

Abstract
Presented here is a method, the hierarchical charge partitioning (HCP) approximation, for speeding up computation of pairwise electrostatic interactions in biomolecular systems. The approximation is based on multiple levels of natural partitioning of biomolecular structures into a hierarchical set of its constituent structural components. The charge distribution in each component is systematically approximated by a small number of point charges, which, for the highest level component, are much fewer than the number of atoms in the component. For short distances from the point of interest, the HCP uses the full set of atomic charges available. For long distance interactions, the approximate charge distributions with smaller sets of charges are used instead. For a structure consisting of N charges, the computational cost of computing the pairwise interactions via the HCP scales as O(N log N), under assumptions about the structural organization of biomolecular structures generally consistent with reality. A proof-of-concept implementation of the HCP shows that for large structures it can lead to speed-up factors of up to several orders of magnitude relative to the exact pairwise O(N2) all-atom computation used as a reference. For structures with more than 2–3 thousand atoms the relative accuracy of the HCP (relative root-mean-square force error per atom), approaches the accuracy of the particle mesh Ewald (PME) method with parameter settings typical for biomolecular simulations. When averaged over a set of 600 representative biomolecular structures, the relative accuracies of the two methods are roughly equal. The HCP is also significantly more accurate than the spherical cutoff method. The HCP has been implemented in the freely available nucleic acids builder (NAB) molecular dynamics package in Amber tools. A 10 ns simulation of a small protein indicates that the HCP simulation is stable, and that it can be faster than the spherical cutoff method. A critical benefit of the HCP approximation is that it is algorithmically very simple, and unlike the PME, the HCP is straightforward to use with implicit solvent models.
Keywords: electrostatics, molecular dynamics, implicit solvent, generalized Born
1 Introduction
Today molecular dynamics simulations are routinely used to study the structure and function of biological molecules.1–4 Traditional all-atom molecular dynamics simulations model the system as a collection of atoms with additive pairwise interactions. Despite several critical approximations employed by this methodology, an all-atom simulation of a typical system with tens of thousands of atoms is limited to tens of microseconds or less, even with the computational capabilities of today’s supercomputers.5–8 Various additional approximations, for example implicit solvent models, can further speed up these simulations, but still not enough to dramatically extend the simulation times, especially for large systems.9 These simulation times are still much shorter than the time scale for many important biomolecular processes, such as ligand binding, folding of most proteins, and enzyme turnover, which have time scales in the range of tens of microseconds to seconds and longer.3, 10–12 The limiting factor in most all-atom simulations is the computation of long range electrostatic interactions.13, 14 For a set of N atomic charges q1 … qN, the long range electrostatic potential, energy, and force can be calculated as
| (1) |
| (2) |
| (3) |
where ϕj is the electrostatic potential at point j, E the electrostatic energy of the system, F⃗j the electrostatic force on atom j, rij the distances from atom i to point j, and ϵij the effective dielectric function of the medium between atom i and point j. Depending on the specific solvent model15 used, the function ϵij may be as simple as constant ϵij = 1 as in the explicit solvent model, or may depend on the geometry of the system in a more complex manner, as in many analytical implicit solvent models such as the distance dependent dielectric16 or the generalized Born models.17–41 However, regardless of the specific mathematical form, the computation of electrostatic energy or forces on all N atoms performed via a pairwise formalism such as equation (2) scales as O(N2), unless additional approximations are made. The cost of such exact computation, incurred for each time step of the simulation, can severely limit the duration of the simulation, or the size of the system being simulated. Several approximations have been developed specifically to reduce the costs associated with computation of long-range pairwise interactions. For biomolecular simulations, the most commonly used approximations are the spherical cutoff and the particle mesh Ewald (PME) methods. Another approximation, the fast multipole method, has also been used for biomolecular simulations. The simplest of these, the spherical cutoff method ignores electrostatic interactions between a given atom and all the atoms outside a predefined cutoff distance from the atom in question, while treating the interactions between atoms within the cutoff distance exactly.42, 43 The “industry standard” PME method uses a mathematical transformation to represent long range electrostatic interactions which decay slowly with distance, as a sum of two rapidly converging series, one in real space and the other in Fourier space. The mathematical transformation assumes a periodic boundary condition, where a central cell containing the molecules of interest is surrounded by an infinite array of images of the central cell.44–47 The fast multipole method partitions the system into a hierarchical set of cubic lattices. Electrostatic interactions between atoms within a lattice and in neighboring lattices are treated exactly, while a truncated multipole expansion is used for electrostatic interactions due to atoms in the more distant lattices. The size of the lattices used in the multipole expansion depends on the distance from the point in question, with a larger lattice size being used for more distant lattices.48–50
The increased speed of simulations based on the approximation methods described above comes at a price of reduced accuracy. For example, the simple and robust spherical cutoff method can produce many artifacts, such as spurious forces or artificial structures around the cutoff distance.51–53 The fast multipole method is more accurate than the spherical cutoff method. It is available, for example, in the CHARMM software package90 and has been applied to biomolecular simulations.89, 91 However, so far it has not been as widely adopted for biomolecular simulations, probably due to its algorithmic complexity and instabilities caused by discontinuities inherent in the method.54 The Particle Mesh Ewald (PME) method, which is much more accurate than the spherical cutoff method, is also significantly more complex and can produce artifacts too, such as biasing the simulation toward the most compact conformations.55, 56 Perhaps the most important limitation of the PME method is that it is not clear if it can be extended to practical implicit solvent models where the effective dielectric constant, ϵij, between atoms can be a complex (non-pairwise) function of the position of all the atoms in the system.57 For example, in the generalized Born model interactions between any two atoms depend, through the effective Born radii, on the position of all other atoms in the molecule. A clean decomposition of the atom-atom interaction into the sums in direct and reciprocal space, required for the PME formulation may be impossible for such potential functions. The PME method has therefore not been implemented to speed up practical implicit solvent models, such as the generalized Born model that offers a number of potential advantages including lower computational cost and faster conformational search. Despite these limitations, through years of refinement the PME method has become the de facto standard for molecular dynamics simulation of biomolecular systems.
A very different class of approximation methods for speeding up molecular dynamics simulations are the coarse grain models. These models replace collections of atoms with a single particle.58 Examples of these methods include united atom,59 united residue,60 segment chain,61 and lattice representation62 models. These models can simulate very large systems over millisecond and longer timescales, however at the cost of needing approximate coarse-grained force fields and non-trivial mechanics for moving complex shapes.63
Our goal is to develop a systematic approximation for pairwise long-range interactions that (i) has the algorithmic simplicity and robustness of the spherical cutoff method, (ii) has accuracy approaching that of the PME method, (iii) can be used with implicit solvent models, and (iv) uses the high quality, extensively tested, all-atom force fields.
The hierarchical charge partitioning (HCP) approximation presented here exploits the natural partitioning of biomolecules into its constituent components to speed up the computation of electrostatic interactions with limited and controllable impact on accuracy. Biomolecules can be systematically partitioned into multiple molecular complexes, which consist of multiple polymer chains or subunits, which in turn are made up of multiple amino acid or nucleotide groups. These components form a hierarchical set with, for example, complexes consisting of multiple subunits, subunits consisting of multiple groups, and groups consisting of multiple atoms. Atoms represent the lowest ”mandatory” level in the hierarchy while the highest level depends on the problem. We assume that this hierarchy is approximately preserved during dynamics, eliminating the need to recompute the hierarchical partitioning at every step of the simulation. This is one of the key advantages of the HCP, compared to some of the more traditional approaches based on space partitioning, such as the fast multipole method. The distribution of charges for each component used in the computation varies depending on distance from the point in question: the farther away a component, the fewer charges are used to represent the component. This study systematically explores the accuracy and speed of this simple idea in the context of molecular dynamics.
The remainder of this paper is organized as follows. First we provide a detailed description of the HCP. To assess its accuracy and speed, the approximation is then applied to the computation of electrostatic interactions for a representative sample set of 600 realistic biomolecular structures. Next, we examine other practical factors that may effect the accuracy of the HCP in the context of molecular dynamics simulations. As a proof-of-concept the HCP is then implemented with some basic molecular dynamics capability and tested in a 10 ns implicit solvent simulation of a protein. In conclusion, we discuss our findings along with possible refinements.
2 Hierarchical charge partitioning (HCP)
The concept of the HCP, as described here, is formalized for the specific case of speeding up electrostatic computations in classical molecular dynamics simulations using non-polarizable force fields. The atoms are treated as single point charges located at the atomic center-of-mass. Possible extensions to other types of simulations, such as QM/MM64 and polarizable force fields,65 are briefly discussed in the “Conclusions”.
