

Abstract
Vascular endothelial (VE)-cadherin is the major adhesion molecule of endothelial adherens junctions. It plays an essential role in controlling endothelial permeability, vascular integrity, leukocyte transmigration, and angiogenesis. Elevated levels of soluble VE-cadherin are associated with diseases like coronary atherosclerosis. Previous data showed that the extracellular domain of VE-cadherin is released by an unknown metalloprotease activity during apoptosis. In this study, we used gain of function analyses, inhibitor studies and RNA interference experiments to analyze the proteolytic release of VE-cadherin in human umbilical vein endothelial cells (HUVECs). We found that VE-cadherin is specifically cleaved by the disintegrin and metalloprotease ADAM10 in its ectodomain releasing a soluble fragment and generating a carboxyterminal membrane bound stub, which is a substrate for a subsequent γ-secretase cleavage. This ADAM10-mediated proteolysis could be induced by Ca2+-influx and staurosporine treatment, indicating that ADAM10-mediated VE-cadherin cleavage contributes to the dissolution of adherens junctions during endothelial cell activation and apoptosis, respectively. In contrast, protein kinase C activation or inhibition did not modulate VE-cadherin processing. Increased ADAM10 expression was functionally associated with an increase in endothelial permeability. Remarkably, our data indicate that ADAM10 activity also contributes to the thrombin-induced decrease of endothelial cell-cell adhesion. Moreover, knockdown of ADAM10 in HUVECs as well as in T cells by small interfering RNA impaired T cell transmigration. Taken together our data identify ADAM10 as a novel regulator of vascular permeability and demonstrate a hitherto unknown function of ADAM10 in the regulation of VE-cadherin-dependent endothelial cell functions and leukocyte transendothelial migration.
Keywords: endothelium, metalloprotease, endothelial permeability, VE-cadherin
Introduction
The endothelium represents the major physical barrier in the extravasation of blood components and leukocytes to the surrounding tissue. Impairment of this barrier by inflammatory mediators leads to disintegration of endothelial junctions and an increase in permeability and formation of edema. However, the molecular mechanisms regulating the cohesion of endothelial junctions are still poorly understood. Vascular-endothelial (VE)-cadherin, which represents the major component of endothelial adherens junctions, is a crucial determinant of vascular integrity. This 130kDa cell surface glycoprotein that mediates homotypic Ca2+-dependent cell adhesion also plays a central role in vasculogenesis, angiogenesis and in the regulation of macromolecular permeability.1 Several inflammatory mediators and vasoactive factors including thrombin2 have the potential to disrupt the VE-cadherin complex and to increase vascular permeability providing the basis of edematous tissue injury in many diseases states including sepsis, ischemia-reperfusion and acute respiratory distress.3,4
The vascular endothelium also forms an important barrier for circulating leukocytes. Endothelial cells actively control the efficiency of leukocyte transmigration by regulating the structure of intercellular junctions.5 It has been proposed that VE-cadherin acts as a gatekeeper for the passage of leukocytes, which induce delocalization of VE-cadherin away from adherens junctions.6 Recently, it was shown that the extracellular domain of VE-cadherin is released by an unidentified metalloprotease activity during apoptosis of endothelial cells suggesting that shedding of VE-cadherin may play a role in several biological settings that involve reorganisation of adherens junctions.7 Moreover, elevated levels of soluble VE-cadherin and increased VE-cadherin processing are associated with coronary atherosclerosis and diabetic retinopathy.8,9 These findings suggest that the proteolytic release of VE-cadherin represents an important mechanism for the regulation of VE-cadherin functions. However, the protease responsible for this process has not been identified.
The majority of ectodomain sheddases belong to the family of zinc-dependent transmembrane disintegrin metalloproteinases (ADAMs). ADAMs play an important role in diverse biological processes such as fertilization, myogenesis, neurogenesis and the activation of growth factors and immune regulators.10 Several ADAMs have been implicated in the development of the cardiovascular system and the remodelling of cardiac tissue.11 In particular ADAM10 and its close relative ADAM17 (TNF-α converting enzyme (TACE)) have been studied in the context of ectodomain shedding. Disruption of ADAM17 in mice results in perinatal lethality.12 The mice show similar heart defects as heparin binding EGF-like growth factor (HB-EGF)-deficient mice, probably due to the role of ADAM17 for processing and activation of HB-EGF, transforming growth factor-α, and amphiregulin.13 ADAM10-deficient mice die at day 9.5 of embryogenesis with multiple defects of the developing central nervous system and cardiovascular system.14 ADAM10 plays a critical role during neurogenesis, in Notch signaling and in the regulation of neuronal cell adhesion.14,15 However, the molecular function of ADAM10 in the vascular system has not been analysed so far. The purpose of the present study was to assess the role of ADAM10 in the regulation of vascular permeability and its influence on the expression and function of the endothelial cell adhesion molecule VE-cadherin.
Materials and Methods
Sources of reagents, immunocytochemistry staining, cell culture methods, transfection methods, and statistical analysis are listed in the expanded Materials and Methods section in the online data supplement available at http://circres.ahajournals.org. Western blotting was performed as described elsewhere.15 Quantification of soluble VE-cadherin was performed in triplicates using Bender MedSystems® (Vienna, Austria) ELISA according to manufacturer’s instructions.
