [PubMed] [Google Scholar]
- 15.Bourke S, Kohn J. Adv Drug Delivery Rev. 2003;55:447. doi: 10.1016/s0169-409x(03)00038-3. [DOI] [PubMed] [Google Scholar]
- 16.Schmeltzer R, Uhrich K. J Bioact Compat Polym. 2006;21:123. doi: 10.1177/0883911506062976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erdmann L, Macedo B, Uhrich K. Biomaterials. 2000;21:2507. doi: 10.1016/s0142-9612(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 18.Bryers J, Jarvis R, Lebo J, Prudencio A, Kyriakides T, Uhrich K. Biomaterials. 2006;27:5039. doi: 10.1016/j.biomaterials.2006.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dashper S, Liu S, Reynolds E. Int J Pept Res Ther. 2007;13:505. [Google Scholar]
- 20.Harten R, Svach D, Schmeltzer R, Uhrich K, Biomed J. Mater Res, Part A. 2005;42A:354. doi: 10.1002/jbm.a.30184. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg L, Carbone A, Uhrich K, Chikindas M. Lett Appl Microbiol. 2008;46:593. doi: 10.1111/j.1472-765X.2008.02356.x. [DOI] [PubMed] [Google Scholar]
- 22.Whitaker-Brothers K, Uhrich K. J Biomed Mater Res. 2004;70A:309. doi: 10.1002/jbm.a.30083. [DOI] [PubMed] [Google Scholar]
- 23.Yeagy B, Prudencio A, Schmeltzer R, Uhrich K, Cook T. J Microencapsulation. 2006;23:643. doi: 10.1080/02652040600776481. [DOI] [PubMed] [Google Scholar]
- 24.Jabara R, Chronos N, Robinson K. Catheter Cardiovasc Interv. 2008;72:186. doi: 10.1002/ccd.21607. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds M, Prudencio A, Aichelmann-Reidy M, Woodward K, Uhrich K. Curr Drug Delivery. 2007;4:233. doi: 10.2174/156720107781023866. [DOI] [PubMed] [Google Scholar]
- 26.Muller E, Al-Attar J, Wolff A, Farber B. J Infect Dis. 1998;177:501. doi: 10.1086/517386. [DOI] [PubMed] [Google Scholar]
- 27.WO 2006127667 (2006), invs: K. Uhrich, Y. Kim. Fast degrading polyanhydrides, anhydride preparation, compositions to prepare medicament, topical administration of drug.
- 28.Carbone A, Uhrich K. Polym Prepr, ACS Div Polym Chem. 2008;49:661. [Google Scholar]
- 29.Vogelson C. Mod Drug Discovery. 2001;4:49. [Google Scholar]
- 30.Huang X, Brazel C, Controlled J. Release. 2001;73:121. doi: 10.1016/s0168-3659(01)00248-6. [DOI] [PubMed] [Google Scholar]
- 31.Monafo W, West M. Drugs. 1990;40:364. doi: 10.2165/00003495-199040030-00004. [DOI] [PubMed] [Google Scholar]
- 32.Loke W, Lau S, Yong L, Khor E, Sum C. J Biomed Mater Res. 2000;53:8. doi: 10.1002/(sici)1097-4636(2000)53:1<8::aid-jbm2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 33.Thakur R, Florek C, Kohn J, Michniak B. Int J Pharm. 2008;364:87. doi: 10.1016/j.ijpharm.2008.07.033. [DOI] [PubMed] [Google Scholar]
- 34.Peppas N, Peppas L. J Controlled Release. 1996;40:245. [Google Scholar]
- 35.Schmeltzer R, Johnson M, Griffin J, Uhrich K. J Biomater Sci, Polym Ed. 2008;19:1295. doi: 10.1163/156856208786052362. [DOI] [PubMed] [Google Scholar]
- 36.Carbone A, Song M, Uhrich K. Biomacromolecules. 2008;9:1604. doi: 10.1021/bm8000759. [DOI] [PubMed] [Google Scholar]
- 37.Domb A, Ron E, Langer R. Macromolecules. 1988;21:1925. [Google Scholar]