There are three types of biomolecules that control the functioning of biological organisms: DNA, RNA and proteins.66 These biomolecules are typically organized into complexes of multiple molecular subunits. The subunits are protein, DNA or RNA polymer chains, consisting of nucleotide or amino acid groups (nucleotides in DNA and RNA subunits, and amino acids in proteins subunits). This hierarchical grouping of atoms into groups, groups into subunits, and subunits into complexes, offers a natural hierarchical partitioning of biomolecular structures that is exploited by the method presented here. An example of such a partitioning is illustrated in figure 1. The figure shows a chromatin fibre made up of 100 nucleosome complexes, constructed as described by Wong et. al.67 Each complex consists of 13 subunits†. The subunits are DNA and protein polymer chains each consisting of 49 to 142 groups. And each group consists of 7 to 32 atoms. In total this chromatin fibre is made up of approximately 3 million atoms.
Figure 1.

Example of the natural partitioning of a chromatin fibre. (a) The fibre is made up of 100 nucleosome complexes. The individual nucleotide groups in the fibre are represented as red beads and amino acid groups as grey beads. (b) Each complex (level 3) is made up of 13 subunits with the segments of DNA linking nucleosome complexes being treated as separate subunits. A complex is shown here with each subunit represented in a different color. (c) Each subunit (level 2) is made up of 49–142 groups. The linker histone subunit is shown here with the groups colored by the type of amino acid. (d) Each group (level 1) is made up of 7–32 atoms (level 0). A histidine amino acid group is shown here with atoms represented as small spheres and covalent bonds between the atoms represented as links. The atoms are colored by the type of atom. The total fibre consists of approximately 3 million atoms. The images were rendered using VMD.68 For clarity only 10 of the 13 subunits are shown in (a) and (b).
The HCP exploits this natural partitioning of biomolecular structures into hierarchical levels of structural components to approximate electrostatic potential, energy, and force. The atomic charges for each of these components, other than at the atomic level (level 0), is approximated by a much smaller number of point charges than the original all-atom representation. For a given point in space, the electrostatic effect of distant components is calculated using this smaller set of point charges, while the full set of atomic charges are used for nearby components. The HCP algorithm determines the level of approximation to be used in the computation, starting from the top level down to the lowest level. If a component is beyond the pre-specified threshold distance, the approximate distribution of charges for the component are used in the computation of electrostatic interactions and no further computations are performed involving its constituent lower level components. The threshold distance also ensures that electrostatic interactions between nearby atoms are treated exactly even if they belong to different components. Figure 2 illustrates the flowchart of the HCP algorithm for a biomolecular structure consisting of two levels of hierarchical organization. Although the example flowchart is for two levels of hierarchical organization, the algorithm can be extend to additional levels, e.g. to three levels in the case of the chromatin fibre shown in figure 1. The general HCP algorithm is described below. The computation of magnitude and location of the smaller set of point charges, that approximate the charge distribution of a component in step 2 of the HCP algorithm is described in the following subsection. To determine the level of approximation to be used in the computations, we define a threshold distance hk for each structural level k: h1 for groups (level 1), h2 for subunits (level 2), h3 for complexes (level 3), etc. Then, the HCP algorithm proceeds as follows:
Partition the structure into a hierarchical set of components. The partitioning may depend on the type of problem. For example, while the chromatin fibre shown in figure 1 is naturally partitioned into three levels, smaller structures may only consist of one or two levels, and larger structures may be partitioned into more than three levels. The partitioning may also depend on the expected dynamics. For example, where the dynamics may result in the separation of subunits that initially form a complex, one may not want to treat the complex as a single next level component.
Compute the approximate distribution of point charges for each component, as described in the subsection below, other than at the atomic level (level 0) which is assumed to be given.
Initially set the current component, C, to be the entire system. The current component C is updated in step 5.3 as the algorithm proceeds from the top (entire system) down to the atomic level.
Set k to be the level of the next lower level components within the current component C, and hk the corresponding threshold distance. For example, for the chromatin fibre shown in figure 1, if the current component, C, is the entire system (the chromatin fibre), then the next lower level components are the nucleosome complexes (level 3), and the corresponding threshold distance is h3. Similarly if the current component is a nucleosome complex (level 3), then the next lower level components are DNA and protein subunits (level 2) and the corresponding threshold distance is h2. For the atomic level (level 0) set the threshold distance h0 to 0.
- For each next lower level component, S, within the current component C:
- 5.1 Calculate the distance from the point in question to the next lower level component S. The distance can be suitably defined, for example as the center-of-mass or center-of-charge.
- 5.2 If the distance to the next lower level component S is greater than the threshold distance hk, the approximate distribution of charges for S are used for the computation of electrostatic interactions in equation (1)–equation (3). In the case where S is a lowest level (level 0) component the given atomic charge is used in the computation.
- 5.3 Otherwise set the current component C = S and return to step 4.
Figure 2.

Illustration of hierarchical partitioning and the HCP algorithm for two levels of approximation. The graph on the left shows the partitioning of the whole structure into subunits which are partitioned into groups, which in turn are partitioned into atoms. The flowchart on the right shows the top-down algorithm for the computation of long range interactions. Here h1 and h2 are the level 1 (group) and level 2(subunit) threshold distances respectively.
The top down approach described by the algorithm above can result in significant computational savings. For example, for a typical amino acid group consisting of 16 atoms, if the group is beyond the threshold distance for groups, h1, the 16 all-atom computations are reduced to a much smaller number of computations, for example one or two, corresponding to the reduced number of charges that approximate the charge distribution at level 1. A detailed analysis of computational complexity in the general case is beyond the scope of this proof-of-concept study. However, one can get a general idea of the scaling of computational cost by considering the following hypothetical example. Consider a hypothetical structure consisting of N atomic charges distributed within the entire structure in such a way that for each atomic charge there are exactly k atomic charges (level 0) within the level 1 threshold distance, k groups (level 1) between the level 1 and 2 threshold distances, k subunits (level 2) between the level 2 and 3 threshold distances, and so forth. Now assume that each component is represented by a single charge. For such a hypothetical structure consisting of L levels, for each atomic charge the HCP calculates k pairwise interactions per level, giving a total of NLk interactions to compute. To express computational cost in terms N we use the relationship between N and L. Note that the structure is a hierarchical tree where each node represents a component with k + 1 nodes below it. The lowest level zero is L levels from the top and thus has exactly (k + 1)L = N charges. Therefore L = logk N which gives a O(Nk logk N) ~ O(N log N) scaling for the HCP, just like the PME method. The above estimate assumes that the computational cost of calculating the approximate distribution of charges in step 2, scales equally or better than O(N log N) which is the case as will be shown in the subsection below. It is interesting to note that similar hierarchical structures occur in nature at vastly different scales, such as in astrophysics where similar hierarchical partitioning methods have been applied to solve an O(N2) gravitational interaction problem with an O(NlogN) approximation.93
Approximating the charge distribution of components
There are many different ways to approximate a larger set of atomic charges with a smaller set. For example, one could identify charges that most closely approximate the electrostatic potential distribution at the surface of a group, subunit or complex, similar to the methods used by the restrained electrostatic potential (RESP) fit69 or by the discrete surface charge optimization (DiSCO) approach.70 For this proof-of-concept study we have chosen a simpler approach, and defer the optimization of this method to future studies. The approach presented here uses a generalization of the “center-of-charge” to compute locations of the smaller set of point charges. The center-of-charge for a set of point charges is defined in a manner similar to the more familiar center-of-mass. To approximate a structural component by more than one charge we group the charges into subsets, and then compute the “center-of-charge” for each subset. Specifically, where we seek to approximate a structural component by m point charges, the n original atomic charges of the structural component are grouped into m subsets. The location of the m charges is then represented by the center-of-charge for each of these m subsets, and their magnitude is given by the sum of charges in the corresponding subset. The grouping of the original atomic charges of a structural component into m subsets is based on the magnitude of the charges. For example, for a 2-charge approximation (m = 2), the charges are separated into two subsets, one for all positive charges and another for all negative charges. More generally, the charges are separated into subsets such that subset j contains all charges qi such that , i = 1 … n, j = 1 … m, where are predefined ranges for each subset and n is the number of the original atomic charges in the component. For example, the ranges for a 2-charge approximation may be defined as, . The location of the approximate point charge, Qj is then calculated as the center-of-charge for the atomic charges in subset j. For a component with n charges, q1 … qn located at r1 … rn, the m approximate point charges Q1 … Qm located at R1 … Rm are calculated as follows:
| (4) |
| (5) |
where represents the sum over the subset of atomic charges within a component with charge values qi in a predefined range through . The computational cost of calculating the magnitude and location of these approximate point charges scales as O(N).