Permeability assay
HUVECs (3×105 normally, 6×105 cells for ADAM10 overexpression or siRNA experiments) were seeded on collagen-coated transwell filters (0.4 μm pore size, Costar) in 24-well dishes and grown until they reached confluence. For the assay cells were preincubated for 30 min in the presence of metalloproteinase inhibitors GI254023X (10 μM), GM6001 (10 μM) or DMSO in RPMI. Afterwards FITC-dextran (Mr 40,000; Sigma) at a final concentration of 1 mg/ml was added to the upper chamber. At the indicated time points 50 μl samples were taken from the lower compartment and replaced with the same volume of growth medium. The fluorescent content of samples was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a fluorescence plate reader (Lambda Fluoro 320, MWG Biotech.).
Preparation of phytohemagglutinin-stimulated T cells (PHA-blasts)
Peripheral blood mononuclear cells were isolated from buffy coat preparations from healthy volunteers and stimulated with PHA (0,5 μg/ml) for four days. Viable cells were further propagated in RPMI 1640 in the presence of rIL-2 (10 U/ml). After two days the cells were used for transmigration assays.
Transmigration assay
HUVECs were grown to confluence on collagen-coated transwell tissue culture inserts (Costar, Cambrige, MA) of 3 μm pore size. PHA-blasts were centrifuged over Ficoll to remove dead cells and debris. Cells were resuspended in Endothelial Cell Growth Medium and 5×105 cells added into each insert and left to migrate through the monolayers. Triplicate wells were used for each data point. After 24 hours incubation, cells in the lower chamber were counted. Results are expressed as percentage of transmigrated cells of 3 independent counts.
Results
ADAM10 Mediates VE-Cadherin Proteolysis and Modulates Vascular Permeability
To analyze whether ADAM9, 10 or 17 are feasible candidates for VE-cadherin cleavage, we cotransfected these proteases together with a carboxy (C)-terminal HA-tagged VE-cadherin construct in COS-7 cells. Using antibodies against the HA-tag, we found that only ADAM10 overexpression increased in the generation of the VE-cadherin C-terminal fragments (CTFs) (Figure 1A).
Figure 1. ADAM10 mediates VE-cadherin shedding and modulates endothelial permeability.

A) ADAM10 overexpression increases VE-cadherin proteolysis. Cos-7 cells were cotransfected with HA-VE-cadherin and empty vector (pcDNA3.1), ADAM9, ADAM10 or ADAM17. Cells were analyzed by immunoblot using anti-HA antibody (lower panel). The same blot was reprobed with an anti-ADAM10 antibody. FL: full length; CTF: C-terminal fragment; p: precursor of ADAM10; m: mature form of ADAM10. B) HUVECs were treated with DMSO, γ-secretase inhibitor X (1 μM) or γ-secretase inhibitor DAPT (10 μM) for 18 hours, followed by lysis and immunoblot analysis using C-terminal VE-cadherin antibody. C) HUVECs were incubated with DMSO, ADAM10 inhibitor (GI254023X, 10 μM), ADAM10/17 inhibitor (GW280264X, 10 μM), broad-spectrum metalloproteinase inhibitors TAPI (20 μM) or GM6001 (10 μM) for 18 hours. Cell pellets were analyzed for the generation of VE-cadherin CTFs by immunoblotting (left panel) and for the release of soluble VE-cadherin ectodomain by VE-cadherin ELISA (right panel). One representative immunoblot and corresponding ELISA out of three experiments is shown. ELISA data are expressed as mean±SD. *P<0.05 vs DMSO treated cells. D) HUVECs were transiently transfected with control siRNA or hADAM10 siRNA. Cell pellets were harvested 48 hours after transfection and analyzed for VE-cadherin CTF generation and ADAM10 expression by immunoblotting. Tubulin was used as loading control. E) ADAM10 modulates endothelial permeability. HUVECs were grown to confluence on porous collagen-coated transwell filters and pretreated with DMSO, GI254023X (10 μM) or GM6001 (10 μM) for 30 min. Permeability for FITC-dextran (40 kDa) was measured at different time points in a fluorescence plate reader (λEX 485 nm; λEM 525 nm) (upper panel). HUVECs were either transfected with empty vector (1, insert) or with ADAM10 (2, insert) and seeded on transwell filters. 48 hours after transfection and after monolayer formation permeability for FITC-dextran (40 kDa) was measured (lower panel). One representative out of three experiments is shown, respectively. Data are expressed as mean±SD. *P<0.05 GI vs DMSO treated cells (upper panel). *P<0.05 vs mock transfected cells (lower panel).
For the analysis of VE-cadherin processing in an endogenous system, we used human umbilical vein endothelial cells (HUVECs). Only minor amounts of CTFs could be detected under constitutive conditions indicating that these fragments might be substrates for further proteolysis (Figure 1B). In order to assess whether VE-cadherin processing is followed by intramembrane γ-secretase-mediated cleavage, VE-cadherin shedding was analyzed in the presence of the γ-secretase inhibitors DAPT or inhibitor X. The application of these inhibitors increased the amount of CTFs, indicating a role of the γ-secretase in VE-cadherin proteolysis (Figure 1B).