To define the range of charge values, , the entire range of typically encountered charge values is divided into m ranges of equal sizes. For biomolecules with atomic charges typically in the range of −1 to +1 atomic units (au), we have the following ranges for qi in the summation in equation (4). For a 1-charge approximation (all charges), for a 2-charge approximation (negative charges), and (positive charges), for a 3-charge approximation , and so on. For example the amino acid leucine with 19 atoms and a zero net charge, can be approximated by a 1-charge approximation of 0, or a 2-charge approximation consisting of charges −2.0059 and +2.0059 placed at each respective center-of-charge of all negative and positive charges, as calculated by equation (5).
The analysis in the following sections is limited to 1- and 2-charge approximations. Although using a larger number of charges to represent each component can improve accuracy, it also increases computational cost. Preliminary studies (results not shown) indicate that for the proof-of-concept method given by equation (4) and equation (5), the incremental improvement in accuracy is small compared to the additional computational cost. A detailed analysis of the costs and benefits of using 3 or more charges is beyond the scope of this proof-of-concept study.
As described in the HCP algorithm, the approximate distribution of charge for a component, used in the computation of electrostatic interactions, equation (1) – equation (3), depends on the distance from the component to the point of interest. In the current implementation of the HCP, the distance to a component is calculated as the distance to its geometric center which is defined as:
| (6) |
One can avoid calculating the geometric center by using the distance to one of the approximate point charges instead of the distance to the geometric center. Also, one can avoid recomputing the approximate distribution of charges at every single step of a molecular dynamics simulation by placing the approximate point charges at the location of the closest atomic charge. Assuming that the relative location of charges within a component do not change significantly during some limited number of steps of a molecular dynamics simulation, the approximate distribution of charges may not need to be recalculated at every singe step. The analysis of these alternative approaches is deferred to a future study.
3 Accuracy and speed of the hierarchical charge partitioning (HCP)
To assess the accuracy and speed of the HCP, we compute the electrostatic force and energy for a sample set of 600 real biomolecular structures ranging in size from 297 to 2,899,800 atoms and ranging in charge from −20,800 to +236 au. The biomolecular structures consist of 595 subunits ranging in size from 297 to 8,542 atoms, 4 complexes ranging in size from 25,086 to 490,040 atoms, and the chromatin fibre consisting of 2,899,800 atoms. The atomic coordinates for these biomolecular structures are obtained from the protein databank (PDB).71 The atomic charges for the biomolecular structures are assigned using the H++ system (www.cs.vt.edu/biophysics/H++)72 which uses the standardized continuum solvent methodology73 to compute protonation states of ionizable groups. For the 595 subunits in the test set, the HCP uses only the group (level 1) approximation because each of these structures consist of a single subunit. Similarly, for the 4 complexes in the test set, only the group and subunit (level 1 and 2) approximations are used. For the chromatin fibre, figure 1, all three levels of the approximation, – group, subunit and complex – are used.
The accuracy of the approximation is measured by the relative force error and relative energy error. Relative force error is defined as the root mean square (RMS) error in force divided by the mean force, where F⃗e is the force from the exact all-atom calculation, F⃗a the force from an approximate calculation, N the total number of atoms, and Fm the mean value for ‖F⃗e‖.74 Relative energy error is defined as the RMS error in energy divided by the mean energy, where Ee is the energy from the exact all-atom calculation, Ea the energy from the approximate calculation, Ns the number of structures, and Em the mean value for Ee. The exact all-atom calculation of electrostatic energy and force uses the Coulomb equation (2) and equation (3). Unless otherwise specified, we use a fixed value of 1 for the dielectric constant. As we will show in the next section, including the solvent effect via an implicit solvent model, may not significantly change the relative force error for the HCP. The speed-up factor is calculated as te/ta, where te is the CPU time for the exact all-atom calculation and ta the CPU time for the approximation.
For comparison we also calculate the error in electrostatic force and energy for the spherical cutoff and the particle mesh Ewald (PME) methods. While comparison between the HCP and the spherical cutoff method is straightforward, this is not the case for the PME which assumes periodic boundary conditions. Within the readily available implementations of the PME, such as the Amber software package75 which is used here for the error analysis, it is not straightforward‡ to separate the error resulting from the artificial periodic boundary and all other errors inherent to the PME. Since most biomolecular systems are non-periodic we use the following strategy to compare the HCP and the PME. We use PME parameter settings that are suggested for typical biomolecular simulations. In particular we assume that the buffer between the biomolecule and the edge of the periodic cell is 10 Å. The dielectric constant is set to 1 for both the HCP and PME calculations. Other parameters are set to their defaults for the Amber version 9 software package. Error for the PME approximation is calculated as the difference between Amber PME energy/force and the corresponding Amber value calculated without the PME (spherical cutoff distance greater than the system size). Due to the limitations of our system configuration the computation of error for the PME approximation is limited to less than 50,000 atoms.
Figure 3 shows the accuracy and speed-up comparison for our sample set of 600 biomolecular structures in vacuum. The HCP calculations use threshold distances of h1 = 10 Å, h2 = 70 Å, and h3 = 125 Å, for groups (level 1), subunits (level 2), and complexes (level3) respectively. The spherical cutoff and PME methods use a cutoff distance of 10 Å. For this sample set of biomolecular structures, the HCP results in a relative force error of 0.024 for the 1-charge HCP and 0.011 for the 2-charge HCP, which is on average significantly more accurate than the spherical cutoff method that has a relative force error of 0.089. When averaged over the entire test set, the 1-charge approximation approaches the accuracy of the more sophisticated PME method that has a relative force error of 0.021, and the 2-charge approximation is on average slightly more accurate than the PME method. However, unlike the relative accuracy of the HCP, the relative accuracy of the PME method is not uniform across the range of molecular sizes, Figure 3. For structures in our test set that are smaller than a few thousand atoms, we find that the PME accuracy is very high compared to its accuracy for larger structures, and it is also significantly higher than that of the HCP. As the structure size increases, the force error of PME becomes comparable to that of the HCP, and for even larger structures the PME eventually becomes less accurate. We stress that in these estimates we have used the same parameter settings for the PME for all the structures, including a constant (10 Å) “solvent buffer” size, that is the smallest distance from the biomolecular structure to the cell boundary. For larger biomolecular structures, the structure in the central cell is therefore closer, relative to the structure size, to its surrounding periodic images, resulting in larger errors due to the artificial periodicity. Although the 10 Å (direct sum) cutoff distance is typically plentiful for high accuracy of the PME, larger values of the spherical cutoff are often used. Therefore we also compare the spherical cutoff method with cutoff distances of 15, 20 and 25 Å to the HCP with level 1 threshold distances of 15, 20 and 25 Å. The 1-charge HCP with corresponding relative force errors of 0.01, 0.005, and 0.003 is significantly more accurate than the spherical cutoff method with relative force errors of 0.05, 0.03 and 0.02.
Figure 3.

Accuracy and speed-up comparison for a sample set of 600 biomolecular structures in vacuum. (a) Relative force error shows that for electrostatic force the hierarchical charge partitioning (HCP) is more accurate than the spherical cutoff method, and on average approaches the accuracy of the particle mesh Ewald (PME) method for all but very small structures. (b) Relative error in energy shows that for electrostatic energy the HCP is on average more accurate than both the spherical cutoff and PME methods. (c) Speedup factor compared to the exact all-atom calculation shows that the HCP can be multiple orders of magnitude faster than the exact O(N2) computation for large structures. The above computations use the following parameters. Spherical cutoff distance = 10 Å, the HCP threshold distances of h1 = 10 Å, h2 = 70 Å, and h3 = 125 Å, and PME cutoff distance = 10 Å. RMS error and speed-up factor for individual structures are grouped into bins, based on structure size in increments of 1000 atoms, and plotted at the midpoint of each bin.
The electrostatic energy calculated by the HCP results in a relative energy error of 0.001 for the 1- and 2-charge HCP. This is on average much more accurate than both the PME and spherical cutoff methods that have relative energy errors of 0.097 and 0.111 respectively. Electrostatic energy calculated by the particle mesh Ewald method is on average less accurate than the HCP due to the contributions of artificial periodic images of the central cell. However, for many applications, such as molecular dynamics, the value of energy in a given state is less important than the difference in energy between states, and so the force error is the more relevant metric.
Figure 3(c) shows that the HCP can be multiple orders of magnitude faster than the corresponding exact all-atom computation; the amount of speed-up depends on the size of the structure. For the chromatin fibre with 2,899,800 atoms the 1-charge HCP is 906 times faster than the exact all-atom calculation, while for a protein of 287 atoms (protein G) the 1-charge HCP is 2 times faster. These results are consistent with the O(NlogN) scaling of the HCP discussed in section 2. We do not attempt a direct comparison of computational times between the unoptimized, proof-of-concept implementation of the HCP with that of optimized implementations of the PME and the spherical cutoff methods, because these timings are highly implementation dependent. However, for a molecular dynamics implementation, described below, we show that the HCP can be faster than the spherical cutoff method.
The above results are based on the simple proof-of-concept approach described in the previous section. Specifically, we use the center-of-charge method for calculating the approximate distribution of charges and a fixed threshold distance for each level. We speculate that using a more sophisticated approach, such as calculating the approximate distribution of charges by optimizing fit to surface potential using methods such as restrained electrostatic potential (RESP) fits,69 or varying the threshold distance based on component size, should result in greater accuracy.
4 The HCP in more detail
The preceding analysis using real biomolecular structures shows that the HCP can be considerably faster than an exact all-atom computation with accuracy approaching that of the “industry standard” PME method. However, the accuracy of the approximation when applied to practical molecular dynamics problems depends on a number of factors. We examine here the effect of the following factors on the accuracy of the HCP: (a) molecular size and charge, (b) parameter values such as the number of charges and threshold distance, (c) discontinuity at threshold boundaries, (d) effect of the violation of Newton’s third law due to mixing of several spatial resolutions in the computation of pairwise forces, and (e) treatment of solvent.
Dependence on molecular size and charge
Although not apparent from the log-log scale plot in figure 3, there is a weak correlation between the accuracy of the HCP, as assessed by relative force error, and the size of the molecular structure. The correlation coefficient is 0.49 for a 1-charge approximation and 0.47 for a 2-charge approximation. The correlation is more evident in the linear scale plot of relative force error as a function of size in figure 4(a). For smaller structures a larger percentage of atoms are treated exactly therefore one would expect the error to be lower. A similar comparison for the particle mesh Ewald (PME) method shows a correlation coefficient of 0.49. For the test set used here, the spherical cutoff method has a slightly weaker correlation with size, with a correlation coefficient of 0.36. Relative force error as a function of molecular charge, figure 4(b), shows that accuracy of the spherical cutoff and PME methods are weakly correlated to molecular charge with correlation coefficients of 0.57 and 0.59 respectively. However, the accuracy of the HCP is not correlated to molecular charge. The correlation coefficient for a 1-charge approximation is 0.11, and 0.19 for a 2-charge approximation. A detailed theoretical analysis of these trends is beyond the scope of this proof-of-concept study.
Figure 4.

Correlation between accuracy and size/charge for the spherical cutoff, HCP and PME methods. Accuracy, as measured by relative force error, is plotted as a function of (a) molecular size as measured by the number of atoms, and (b) absolute molecular charge. The HCP uses a 1-charge approximation with group threshold h1 = 10 Å. The PME and spherical cutoff methods use a 10 Å cutoff distance. The correlations are computed using the group level (level 1) approximation for the 595 subunits out of the 600 biomolecular structures in our sample set.
The above correlation analysis represents the group (level 1) approximation for the 595 subunits, out of the 600 biomolecular structures in our sample set. Correlation analysis for the subunit and complex (level 2 and 3) approximation would not be statistically meaningful due to the small sample size of only four complexes and one multi-complex structure. The preceding analysis uses a group (level 1) threshold distance of h1 = 10 Å for the HCP, and a 10 Å cutoff distance for the PME and spherical cutoff methods.
Choice of parameter values: accuracy/speed trade-off
Accuracy and speed of the HCP is controlled by two sets of parameters: the number of charges in the approximate distribution of charges per component, m, and threshold distances, h1 … hk, where k is the number of hierarchical levels of natural partitioning of biomolecular structures. The trade-off between accuracy and speed for our sample set is shown in figure 5. The 2-charge HCP is more accurate on average, but slower, than the 1-charge approximation. And a larger threshold distance results in a more accurate approximation, on average. It is interesting to note that interactions between atoms at very large distances can affect the accuracy of the computations. For example, changing the subunit threshold distance, h2, from 50 Å to 75 Å significantly improves accuracy.
Figure 5.
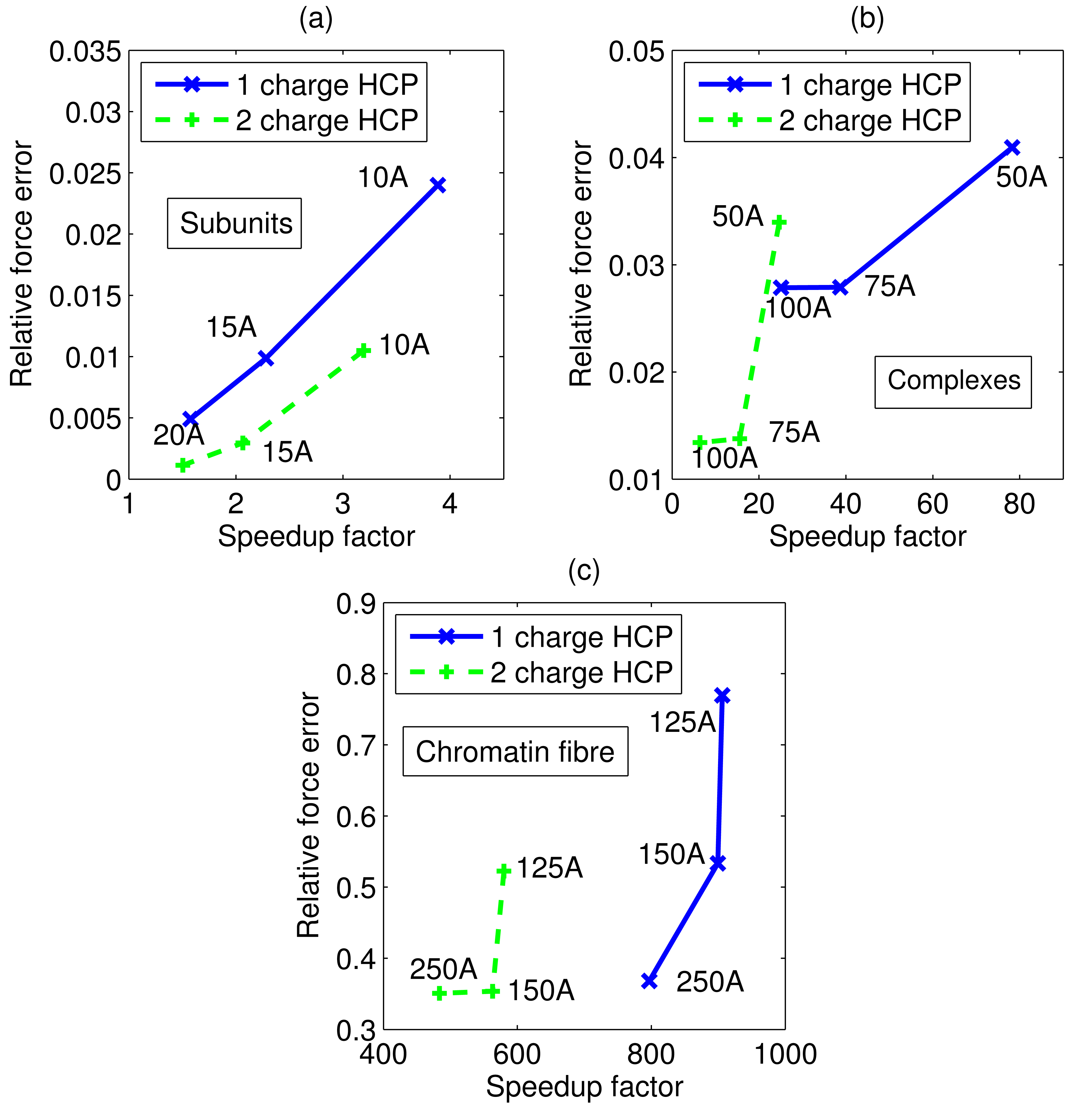
Trade-offs between accuracy (relative force error) and speed for the HCP. (a) Average trade-off for our sample set of 595 real biomolecular subunits. Group (level 1) threshold distance for each data point shown next to the data point. (b) Average trade-off for our sample set of 4 complexes, using group (level 1) threshold distance h1 = 10 Å. Subunit (level 2) threshold distance for each data point shown next to the data point. (c) Average trade-off for the chromatin fibre, using h1 = 10 Å and h2 = 70 Å group (level 1) and subunit (level 2) threshold distances. Complex (level 3) threshold distance for each data point shown next to the data point. Connecting lines shown to guide the eye.
Figure 5 also suggests how one might select parameter values for the HCP. The trade-off shown in these figures is the average value for our sample set described in the previous section. For any given structure the trade-off may be different, therefore a similar analysis for the specific structure may be performed to identify the best set of parameters. Alternatively, one can consider a more flexible mechanism where threshold distance is a function of the size and charge of the component. We do not consider these more elaborate schemes in this proof-of-concept work.