In order to clarify the involvement of the most likely candidate sheddase ADAM10 and its close homologue ADAM17 for VE-cadherin proteolysis in HUVECs we performed inhibitor studies with two hydroxamate based compounds that differ in their capacity to block the activities of these two proteases. The inhibitor GW280264X has been shown to block ADAM17 and ADAM10, while the compound GI254023X blocked ADAM10 with more than 100-fold increased potency compared to ADAM17.16 To avoid the rapid processing of the CTFs by γ-secretase, we included the γ-secretase inhibitor X in our assays. As shown in Figure 1C, both the VE-cadherin-CTF generation (left panel) and the release of soluble VE-cadherin (right panel) could be strongly diminished by the preferential ADAM10 inhibitor GI254023X. The inhibitor led to a dose-dependent decrease of VE-cadherin proteolysis (supplemental Figure 1), indicating that the majority of metalloprotease-released VE-cadherin in HUVECs can be attributed to ADAM10. This result was verified using siRNA experiments. Transfection of HUVECs with ADAM10 siRNA led to 80% reduction of ADAM10 protein expression, which resulted in a 60% reduction of VE-cadherin processing as shown in Figure 1D (see also supplemental Figure 2). Since metalloproteases might also exert indirect effects by activating each other, we stimulated HUVECs with APMA, a potent activator of both MMPs and ADAMs.17 APMA treatment increased VE-cadherin proteolysis only in the presence of ADAM10, indicating that ADAM10 is directly responsible for VE-cadherin shedding (supplemental Figure 3A). Our observation that recombinant ADAM10 can cleave recombinant VE-cadherin in vitro provides additional evidence for a direct interaction of these proteins (supplemental Figure 3B).
Endothelial cell-cell junctions control the intercellular permeability to plasma solutes and their integrity depends on the structure and function of VE-cadherin.18,19 To analyze whether ADAM10 would affect the integrity of intercellular junctions, we measured the permeability of a confluent endothelial monolayer for 40 kDa FITC-dextran. Cells were cultivated on transwell filter inserts in the presence of the preferential ADAM10 inhibitor GI254023X or the broad-spectrum metalloprotease inhibitor GM6001. ADAM10 inhibition led to a significant decrease of endothelial permeability compared to the mock treated cells (Figure 1E, upper panel).
Previously, it has been described that endothelial activation by LPS, TNF-α or antigraft antibodies induced an upregulation of ADAM10 at the endothelial cell surface.20 To evaluate whether increased ADAM10 expression would also alter endothelial permeability, HUVECs were transfected either with ADAM10 or empty vector and the endothelial permeability for FITC-dextran was measured 48 hours after transfection. Indeed, overexpression of ADAM10 led to increased endothelial permeability (Figure 1E, lower panel). These results indicate that ADAM10-dependent regulation of VE-cadherin expression is of functional relevance for vascular permeability.
Calcium Influx but not PKC Activation Induces ADAM10-Mediated VE-Cadherin Proteolysis
The proteolytic release of transmembrane proteins does not only occur constitutively, but might also be enhanced by stimulation. Therefore, we set out to assess which stimuli might activate ADAM10-mediated VE-cadherin shedding. Previously, Herren and colleagues showed that serum starvation-induced endothelial apoptosis correlates with a dramatic decrease of VE-cadherin at the cell surface.7 When HUVECs were deprived of growth factors for 16 hours in the presence or absence of the ADAM10 inhibitor GI254023X we found that VE-cadherin CTF formation did not significantly increase (Figure 2A). This became even more apparent, when shedding was calculated as the percentage of VE-cadherin CTFs in relation to total VE-cadherin (full-length protein and VE-cad fragment) by densitometric quantification of three experiments. In contrast, staurosporine, a general protein kinase inhibitor, which is also well known to induce endothelial cell apoptosis, significantly increased ADAM10-dependent VE-cadherin proteolysis (Figure 2B). This effect was rather due to the apoptotic signaling cascade than to protein kinase C (PKC) inhibition since two PKC inhibitors, GF109203X and GÖ6976, did not affect VE-cadherin shedding (supplemental Figure 4).
Figure 2. Staurosporine induced-apoptosis activates ADAM activity.

A) HUVECs were serum starved (SS) for 16 hours or B) treated with staurosporine (ST, 1 μM) for 6 hours combined with DMSO or GI254023X (10 μM) in the presence of the γ-secretase inhibitor. Cells were lysed and analyzed by immunoblot using anti-VE-cadherin antibody. Tubulin was used as loading control. CTF generation was calculated as percentage of total VE-cadherin (full length VE-cadherin plus CTF) by densitometric analysis. Data are expressed as mean±SEM; n=3 independent experiments, respectively. *P<0.05 vs DMSO treated cells. °P<0.05 vs SS+DMSO or ST+DMSO treated cells.
ADAM-mediated shedding of diverse membrane proteins occurs in response to PKC activation by phorbol esters like PMA. PKC activation has also been discussed to influence endothelial permeability,21 but the results in HUVECs are conflicting so far. While Yamada et al22 showed that activation of PKC caused a decrease in endothelial permeability, Bussolino et al23 found an increase in endothelial permeability. As shown in figure 3A, PMA stimulation of HUVECs did not result in increased VE-cadherin proteolysis suggesting that ADAM10-mediated VE-cadherin processing is independent of PKC activation.
Figure 3. Calcium-influx but not PKC activation induces ADAM10-dependent VE-cadherin proteolysis.