Removing discontinuity at threshold boundaries
At threshold boundaries between levels of the HCP, there is a switch in the level of approximation used by the HCP for the calculation of energy and forces. The use of two different levels of approximation across the threshold boundary introduces discontinuities in the calculated values for energy and forces at the threshold boundary. Due to these discontinuities energy may not be conserved during the course of molecular dynamics simulation.76 The discontinuity in calculated energy and forces can be eliminated using a smoothing function.77, 78 To demonstrate the effect of such a smoothing function on the HCP, we calculate electrostatic potential along a line passing through the hollow core of the chromatin fibre shown in figure 6. To highlight the effect of the discontinuity, the line through the central core is offset from the center to pass as close as 3 Å – much less than the HCP threshold distances – from some of the atoms surrounding the core.
Figure 6.

Geometric setup used for estimates of discontinuities at the threshold boundaries. Left: Chromatin fibre showing the line along which electrostatic potential is calculated. Right: Cross-section of chromatin fibre looking down the fibre axis. The line passes through the central core as close as 3 Å from some of the atoms surrounding the core. Image rendered using VMD68
The smoothing function used here makes the electrostatic potential and its derivative continuous by gradually switching from one level of approximation to another over a small switching distance beyond the threshold distance. The function is adapted from a similar function used for the spherical cutoff approximation as described by Loncharich and Brooks.52 For an HCP threshold distance hk and a smoothing distance sk, where the subscript k refers to the HCP approximation level, the potential, ϕ at distance r within the smoothing region, hk < r < hk + sk, is calculated as:
| (7) |
where the smoothing function S(r) is defined as
| (8) |
The smoothing region in the above equations starts at the HCP threshold distance, hk, and extends out to hk + sk.
Figure 7 shows the effect of applying the smoothing function to the HCP. The figure shows the electrostatic potential, ϕ, and the z-component of force, fz, acting on a unit charge. The force, fz, for the HCP with smoothing is numerically calculated as −∂ϕ/∂z, along the line passing through the central core of the chromatin fibre, whereas fz for the HCP without smoothing, the exact all-atom calculation and the spherical cutoff methods are calculated by the Coulomb equation (3). For the chromatin fibre, electrostatic potential and force calculated by the HCP, with and without smoothing, is significantly more accurate than the calculations based on the spherical cutoff method. The HCP calculations use threshold distances of h1 = 10 Å, h2 = 70 Å, and h3 = 125 Å, for groups (level 1), subunits (level 2) and complexes (level 3). The smoothing function uses a smoothing region, sk, that is 30% of the threshold distance, hk.
Figure 7.

Effect of the smoothing function used to eliminate threshold discontinuities in the 1-charge HCP. Figures show electrostatic potential and force calculated using the spherical cutoff method, the exact all-atom computation, and the HCP with and without smoothing. (a) Electrostatic potential, ϕ, along the z-axis passing through the central core. (b) Expanded view of potential for a 100 Å region. (c) z-component of electrostatic force, fz, acting on a unit charge along the z-axis. (d) Expanded view of the force for a 100 Å region. For the HCP with smoothing, force is numerically calculated as fz = −∂ϕ/∂z. The spherical cutoff method uses a 10 Å cutoff distance. The HCP uses h1 = 10, h2 = 70 and h3 = 125 Å for level 1, 2 and 3 threshold distances. The smoothing region for each threshold is 30% of the corresponding threshold distance.
Effect of the violation of Newton’s Third Law
Adaptive resolution methods, such as the spherical cutoff method with a switching function and multi-scale models with a mixing function, are known to result in a net non-zero force (and torque) on a closed system, in violation of Newton’s third law.76, 79 This is also true for the HCP. This residual force is due to the mixing of two different levels of approximation. See figure 8 for an illustration. During molecular dynamics simulations these residual forces introduce an artificial motion of the center-of-mass of the system and an overall rotation of the structure.
Figure 8.

Violation of Newton’s third law for an illustrative 3-atom system approximated by the HCP. The three atoms have charges q1, q2, and q3. Atoms 2 and 3 have equal and opposite charges, q2 = −q3 = e. Calculated forces between atoms i and j are shown as fij. For the exact all-atom calculation, the total force acting on the center-of-mass, ∑i,j fij = 0 since fij = fji per Newton’s third law. For the HCP, let atoms 2 and 3 belong to the same group while atom 1 does not belong to the group. The approximate point charge for the group represented by a, is qa = q2 + q3 = 0. For such a system the force acting on atom 1 due to the group is zero, while, in general, the net force on atoms 2 and 3, f12 + f13, is not zero. Therefore the HCP can result in a non-zero net force on a closed system, in violation of Newton’s third law.
To estimate the effect of the residual force due to the HCP, we calculate the linear and rotational displacement and kinetic energies for our sample set of 600 biomolecular structures described above. For these calculations we treat the molecule as a rigid body. The rotational velocity is approximated assuming the principle axis of rotation passes through the center-of-mass. For our sample set of biomolecular structures in vacuum the average net residual force is 1.4 × 10−8 N, and the average net residual torque about the center-of-mass is 2.3 × 10−17 N. After 10 steps of a typical molecular dynamics simulation (10 fs), the average linear center-of-mass displacement due to the net residual force, would be 1.4 × 10−5 Å, with an average linear kinetic energy of 0.0358 kT, and the average rotation about the center-of-mass due to the net residual torque would be 8.5 × 10−7 rad, with an average rotational kinetic energy of 0.0002 kT. These displacements are very small, so one can expect that the standard techniques used to eliminate center-of-mass motion and rotation in molecular dynamics simulations will work for the HCP. As we shall see in the next section, these spurious motions might also be “eliminated” by the use of stochastic collisions employed in constant temperature simulations. The chromatin fibre in figure 1 represents the worst case with a net force of 8.2 × 10−6 N, and net torque about the center-of-mass of 1.4 × 10−14 N. After 10 steps of a typical molecular dynamics simulation (10 fs), the linear displacement for the chromatin fibre would be 1.1 × 10−4 Å, linear kinetic energy 21.3555 kT, rotation about the center-of-mass 6.6 × 10−6 rad, and rotational kinetic energy 0:0595 kT. We speculate that the large number of highly charged components in the chromatin fibre give rise to a larger net residual force on the molecule compared to the other structures in the sample set.
Treatment of solvent
The HCP exploits the natural partitioning of biomolecular structures into a hierarchical set of components: complexes, subunits, groups and atoms, to reduce the computational cost of molecular dynamics simulations. This concept could in principle be extended to treat solvent molecules as subunits. However, since the most important solvent is composed of 3-atom water molecules, the computational cost reductions obtained through the HCP would be small, while introducing significant additional errors. For example a water molecule represented by three point charges, e.g. the TIP3P water model,80 could be treated as a single subunit and approximated by one or two point charges. The 1-charge HCP for water molecule would result in an approximate point charge of zero, effectively ignoring water molecules beyond the threshold distance. This is similar to the spherical cutoff method which has been shown to produce severe artifacts.51 The 2-charge HCP, on the other hand, would not result in any tangible computational cost savings while still most likely producing serious artifacts.
We speculate that the HCP may still be of benefit in explicit water simulations of very large solutes, such as viral particles, surrounded by a relatively thin layer of solvent. In this case, one can treat the solvent atoms exactly (explicit solvent model) and still benefit from the HCP being applied to the solute. Another type of explicit solvent simulations where the HCP may be of benefit is where artificial periodicity must be avoided. However, we expect the HCP to make the most impact in computations that utilize pairwise implicit solvent models.
The implicit solvent model treats the solvent as a continuum and calculates the electrostatic effect of the solvent on individual solute atoms using various approximations, including those based on analytical expressions. Although not as accurate as the explicit solvent model, there are a number of potential benefits from using the implicit solvent model.9, 18, 24, 25, 34, 81–83 One of the key benefits is the computational cost reduction from treating only the solute atoms explicitly. However, for the fast analytical implicit solvent models based on pairwise summation schemes popular in molecular dynamics simulations, such as the generalized Born (GB) model, the computation of the electrostatic effect of the solvent on solute atoms scales poorly as O(N2), where N is the number of solute atoms. Therefore the HCP, with its O(N log N) computational complexity, is poised to make a large impact in these types of simulations. The key to implementing the HCP for implicit solvent models, is handling of the dielectric function ϵij in equation (1)–equation (3), where j can represent an HCP component instead of an atom. For “simple” implicit solvent models, such as the distance dependent dielectric model, ϵij is simply a function of distance, from atom i to atom j only. To implement HCP for such a model, one can simply replace the distance to individual atomic charges within a component by the distance to the approximate charges for a component. This is how it is done in the proof-of-concept simulation described in section 5 below. For more complex implicit solvent models, such as the GB model, ϵij is a non-trivial function of the entire structure, related to the so-called effective Born radii of both atoms.23 The Born radius for an atom is generally estimated as a function of it’s relative location within the molecule.9, 18 To implement HCP for the GB model, one can define the appropriately averaged effective Born radius for each hierarchical component. The residue-level effective radii defined by Archontis and Simonson31 give an example of one such averaging scheme that might also be useful here. We stress that any specific definition will have to be thoroughly tested in the specific context of the HCP. Then, similar to using approximate charge distribution for components, the HCP can use the “component Born radii” for GB computations involving interactions with distant components. Similarly, we envision an O(N log N) hierarchical partitioning method for the calculation of individual atomic effective Born radii. At present an immediate speed-up of the computation of individual effective Born radii can be achieved by limiting the maximum distance between atom pairs used in the computation of effective Born radii, the RGBMAX option already implemented in Amber.75 A detailed analysis of implicit solvent implementations of HCP is deferred to a future study.