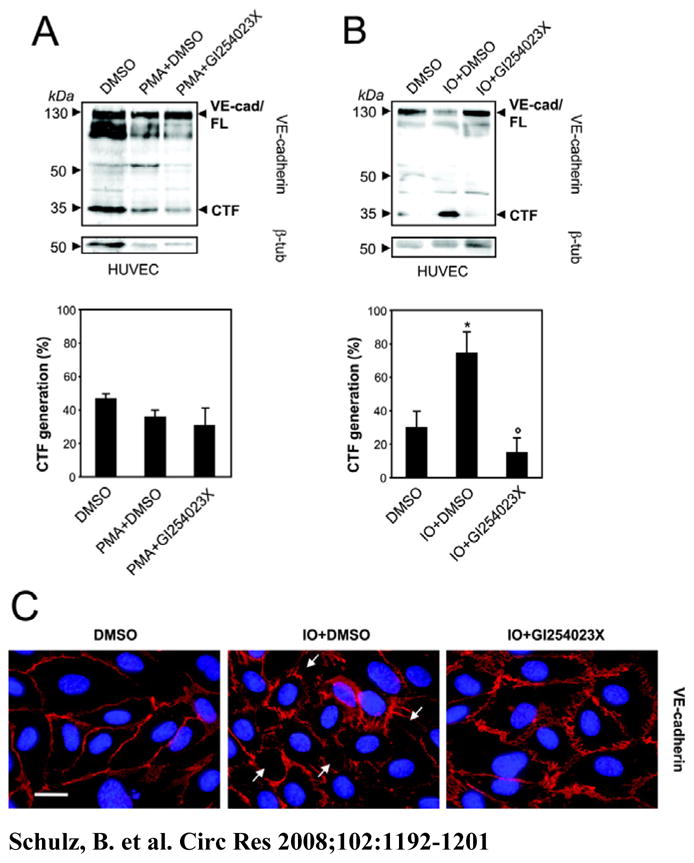
A) HUVECs were treated with PMA (200 ng/ml) for 2 hours or B) ionomycin (IO, 10 μM) for 30 min, combined with DMSO or GI254023X (10 μM) in the presence of the γ-secretase inhibitor. VE-cadherin proteolysis was quantificated by densitometric analysis of three independent experiments. Tubulin was used as loading control. Data represent mean±SEM of these experiments. *P<0.05 vs DMSO treated cells. °P<0.05 vs IO+DMSO treated cells. C) HUVECs were grown to confluence on collagen-coated glass coverslips and stimulated with ionomycin (IO, 10 μM) combined with DMSO or GI254023X (10 μM). Cells were fixed and immunostained with VE-cadherin (red). Nuclei were counterstained with DAPI. Bar: 20 μm.
Endothelial dysfunction can be induced by several vasoactive substances, but also by Ca2+ ionophores such as ionomycin, which are known to elevate endothelial permeability. Ionomycin stimulation strongly increased the generation of VE-cadherin CTFs and this effect was abrogated in the presence of the preferential ADAM10 inhibitor GI254023X (Figure 3B) or by ADAM10 siRNA transfection (supplemental Figure 5A). Stimulation with ionomycin also led to loss of VE-cadherin from the cell surface (Figure 3C). Accordingly, cells lost cell-cell contacts and exhibited a slightly rounded appearance. In contrast, co-treatment with the ADAM10 inhibitor GI254023X retained VE-cadherin at the cell surface and prevented gap formation. This observation confirms our biochemical data and strongly suggests that ADAM10-dependent changes in VE-cadherin localisation and endothelial cell-cell adhesion can be induced by Ca2+-influx.
Thrombin Stimulation Leads to ADAM10-Mediated VE-Cadherin Shedding
Thrombin induces endothelial permeability by stimulation of cytoskeletal signaling pathways resulting in an increase of intracellular Ca2+.24 To test whether thrombin induces VE-cadherin proteolysis, HUVECs were stimulated with thrombin for 5 to 120 minutes and analyzed by Western blot analysis. Thrombin rapidly induced VE-cadherin CTF formation (Figure 4A). This effect started 5 minutes after stimulation and was maximal after 30 minutes. This increase was abolished in the presence of a broad-spectrum metalloprotease inhibitor or by ADAM10 siRNA transfection (Figure 4B and supplemental Figure 5B), suggesting that thrombin leads to an ADAM10-mediated VE-cadherin proteolysis.
Figure 4. Thrombin m odulates endothelial permeability partially by activation of ADAM10-dependent VE-cadherin shedding.
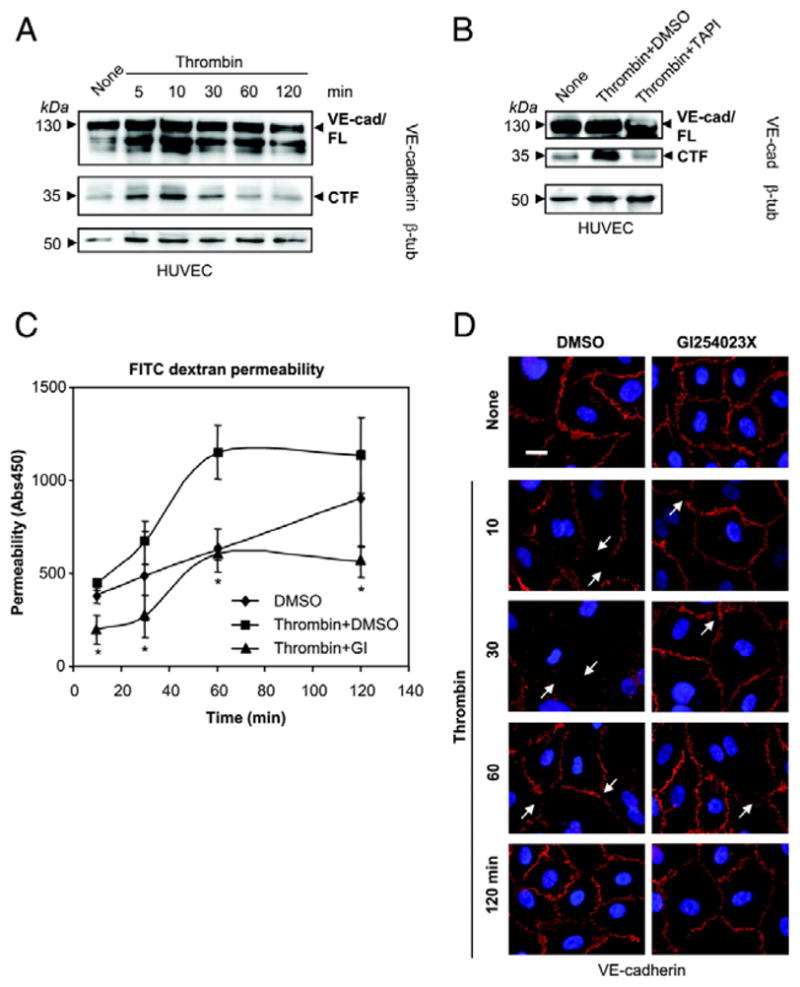
A) Time course of thrombin-induced VE-cadherin proteolysis. HUVECs were incubated with thrombin (1 U/ml) and examined for the generation of VE-cadherin CTFs by immunoblotting. The blot was reprobed with β-tubulin antibody. B) HUVECs were incubated with thrombin (1 U/ml) for 30 min in the presence of DMSO or metalloproteinase inhibitor TAPI (20 μM).
C) HUVECs were grown to confluence on collagen-coated transwell filters and pretreated with DMSO or ADAM10 inhibitor GI254023X (10 μM) for 30 min. Then, cells were stimulated with thrombin (1 U/ml) and permeability for FITC-dextran (40 kDa) was measured at different time points in a fluorescence plate reader (λEX 485 nm; λEM 525 nm). One representative out of three experiments is shown. Data are expressed as mean±SD. *P<0.05 Thrombin+GI vs Thrombin+DMSO.
D) HUVECs were grown to confluence on collagen-coated glass coverslips and pretreated with DMSO or ADAM10 inhibitor GI254023X (10 μM) for 30 min. Afterwards cells were stimulated with thrombin (1 U/ml) for 120 min. Cells were fixed and immunostained with anti VE-cadherin antibodies (red). Nuclei were counterstained with DAPI. Bar, 20 μm.
Thrombin stimulation also induced a rapid increase in endothelial permeability, which was sustained for 1 hour (Figure 4C). Preincubation with the preferential ADAM10 inhibitor GI254023X reduced the constitutive shedding and led to 40% reduction of the thrombin effect. Nevertheless, thrombin also slightly induced endothelial permeability in the presence of the inhibitor, indicating that ADAM10 activity contributes to the thrombin-induced decrease of endothelial cell-cell adhesion but acts in concert with other mechanisms.
This assumption was further confirmed using immunocytochemical analyses. VE-cadherin immunoreactivity was very prominent at cell-cell junctions of mock-treated cells (Figure 4D). After 30 minutes Thrombin stimulation led to nearly complete loss of VE-cadherin from the cell surface and to gap formation due to cell rounding and cell contraction. Even though thrombin also affected HUVECs pretreated with the ADAM10 inhibitor GI254023X, gap formation and loss of cell-cell contacts was less pronounced compared to mock treated cells. Instead, VE-cadherin immunoreactivity was mostly preserved at cell-cell borders. While mock treated cells recovered slowly, VE-cadherin expression and cell contact formation was completely restored after 120 minutes in the presence of the ADAM10 inhibitor. These data confirm that the ADAM10 inhibitor stabilizes the surface expression of VE-cadherin and counteracts the thrombin-induced cell dissociation.
Transmigration of Activated T Cells Through Endothelial Monolayer is ADAM10-Dependent
Activated leukocytes have to passage through the endothelial barrier before they get to the sites of inflammation.25 Migrating leukocytes induce delocalisation of VE-cadherin resulting in a gap through which the cells can pass.6 To analyze whether ADAM10 activity would affect leukocyte transmigration, HUVECs were preincubated with different metalloprotease inhibitors and T cell transmigration was monitored after 16 hours in a transwell system. The broad-spectrum metalloprotease inhibitors GM6001 and TAPI significantly reduced transendothelial migration (TEM) compared to mock treated cells (Figure 5A). Interestingly, the preferential ADAM10 inhibitor GI254023X showed the most pronounced effect. This result was further confirmed in a similar assay using ADAM10 siRNA transfection, which led to decreased ADAM10 protein expression in HUVECs (Figure 5B, insert) and to a decreased T cell transmigration (Figure 5B). These data demonstrate that ADAM10 activity does not only modulate endothelial cell-cell adhesion but also leukocyte transmigration. In order to analyze whether ADAM10 activity would also be required on the surface of the transmigrating T cells, we additionally transfected the T cells with ADAM10 siRNA. As shown in Figure 5B, ADAM10 reduction in T cells also led to a decreased transmigration rate. These findings indicate that ADAM10 plays an additional role for the regulation of adhesion and migration capacities of activated human T cells.
Figure 5. Inhibition of ADAM10 decreases transendothelial migration of T cells.

A) HUVECs were grown to confluence on 3 μm pore size transwell filters and pretreated with DMSO, broad-spectrum metalloproteinase inhibitor GM6001 (10 μM), GI254023X (10 μM) or GW280264X (10 μM) for 1 hour. After medium changes in the upper chamber, HUVECs were incubated with 5×105 T cells (PHA blasts) per filter. T cell transmigration was measured by counting cells in the lower chamber after 24 hours. Data are expressed as mean±SEM; n=3 independent experiments. *P<0.05 vs DMSO treated cells. B) Either T cells or HUVECs were transfected with control siRNA (1, insert) or ADAM10 siRNA (2, insert) and analyzed for T cell transmigration. The downregulation of ADAM10 (A10, insert) expression was confirmed using immunoblot analyses. p: precursor of ADAM10; m: mature form of ADAM10. Data are mean±SEM; n=3 independent experiments. *P<0.05 vs mock transfected cells.