An additional comment is warranted on the effect of solvent on the accuracy of the HCP. Since effective pairwise interactions in the presence of solute/solvent boundary fall off faster than 1/r, one expects84 that the HCP will perform at least as well for solvated molecules. To be specific, we have estimated the influence of solvation on HCP accuracy by using the analytical linearized Poisson-Boltzmann (ALPB) approach,92 adapted to calculate the electrostatic potential near the molecular surface, and available in the GEM software.85, 86 For this proof-of-concept analysis we use only one level of approximation for HCP, which is applicable to 595 biomolecular subunits out of the full sample set of 600 biomolecular structures. The error in surface potential is calculated with solvent dielectric ϵ = 80 (water), and solute dielectric ϵ = 1. The relative RMS error is found to be similar to the vacuum case analyzed above, which suggests that the findings from the preceding assessment of accuracy, in vacuum, should be similar to what one might expect to find in implicit solvent.
In the following section we perform the most relevant test of the HCP: test it directly in the context of a molecular dynamics simulation based on an implicit solvent model.
5 A molecular dynamics (MD) implementation of the HCP
As a final proof-of-concept – to demonstrate stability of the HCP algorithm – it is tested in the context of a realistic molecular dynamics simulation. For this implementation the nucleic acid builder (NAB) MD component of the Amber tools software package is modified to incorporate the HCP. In the context of MD simulations, the NAB is a minimal version of the more comprehensive Amber molecular modeling software package.75 For example, NAB includes the spherical cutoff method but not the PME method.
A 10 ns simulation of a 36-residue protein using the HCP for computing long-range electrostatic interactions, is compared to two reference simulations: one using the exact all-atom (infinite cutoff) computation, and the other using the spherical cutoff method. The simulations use h1 = 10 Å group level (level 1) threshold distance for the HCP and a 10 Å cutoff distance for the spherical cutoff method. The structure was protonated as described in Section 3. Similar to the spherical cutoff method, the 6–12 van der Waals interactions for the HCP are computed using only the atoms of components within the threshold distance, that is those that are treated exactly. The simulations use the Amber ff99SB force field.87 The sigmoidal distant dependent dielectric implicit solvent model94 is used along with temperature control using Langevin dynamics with a 50 ps−1 collision frequency. The integration time step is 1 fs. Default values are used for all other parameters. The conjugate gradient method with a restraint weight of 5.0 is used to first minimize the crystal structure. The minimized structures is then heated to 300 K over 10 ps with a restraint weight of 0.1. The resulting structure is then equilibrated at 300 K for 10 ps with a restraint weight of 0.01. The smoothing function was not implemented.
Figure 9 shows the energy, temperature and root mean square deviation (RMSD) of backbone atoms from the initial structure, as a function of simulation time. The results show that the HCP is in reasonable agreement with the exact all-atom computation for this simulation, The results also show that for this single-CPU simulation, the 1-charge and 2-charge HCP are 40% and 14% faster respectively than the spherical cutoff method, and that the 3× speedup compared to the exact all-atom simulation is consistent with the speedup for the single point computation described earlier. Curiously, the stochastic collisions with the thermal bath were enough to “eliminate” the drift resulting from spurious center-of-mass motion due to the effects discussed in the earlier section. We stress that our goal here was only to demonstrate stability of the basic algorithm.
Figure 9.

A 10 ns simulation of villin headpiece protein, PDB id 1VII, using the HCP for long-range electrostatic computations, is compared to simulations using the exact all-atom computation and the spherical cutoff method. (a) RMS deviation of backbone atoms from the initial structure, (b) total energy, (c) temperature, and (d) potential energy, are shown as a function of simulation time. Data is plotted every 0.5 ns. Connecting lines are shown to guide the eye.
6 Conclusions
Molecular simulations are an important tool for biomolecular modeling. However, the computational cost of calculating long range electrostatic interactions limits the accessible time scales: a number of approximation methods have been developed to speed up these computations. The most commonly used of these are the spherical cutoff and the particle mesh Ewald (PME) methods. The PME method, which scales as O(N log N) with the number of particles, N, is the de facto “industry standard” for explicit solvent calculations due to its accuracy that far exceeds that of the spherical cut-off method. Although widely used, the PME has several drawbacks, including inapplicability to many practical implicit solvent models such as the generalized Born approximation. We present here an O(N log N) method, the hierarchical charge partitioning (HCP) approximation, which retains the algorithmic simplicity of the basic spherical cutoff method, can be multiple orders of magnitude faster than an exact all-atom computation, and approaches the accuracy of the much more sophisticated PME method. The HCP exploits the natural partitioning of biomolecular structures into a hierarchical set of structural components, such as molecular complexes, DNA or RNA subunits, and amino and nucleic acid groups. By systematically approximating the charge distribution within larger but more distant components by fewer point charges than in the original distribution, the HCP needs to compute fewer pairwise interactions than the much larger number needed by the corresponding exact all-atom computation which scales poorly as O(N2). To understand the suitability of this simple idea for molecular dynamics, we have systematically explored the accuracy and speed of the HCP. We tested the approximation on a sample set of 600 representative biomolecular structures ranging in size from approximately 300 to 3 million atoms and ranging in charge from −20,800 to +236 a.u. For a mini-protein of ~ 300 atoms, a proof-of-concept implementation of the HCP is at least a factor of 2 faster than the corresponding exact all-atom computation, and can be multiple orders of magnitude faster for larger structures, with the speed-up approaching three orders of magnitude for the chromatin fibre consisting of approximately 3 million atoms. To assess the accuracy of the HCP, we compared it with two other popular methods for speeding-up computations of long range interactions in molecular simulations: the spherical cut-off and the particle mesh Ewald (PME) methods. To make such comparisons between very different methods meaningful, we have assumed parameter settings typical of biomolecular simulations. Our primary metric is the root-mean-square error of the per atom force, estimated relative to the corresponding exact computation, and averaged over groups of molecules of similar size. For all the size groups, the HCP is considerably more accurate than the spherical cutoff method. Compared to the the particle mesh Ewald (PME) method, the HCP is as accurate or nearly as accurate for structures larger than about 2–3 thousand atoms, and can be even more accurate than the PME for very large structures (N > 104 atoms). Perhaps the most critical advantage of the HCP relative to the PME method is that it can be straightforwardly applied to speed-up analytical pairwise implicit solvent models (e.g. the GB, the distance-dependent dielectric, etc.) to further enhance their advantages for biomolecular simulations.
Use of the natural hierarchical partitioning of biomolecules, which is largely preserved during dynamics, offers several important advantages. The natural partitioning provides the basis for an algorithmically simple yet accurate approach for approximating the charge distribution of biomolecular structures. This hierarchical nature of the partitioning is exploited by the top down HCP algorithm to significantly reduce computational costs. The HCP offers significant additional savings for proteins: 80% of the amino acids in proteins typically have a zero net charge. Thus, within the simplest “1-charge” HCP that approximates the actual charge distribution of a structural component by a single charge equal to the total charge of the component, there is no computational cost for approximating the distant electrostatic contributions of the net neutral groups. Finally, sets of atoms that make up each component do not change during dynamics thus eliminating one possible source of discontinuity.
The HCP is a new method, although it may appear to have similarities to some other approximations also aimed at increasing the speed of computing the long-range interactions. For example, similar to the fast multipole method, the HCP replaces an exact charge distribution with an approximate one to estimate the contribution due to a distant group of charges. However, unlike the fast multipole method based on an artificially imposed regular mesh, the HCP uses the natural hierarchical organization of biomolecular structures for grouping charges. This results in a much simpler algorithm. In addition, the grouping of charges do not need to be recomputed during dynamics since within the HCP there is no movement of atoms between components, unlike the movement of atoms across artificial spatial boundaries in the fast multipole method, which is a potential source of discontinuities. Also, we speculate that the HCP is more accurate in the near-field, where the multipole expansion is not intended to work. Consequently, the same level of accuracy can presumably be achieved at a smaller computational cost. Implementation of the HCP in existing MD codes is also more straightforward since the mathematical form of the existing energy functions need not be changed. For electrostatic computations, the HCP approximates biomolecular components (e.g. groups, subunits and complexes), with a small number of point charges. This may appear similar to some coarse grain approximation methods where groups (e.g. united residue method) and subunits (e.g. segment chain method) are replaced by a single particle.60, 61 However, unlike these methods that calculate forces on coarse-grained components treated as single particles, the HCP still calculates the force on each individual atom. Thus, another critical advantage for the HCP is that it does not require additional approximations to map all-atom force fields onto a less accurate coarse grained description. The HCP uses the same force field as the underlying the exact all-atom computation.