Discussion
In this study, we have identified a critical role for ADAM10 in the regulation of vascular permeability and transendothelial T cell migration. Our data demonstrate that ADAM10-mediated cleavage of VE-cadherin is an important mechanism to regulate endothelial cell-cell adhesion. Using HUVECs, we demonstrate here that pharmacological inhibition and siRNA knockdown of ADAM10 reduces VE-cadherin proteolysis, thus leading to increased amounts of VE-cadherin on the cell membrane and decreased vascular permeability and leukocyte transmigration.
Interestingly, our data demonstrate for the first time, that the metalloprotease generated C-terminal VE-cadherin fragment is a substrate for regulated intramembrane proteolysis. This proteolytic sequence has been implicated in the processing of an increasing number of proteins, which are able to directly regulate signal transduction via translocation to the nucleus. For other proteins, like N-cadherin, the intramembrane cleavage has been shown to indirectly influence cell signaling by enhancing the degradation of transcription factors and β-catenin signaling.15,26 In this context, our data suggest the interesting possibility that VE-cadherin might also directly contribute to cellular signaling.
The intercellular junctions of endothelial cells have an important barrier function regulating the permeability to small molecules. VE-cadherin is critically involved in the modulation of endothelial permeability. Pharmacological inhibition of ADAM10 activity correlated with decreased paracellular permeability as evidenced by a decreased passage of 40 kDa dextran. Recently, it has been described that activation of endothelial cells with LPS or TNF-α led to upregulation of ADAM10 in endothelial cells.20 Together with our finding of increased endothelial permeability due to ADAM10 overexpression these data indicate that misregulation of ADAM10 activity might contribute to inflammation induced changes in vascular permeability.
However, we can not exclude that ADAM10 is additionally involved in the shedding of other endothelial cell adhesion molecules, which are also important for vascular permeability and might directly or indirectly contribute to the observed effect.
In contrast to the junctional distribution of VE-cadherin, the ADAM10 substrate N-cadherin15 shows a diffusive expression over the whole cell membrane of endothelial cells. It has been speculated that N-cadherin may be responsible for the anchorage of the endothelium to other N-cadherin expressing surrounding cell types such as smooth muscle cells and pericytes.27,28 Therefore, it is more likely that other junctional molecules might contribute to the observed findings. Besides VE-cadherin five other molecules are concentrated at the lateral borders of endothelial cells that have been implicated in the process of endothelial permeability and leukocyte transmigration, namely PECAM (Platelet Endothelial Cell Adhesion Molecule), CD99 and JAM-A,-B and -C. While we could not find any evidence for an involvement of ADAM10 in PECAM processing in HUVECs (data not shown), CD99 and JAMs are interesting candidate molecules for ADAM-mediated cleavage and additional modulation of vascular permeability through such shedding events.
ADAM-mediated constitutive shedding of transmembrane proteins can be strongly enhanced by external stimuli, such as phorbol esters, ionophores, and growth factors. Here we demonstrate that ionomycin-induced calcium influx significantly activated ADAM10-dependent VE-cadherin proteolysis. Several vasoactive agents like histamine or thrombin, which lead to endothelial gap formation, induce a transient increase in cytoplasmic Ca2+ concentration and might also lead to activation of ADAM10. In our analyses of VE-cadherin proteolysis we focused on the influence of thrombin, which is known to induce an increase in permeability in endothelial monolayer comparable with the in vivo effect.29,30 Our data confirm that thrombin stimulation leads to an activation of ADAM10-mediated VE-cadherin proteolysis. However, the thrombin-induced increase in endothelial permeability was only partially reduced in the presence of a preferential ADAM10 inhibitor. Since several signaling mechanisms are involved in thrombin-induced hyperpermability including the RhoA/Rho kinase signaling pathway or protein tyrosine kinase pathway,31 it is likely that the activation of ADAM10 is part of several associated thrombin effects.
The vascular endothelium forms a barrier for macromolecules but also for circulating leukocytes. Previous studies have demonstrated that through their adhesion, leukocytes could transfer intracellular signals to endothelial cells in different ways. It has been described that transmigration across endothelium resulted in dissociation and loss of the VE-cadherin complex,32,33 but the mechanism by which delocalization of VE-cadherin occurs is still unclear.34 Activated polymorphonuclear cell (PMN) or purified PMN elastase have been reported to cleave VE-cadherin.3 In contrast, Allport et al35 reported that mononuclear leukocytes, which have significantly less proteolytic capabilities as compared to neutrophils, still induce focal changes in VE-cadherin complex that correlate with the location of actively transmigrating leukocytes. These findings suggest that the loss of VE-cadherin is due to an endothelial-dependent mechanism.
According to our data, ADAM10 is involved in the regulation of the transmigration process by controlling endothelial cell adhesion. It is tempting to speculate that transmigrating leukocytes induce endothelial intracellular signaling that activates ADAM10-dependent VE-cadherin cleavage leading to transient gap formation. Noteworthy, leukocyte adhesion is also associated with an increase in intracellular Ca2+.36 This effect might contribute to an activation of ADAM10-dependent VE-cadherin shedding. Our results demonstrating that Ca2+-influx induces the cleavage of VE-cadherin depending on ADAM10 activity offers valuable clues to understand the disappearence of VE-cadherin and the gap formation during the transmigration process (Figure 6).
Figure 6. Schematic model of ADAM10-dependent endothelial gap formation.