The speed and accuracy of the HCP suggests that it may be a practical option for molecular dynamics simulations. As a proof-of-concept, the HCP has been implemented in the NAB molecular dynamics software and tested in a 10 ns long implicit solvent simulation of a 36-residue protein. For this simulation, the HCP produced a stable MD trajectory in reasonable agreement with the reference simulation without the approximation. The simulation also shows that the HCP can be faster than the spherical cutoff method. This indicates that the HCP may be an attractive alternative to the spherical cutoff and PME methods for molecular dynamics simulations, particularly those based on implicit solvent models. However, more extensive testing is needed before the method can be routinely applied in practice. There are also a number of refinements to the HCP that were identified during this study that should be considered in a subsequent study. These refinements include calculating the approximate point charges by fitting to potential distribution in the solvent space, defining threshold distances as a function of component size and charge, and refining the algorithm for placing the approximate point charges of components.
The HCP approximation presented here is designed to speed up electrostatic computations in classical molecular dynamics simulations that use non-polarizable force fields. With some refinements we expect that the HCP can be extended to other types of calculations such as the hybrid quantum mechanical/molecular mechanical (QM/MM) simulations and simulations using polarizable force fields. The general idea is to treat interactions that matter the most, exactly, while using the HCP where the sensitivity to inaccuracies in the estimates of the interactions is lower. For example, for QM/MM models the HCP can be applied outside of the QM region.
Acknowledgments
This work was supported by NIH grant GM076121. We would also like to thank Julien Mozziconacci for providing the coordinate files and transformations required to create the chromatin fibre, and John Gordon and Mayank Daga for their help with software implementation. We also thank Michael Crowley for many helpful suggestions.
Footnotes
In the structure shown here the 49 nucleotide segments of DNA linking nucleosome complexes are treated as separate chains (subunits).
Eliminating the error due to artificial periodicity by using a very large box size is not a realistic strategy for anything but the smallest systems.
Contributor Information
Ramu Anandakrishnan, Email: ramu@cs.vt.edu, Department of Computer Science, Virginia Tech 2050 Torgersen Hall (0106), Blacksburg, VA 24061 USA.
Alexey V. Onufriev, Email: alexey@cs.vt.edu, Departments of Computer Science and Physics, Virginia Tech 2050 Torgersen Hall (0106), Blacksburg, VA 24061
References
- 1.Freddolino PL, Arkhipov AS, Larson SB, Mcpherson A, Schulten K. Structure. 2006 March;14(3):437–449. doi: 10.1016/j.str.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Karplus M, McCammon JA. Nat Struct Biol. 2002 September;9(9):646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 3.Karplus M, Kuriyan J. Proc Natl Acad Sci U S A. 2005 May;102(19):6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W, Donini O, Reyes CM, Kollman PA. Annu Rev Biophys Biomol Struct. 2001;30:211–243. doi: 10.1146/annurev.biophys.30.1.211. [DOI] [PubMed] [Google Scholar]
- 5.Kumar S, Huang C, Zheng G, Bohm E, Bhatele A, Phillips JC, Yu H, Kalé LV. IBM Journal of Research and Development. 2008;52(1/2):177–187. [Google Scholar]
- 6.Ruscio JZ, Kumar D, Shukla M, Prisant MG, Murali TM, Onufriev AV. Proc. Natl. Acad. Sci. USA. 2008;105(27):9204–9209. doi: 10.1073/pnas.0710825105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw DE, Deneroff MM, Dror RO, Kuskin JS, Larson RH, Salmon JK, Young C, Batson B, Bowers KJ, Chao JC, Eastwood MP, Gagliardo J, Grossman JP, Ho RC, Ierardi DJ, Kolossváry I, Klepeis JL, Layman T, Mcleavey C, Moraes MA, Mueller R, Priest EC, Shan Y, Spengler J, Theobald M, Towles B, Wang SC. Communications of the ACM. 2008;51(7):91–97. [Google Scholar]
- 8.Zhou R, Eleftheriou M, Hon CC, Germain RS, Royyuru AK, Berne BJ. IBM Journal of Research and Development. 2008;52(1/2):19. [Google Scholar]
- 9.Onufriev A. volume 4 of Annual Reports in Computational Chemistry. The Netherlands: Elsevier, Amsterdam; 2008. pp. 125–137. [Google Scholar]
- 10.Grossfield A, Feller SE, Pitman MC. Proteins: Structure, Function, and Bioinformatics. 2007;67(1):31–40. doi: 10.1002/prot.21308. [DOI] [PubMed] [Google Scholar]
- 11.Ratner MA. Proc Natl Acad Sci U S A. 2001 January;98(2):387–389. doi: 10.1073/pnas.98.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sagui C, Darden TA. Annu Rev Biophys Biomol Struct. 1999;28:155–179. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 13.Koehl P. Curr Opin Struct Biol. 2006 March;16(6):142–151. doi: 10.1016/j.sbi.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Robertson A, Luttmann E, Pande VS. Journal of Computational Chemistry. 2007;29(5):694–700. doi: 10.1002/jcc.20828. [DOI] [PubMed] [Google Scholar]
- 15.Adcock S, McCammon J. Chemical Reviews. 2006;106(5):1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hingerty BE, Ritchie RH, Ferrell TL, Turner JE. Biopolymers. 1985;24(3):427–439. [Google Scholar]
- 17.Still WC, Tempczyk A, Hawley RC, Hendrickson T. J. Am. Chem. Soc. 1990;112:6127–6129. [Google Scholar]
- 18.Bashford D, Case DA. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 19.Hawkins GD, Cramer CJ, Truhlar DG. Chem. Phys. Lett. 1995;246:122–129. [Google Scholar]
- 20.Hawkins GD, Cramer CJ, Truhlar DG. J. Phys. Chem. 1996;100:19824–19836. [Google Scholar]
- 21.Ghosh A, Rapp CS, Friesner RA. J. Phys. Chem. B. 1998;102:10983–10990. [Google Scholar]
- 22.Lee MS, F. R. Salsbury J, Brooks CL., III J. Chem. Phys. 2002;116:10606–10614. [Google Scholar]
- 23.Onufriev A, Bashford D, Case DA. Proteins. 2004 May;55(2):383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 24.Tsui V, Case D. Journal of the American Chemical Society. 2000;122(11):2489–2498. [Google Scholar]
- 25.Cramer C, Truhlar D. Chemical Reviews. 1999;99(8):2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 26.David L, Luo R, Gilson MK. Journal of Computational Chemistry. 2000;21(4):295–309. doi: 10.1002/jcc.10120. [DOI] [PubMed] [Google Scholar]
- 27.Im W, Lee MS, Brooks CL. Journal of computational chemistry. 2003 November;24(14):1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 28.Schaefer M, Karplus M. J. Phys. Chem. 1996;100:1578–1599. [Google Scholar]
- 29.Calimet N, Schaefer M, Simonson T. Proteins: Structure, Function, and Genetics. 2001;45(2):144–158. doi: 10.1002/prot.1134. [DOI] [PubMed] [Google Scholar]
- 30.Feig M, Im W, Brooks CL. J. Chem. Phys. 2004 January;120(2):903–911. doi: 10.1063/1.1631258. [DOI] [PubMed] [Google Scholar]
- 31.Archontis G, Simonson T. The journal of physical chemistry. B. 2005 December;109(47):22667–22673. doi: 10.1021/jp055282+. [DOI] [PubMed] [Google Scholar]
- 32.Feig M, Brooks CL. Curr Opin Struct Biol. 2004 April;14(2):217–224. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Nymeyer H, Garca AE. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(24):13934–13939. doi: 10.1073/pnas.2232868100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scarsi M, Apostolakis J, Caflisch A. J. Phys. Chem. A. 1997 October;101(43):8098–8106. [Google Scholar]
- 35.Dominy BN, Brooks CL. J. Phys. Chem. B. 1999;103:3765–3773. [Google Scholar]
- 36.Gallicchio E, Levy RM. Journal of Computational Chemistry. 2004;25(4):479–499. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- 37.Grant JA, Pickup BT, Sykes MJ, Kitchen CA, Nicholls A. Phys. Chem. Chem. Phys. 2007;9(35):4913–4922. doi: 10.1039/b707574j. [DOI] [PubMed] [Google Scholar]
- 38.Haberthür U, Caflisch A. J Comput Chem. 2007 October;29:701–715. doi: 10.1002/jcc.20832. [DOI] [PubMed] [Google Scholar]
- 39.Spassov VZ, Yan L, Szalma S. J. Phys. Chem. B. 2002 August;106(34):8726–8738. [Google Scholar]
- 40.Ulmschneider MB, Ulmschneider JP, Sansom MS, Di Nola A. Biophys J. 2007 April;92(7):2338–2349. doi: 10.1529/biophysj.106.081810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanizaki S, Feig M. The Journal of chemical physics. 2005 March;122(12) doi: 10.1063/1.1865992. [DOI] [PubMed] [Google Scholar]
- 42.Beck DAC, Armen RS, Daggett V. Biochemistry. 2005 January;44(2):609–616. doi: 10.1021/bi0486381. [DOI] [PubMed] [Google Scholar]
- 43.Ruvinsky AM, Vakser IA. Proteins: Structure, Function, and Bioinformatics. 2008;70(4):1498–1505. doi: 10.1002/prot.21644. [DOI] [PubMed] [Google Scholar]
- 44.Darden T, York D, Pedersen L. The Journal of Chemical Physics. 1993;98(12):10089–10092. [Google Scholar]
- 45.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. The Journal of Chemical Physics. 1995;103(19):8577–8593. [Google Scholar]
- 46.Toukmaji AY, Board JA. Computer Physics Communications. 1996 June;95(2–3):73–92. [Google Scholar]
- 47.York D, Yang W. The Journal of Chemical Physics. 1994;101(4):3298–3300. [Google Scholar]
- 48.Carrier J, Greengard L, Rokhlin V. SIAM Journal on Scientific and Statistical Computing. 1988;9(4):669–686. [Google Scholar]
- 49.Cai W, Deng S, Jacobs D. Journal of Computational Physics. 2007 May;223(2):846–864. [Google Scholar]
- 50.Lambert CG, Darden TA, Board JA., Jr. Journal of Computational Physics. 1996 July;126(2):274–285. [Google Scholar]
- 51.Mark P, Nilsson L. Journal of Computational Chemistry. 2002;23(13):1211–1219. doi: 10.1002/jcc.10117. [DOI] [PubMed] [Google Scholar]
- 52.Loncharich RJ, Brooks BR. Proteins. 1989;6:32–45. doi: 10.1002/prot.340060104. [DOI] [PubMed] [Google Scholar]
- 53.Schreiber H, Steinhauser O. Chemical Physics. 1992 December;168(1):75–89. [Google Scholar]
- 54.Bishop TC, Skeel RD, Schulten K. Journal of Computational Chemistry. 1997;18(14):1785–1791. [Google Scholar]
- 55.Hunenberger PH, McCammon JA. Biophysical Chemistry. 1999 April;78(1–2):69–88. doi: 10.1016/s0301-4622(99)00007-1. [DOI] [PubMed] [Google Scholar]
- 56.Weber W, Hunenberger PH, McCammon JA. Journal of Physical Chemistry B. 2000;104(15):3668–3675. [Google Scholar]
- 57.Lee MS, Salsbury FR, Olson MA. J Comput Chem. 2004 December;25(16):1967–1978. doi: 10.1002/jcc.20119. [DOI] [PubMed] [Google Scholar]
- 58.Nielsen SO, Lopez CF, Srinivas G, Klein ML. Journal of Physics: Condensed Matter. 2004;16(15):R481–R512. [Google Scholar]
- 59.Mccoy JD, Curro JG. Macromolecules. 1998 December;31(26):9362–9368. [Google Scholar]
- 60.Liwo A, Ołdziej S, Pincus MR, Wawak RJ, Rackovsky S, Scheraga HA. Journal of Computational Chemistry. 1997;18(7):849–873. [Google Scholar]
- 61.Fukunaga H, Takimoto J, Doi M. The Journal of Chemical Physics. 2002;116(18):8183–8190. [Google Scholar]
- 62.Kolinski A, Skolnick J. The Journal of Chemical Physics. 1992;97(12):9412–9426. [Google Scholar]
- 63.Voltz K, Trylska J, Tozzini V, Kurkal-Siebert V, Langowski J, Smith J. Journal of Computational Chemistry. 2008;29(9):1429–1439. doi: 10.1002/jcc.20902. [DOI] [PubMed] [Google Scholar]
- 64.Walker RC, Crowley MF, Case DA. Journal of Computational Chemistry. 2007;29(7):1019–1031. doi: 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- 65.Tan YH, Tan C, Wang J, Luo R. The Journal of Physical Chemistry. B. 2008 June;112(25):7675–7688. doi: 10.1021/jp7110988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lodish H, Berk A, Zipursky S, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology. New York: W.H. Freeman and Company; 2000. [Google Scholar]
- 67.Wong H, Victor J-M, Mozziconacci J. PLOS One. 2007;436(7):e877. doi: 10.1371/journal.pone.0000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Humphrey W, Dalke A, Schulten K. J. Molec. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 69.Cieplak P, Cornell WD, Bayly C, Kollman PA. Journal of Computational Chemistry. 1995;16(11):1357–1377. [Google Scholar]
- 70.Beard DA, Schlick T. Biopolymers. 2001;58(1):106–115. doi: 10.1002/1097-0282(200101)58:1<106::AID-BIP100>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 71.Berman H, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov I, Bourne P. Nucl. Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gordon J, Myers J, Folta T, Shoja V, Heath LS, Onufriev A. Nucl. Acids Res. 2005;33:68–71. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bashford D, Gerwert K. J. Mol. Biol. 1992;224:473–486. doi: 10.1016/0022-2836(92)91009-e. [DOI] [PubMed] [Google Scholar]
- 74.Feller S, Pastor R, Rojnuckarin A, Bogusz S, Brooks B. J. Phys. Chem. 1996;100(42):17011–17020. [Google Scholar]
- 75.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005 December;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Delle Site L. Physical Review E (Statistical, Nonlinear, and Soft Matter Physics) 2007;76(4) doi: 10.1103/PhysRevE.76.047701. 047701. [DOI] [PubMed] [Google Scholar]
- 77.Leach A. Molecular modeling: Principles and applications. Prentice Hall; 2001. [Google Scholar]
- 78.Schlick T. Molecular modeling and simulation, an interdisciplinary guide. New York: Springer-Verlag; 2002. [Google Scholar]
- 79.Praprotnik M, Delle Site L, Kremer K. Annual Review of Physical Chemistry. 2008;59(1):545–571. doi: 10.1146/annurev.physchem.59.032607.093707. [DOI] [PubMed] [Google Scholar]
- 80.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. The Journal of Chemical Physics. 1983;79(2):926–935. [Google Scholar]
- 81.Honig B, Nicholls A. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 82.Luo R, David L, Gilson MK. Journal of Computational Chemistry. 2002 July;23(13):1244–1253. doi: 10.1002/jcc.10120. [DOI] [PubMed] [Google Scholar]
- 83.Baker NA. Curr Opin Struct Biol. 2005 April;15(2):137–143. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 84.Anandakrishnan R, Onufriev A. Journal of Computational Biology. 2008;15(2):165–184. doi: 10.1089/cmb.2007.0144. [DOI] [PubMed] [Google Scholar]
- 85.Fenley AT, Gordon JC, Onufriev A. The Journal of Chemical Physics. 2008;129(7) doi: 10.1063/1.2956499. 075101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gordon JC, Fenley AT, Onufriev A. The Journal of Chemical Physics. 2008;129(7) doi: 10.1063/1.2956499. 075102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins: Structure, Function, and Bioinformatics. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vasilyev V. Journal of Computational Chemistry. 2002;23(13):1254–1265. doi: 10.1002/jcc.10131. [DOI] [PubMed] [Google Scholar]
- 89.Stote RH, Karplus M. Proteins: Structure, Function, and Genetics. 1995;23(1):12–31. doi: 10.1002/prot.340230104. [DOI] [PubMed] [Google Scholar]
- 90.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Journal of Computational Chemistry. 1983;4(2):187–217. [Google Scholar]
- 91.Shimada J, Kaneko H, Takada T. Journal of Computational Chemistry. 1994;15(1):28–43. [Google Scholar]
- 92.Sigalov G, Fenley A, Onufriev A. The Journal of Chemical Physics. 2006;124(12):124902. doi: 10.1063/1.2177251. [DOI] [PubMed] [Google Scholar]
- 93.Appel AW. SIAM J. Sci. Stat. Comput. 1985;6(1):85–103. [Google Scholar]
- 94.Ramstein J, Lavery R. Proceedings of the National Academy of Sciences of the United States of America. 1988 October;85(19):7231–7235. doi: 10.1073/pnas.85.19.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]