Thrombin binding to its receptor (PAR-1) induces intracellular signaling pathways, which activate ADAM10-dependent VE-cadherin proteolysis. This effect contributes to endothelial cell dissociation and gap formation. ADAM10 also regulates the transmigration of T cells through the endothelium. Most likely, the binding of activated T cells to the endothelium induces intracellular signaling cascades in endothelial cells, which lead to ADAM10 activation and increased VE-cadherin proteolysis. T cell-expressed ADAM10 might also contribute to the regulation of the transmigration process through cleavage of cell adhesion molecules on the T cell surface or through trans-shedding of endothelial VE-cadherin.
Since ADAM10, which is also expressed at the T cell surface,37 has also been described to act in trans,38 the leukocytes might in principle also cleave off the endothelial VE-cadherin. This could be one explanation for our finding that ADAM10 reduction in activated T cells led to decreased transendothelial migration. On the other hand, the leukocyte adhesion and transmigration process involves several adhesion molecules like PECAM or JAM, therefore, it is more likely that other unidentified ADAM10 substrates could be responsible for this effect. Further studies will have to show if VE-cadherin trans-shedding is feasible or which additional proteolytic events and molecules are involved in the transmigration process (Figure 6).
In conclusion, our findings on the molecular interactions of ADAM10 and VE-cadherin help to explain an important regulatory mechanism of VE-cadherin-mediated function, such as control of paracellular permeability and leukocyte transmigration. Dysregulation of ADAM10 activity might contribute to VE-cadherin-dependent defects in vascular permeability or angiogenesis associated with diseases like atherosclerosis or chronic edema. Future studies are required to understand how this interaction is integrated into the various signaling networks that regulate vascular permeability and leukocyte TEM.
Supplementary Material
Acknowledgments
Murine VE-cadherin expression plasmid was kindly provided by D. Vestweber. We would like to thank Bjorn Ahrens for excellent technical assistance, Johann Groth for initial experiments in an advanced student course and Maria Sacta for proof-reading the manuscript.
SOURCES OF FUNDING
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 415, TPB9) given to P.S. and K.R., and the Interuniversity Attraction Poles Program P5/19 of the Belgian Federal Science Policy Office and the European Union (APOPIS: LSHM-CT-2003-503330), and NIH EY015719 to CPB. K.R. was supported by the Stiftung zur Forderung der medizinischen Forschung (Faculty of Medicine, University of Kiel) and the Hensel-Stiftung.
Abbreviations
- ADAM
a disintegrin and metalloproteinase
- CTF
C-terminal cleavage fragment
- siRNA
small interfering RNA
Footnotes
DISCLOSURES: None.
References
- 1.Vincent PA, Xiao K, Buckley KM, Kowalczyk AP. VE-cadherin: adhesion at arm’s length. Am J Physiol Cell Physiol. 2004;286:C987–C997. doi: 10.1152/ajpcell.00522.2003. [DOI] [PubMed] [Google Scholar]
- 2.Rabiet MJ, Plantier JL, Rival Y, Genoux Y, Lampugnani MG, Dejana E. Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol. 1996;16:488–496. doi: 10.1161/01.atv.16.3.488. [DOI] [PubMed] [Google Scholar]
- 3.Carden D, Xiao F, Moak C, Willis BH, Robinson-Jackson S, Alexander S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am J Physiol. 1998;275:H385–H392. doi: 10.1152/ajpheart.1998.275.2.H385. [DOI] [PubMed] [Google Scholar]
- 4.Baumgartner I, Rauh G, Pieczek A, Wuensch D, Magner M, Kearney M, Schainfeld R, Isner JM. Lower-extremity edema associated with gene transfer of naked DNA encoding vascular endothelial growth factor. Ann Intern Med. 2000;132:880–884. doi: 10.7326/0003-4819-132-11-200006060-00005. [DOI] [PubMed] [Google Scholar]
- 5.Dejana E, Spagnuolo R, Bazzoni G. Interendothelial junctions and their role in the control of angiogenesis, vascular permeability and leukocyte transmigration. Thromb Haemost. 2001;86:308–315. [PubMed] [Google Scholar]
- 6.Shaw SK, Bamba PS, Perkins BN, Luscinskas FW. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol. 2001;167:2323–2330. doi: 10.4049/jimmunol.167.4.2323. [DOI] [PubMed] [Google Scholar]
- 7.Herren B, Levkau B, Raines EW, Ross R. Cleavage of beta-catenin and plakoglobin and shedding of VE-cadherin during endothelial apoptosis: evidence for a role for caspases and metalloproteinases. Mol Biol Cell. 1998;9:1589–1601. doi: 10.1091/mbc.9.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soeki T, Tamura Y, Shinohara H, Sakabe K, Onose Y, Fukuda N. Elevated concentration of soluble vascular endothelial cadherin is associated with coronary atherosclerosis. Circ J. 2004;68:1–5. doi: 10.1253/circj.68.1. [DOI] [PubMed] [Google Scholar]
- 9.Navaratna D, McGuire PG, Menicucci G, Das A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes. 2007;56:2380–2387. doi: 10.2337/db06-1694. [DOI] [PubMed] [Google Scholar]
- 10.Reiss K, Ludwig A, Saftig P. Breaking up the tie: Disintegrin-like metalloproteinases as regulators of cell migration in inflammation and invasion. Pharmacol Ther. 2006;111:985–1006. doi: 10.1016/j.pharmthera.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Horiuchi K, Zhou HM, Kelly K, Manova K, Blobel CP. Evaluation of the contributions of ADAMs 9, 12, 15, 17, and 19 to heart development and ectodomain shedding of neuregulins beta1 and beta2. Dev Biol. 2005;283:459–471. doi: 10.1016/j.ydbio.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 13.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena IA, von Figura K, Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- 15.Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005;24:742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ludwig A, Hundhausen C, Lambert MH, Broadway N, Andrews RC, Bickett DM, Leesnitzer MA, Becherer JD. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb Chem High Throughput Screen. 2005;8:161–171. doi: 10.2174/1386207053258488. [DOI] [PubMed] [Google Scholar]
- 17.Merlos-Suarez A, Ruiz-Paz S, Baselga J, Arribas J. Metalloprotease-dependent protransforming growth factor-alpha ectodomain shedding in the absence of tumor necrosis factor-alpha-converting enzyme. J Biol Chem. 2001;276:48510–48517. doi: 10.1074/jbc.M103488200. [DOI] [PubMed] [Google Scholar]
- 18.Breviario F, Caveda L, Corada M, Martin-Padura I, Navarro P, Golay J, Introna M, Gulino D, Lampugnani MG, Dejana E. Functional properties of human vascular endothelial cadherin (7B4/cadherin-5), an endothelium-specific cadherin. Arterioscler Thromb Vasc Biol. 1995;15:1229–1239. doi: 10.1161/01.atv.15.8.1229. [DOI] [PubMed] [Google Scholar]
- 19.Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, Dejana E. The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin) J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulday G, Coupel S, Coulon F, Soulillou JP, Charreau B. Antigraft antibody-mediated expression of metalloproteinases on endothelial cells. Differential expression of TIMP-1 and ADAM-10 depends on antibody specificity and isotype. Circ Res. 2001;88:430–437. doi: 10.1161/01.res.88.4.430. [DOI] [PubMed] [Google Scholar]
- 21.Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis CA, Tiruppathi C. Ca(2+) signalling and PKCalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J Physiol. 2001;533:433–445. doi: 10.1111/j.1469-7793.2001.0433a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada Y, Furumichi T, Furui H, Yokoi T, Ito T, Yamauchi K, Yokota M, Hayashi H, Saito H. Roles of calcium, cyclic nucleotides, and protein kinase C in regulation of endothelial permeability. Arteriosclerosis. 1990;10:410–420. doi: 10.1161/01.atv.10.3.410. [DOI] [PubMed] [Google Scholar]
- 23.Bussolino F, Camussi G, Aglietta M, Braquet P, Bosia A, Pescarmona G, Sanavio F, D’Urso N, Marchisio PC. Human endothelial cells are target for platelet-activating factor. I. Platelet-activating factor induces changes in cytoskeleton structures. J Immunol. 1987;139:2439–2446. [PubMed] [Google Scholar]
- 24.Vouret-Craviari V, Boquet P, Pouyssegur J, Obberghen-Schilling E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol Biol Cell. 1998;9:2639–2653. doi: 10.1091/mbc.9.9.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aurrand-Lions M, Johnson-Leger C, Imhof BA. The last molecular fortress in leukocyte trans-endothelial migration. Nat Immunol. 2002;3:116–118. doi: 10.1038/ni0202-116. [DOI] [PubMed] [Google Scholar]
- 26.Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Navarro P, Ruco L, Dejana E. Differential localization of VE- and N-cadherins in human endothelial cells: VE-cadherin competes with N-cadherin for junctional localization. J Cell Biol. 1998;140:1475–1484. doi: 10.1083/jcb.140.6.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 29.Garcia JG, Pavalko FM, Patterson CE. Vascular endothelial cell activation and permeability responses to thrombin. Blood Coagul Fibrinolysis. 1995;6:609–626. doi: 10.1097/00001721-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Horgan MJ, Fenton JW, Malik AB. Alpha-thrombin-induced pulmonary vasoconstriction. J Appl Physiol. 1987;63:1993–2000. doi: 10.1152/jappl.1987.63.5.1993. [DOI] [PubMed] [Google Scholar]
- 31.Nieuw Amerongen GP, Beckers CM, Achekar ID, Zeeman S, Musters RJ, van Hinsbergh VW. Involvement of Rho Kinase in Endothelial Barrier Maintenance. Arterioscler Thromb Vasc Biol. 2007 doi: 10.1161/ATVBAHA.107.152322. [DOI] [PubMed] [Google Scholar]
- 32.Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, Dejana E. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allport JR, Ding H, Collins T, Gerritsen ME, Luscinskas FW. Endothelial-dependent mechanisms regulate leukocyte transmigration: a process involving the proteasome and disruption of the vascular endothelial-cadherin complex at endothelial cell-to-cell junctions. J Exp Med. 1997;186:517–527. doi: 10.1084/jem.186.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Shaw SK, Ma S, Yang L, Luscinskas FW, Parkos CA. Regulation of leukocyte transmigration: cell surface interactions and signaling events. J Immunol. 2004;172:7–13. doi: 10.4049/jimmunol.172.1.7. [DOI] [PubMed] [Google Scholar]
- 35.Allport JR, Muller WA, Luscinskas FW. Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J Cell Biol. 2000;148:203–216. doi: 10.1083/jcb.148.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulte M, Reiss K, Lettau M, Maretzky T, Ludwig A, Hartmann D, de Strooper B, Janssen O, Saftig P. ADAM10 regulates FasL cell surface expression and modulates FasL-induced cytotoxicity and activation-induced cell death. Cell Death Differ. 2007;14:1040–1049. doi: 10.1038/sj.cdd.4402101. [DOI] [PubMed] [Google Scholar]
- 38.Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123:291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.