

Abstract
Cigarette smoke demonstrates a carcinogenic effect through chronic exposure, not acute exposures. However, current cell line models study only the acute effects of cigarette smoke. Using a cell line model, we compared the effects of acute versus chronic cigarette-smoke-extract (CSE) on mitochondria in minimally-transformed oral keratinocytes (OKF6).
OKF6 cells were treated with varying concentrations of CSE for 6-months. Cells were analyzed monthly by flow cytometry for mitochondrial-membrane-potential (MMP), cytochrome-c release, caspase-3 activation and viability after CSE-exposure. At each time point the same assays were performed after 24hrs of valinomycin (MMP depolarizing agent) treatment. The mitochondrial-DNA of chronically CSE-treated cells was sequenced.
After 6-months of CSE-treatment, the cells were increasingly resistant to CSE-mediated and valinomycin induced cell death. In addition, chronic CSE-treatment caused chronic depolarization of MMP, cytochrome c release, and caspase activation. Cells grown in the presence of only CSE vapor also exhibited the same resistance and chronic baseline apoptotic activation. Mitochondrial DNA sequencing found that chronic CSE treated cells had more amino acid changing mitochondrial mutations than acutely treated cells.
CSE treatment of normal cells select for apoptotic dysfunction as well as mitochondrial mutations. These findings suggest that chronic tobacco exposure induce carcinogenesis via selection of apoptosis resistance and mitochondrial mutation in addition to previously known genotoxic effects that were found by acute treatments. Chronic models of tobacco exposure on upper aerodigestive epithelia may be more insightful than models of acute exposure in studying head and neck carcinogenesis
Keywords: Cigarette Smoke Extract, CSE, Apoptosis, Mitochondria, Keratinocytes
Introduction
Head and neck squamous cell carcinoma (HNSCC) is among the most morbid of human cancers with approximately 40,000 new yearly cases in the United States. The primary risk factors for HNSCC is tobacco exposure with alcohol as a co-carcinogen, with a latency period of several decades.(1) As early as 1964, there were published reports of the tumor promoting activity of tobacco extracts in mice.(2, 3) Despite these early studies, there is incomplete knowledge of how cigarette smoke induces the early cellular changes that lead to malignancy.
Impaired apoptosis is a central characteristic of neoplastic and malignant transformation.(4, 5) Mitochondria have a central role in the signal transduction and coordination of apoptosis.(6-8) Apoptosis at the mitochondrial level is initiated by depolarization of the mitochondrial membrane, and proceeds via release of cytochrome c and other apoptogenic factors from the intermembraneous space of mitochondria.(8) Valinomycin, a potassium ionophore which facilitates the selective transport of K+ ions across the inner membrane of mitochondria,(9) induces apoptosis in various mammalian cell lines(6, 10-12) by disrupting the ΔΨm.(13) Previous study by our lab showed that HNSCC cell lines are resistant to ΔΨm depolarization induced apoptosis (i.e. valinomycin treatment).(14) This finding, in conjunction with the knowledge that mitochondria are uniquely susceptible to oxidative damage and that cigarette smoke mediate its effects through oxidative damage,(15-19) imply that the effects of tobacco exposure on mitochondrial mechanisms of apoptosis are of significant interest in HNSCC carcinogenesis.
There are two canonical apoptotic caspase pathways involved in apoptosis: the mitochondrion (intrinsic) pathway and the death receptor (extrinsic) pathway. The mitochondrial apoptotic pathway is initiated via signaling from pro-apoptotic proteins from the Bcl-2 family such as Bax, which trigger the release of cytochrome c. Cytochrome c then associates with Apaf-1, caspase-9, and ATP to form an apoptosome that activates caspase-3, which in turn activates the caspase cascade and the degradation phase of apoptosis (20). The death receptor apoptotic pathway is initiated by binding of death activators (i.e. FasL, TNF) to their respective transmembrane death receptors. This binding enables the death receptor to interact with the cytoplasmic adaptor protein FADD, which then activates caspases 8. Similarly to the function of caspase 9 in the intrinsic pathway, caspase 8 also leads to effector caspases activation and the degradation phase of apoptosis.(8, 21)
Prior studies have attempted to model the acute effects of cigarette smoke extract (CSE) in cell lines. Investigators interested in obstructive pulmonary disease treated primary lung cells with CSE at high concentrations and short exposures in order to determine the mechanism by which cigarette smoke induces apoptosis. The CSE was created using methods developed by Carp and Janoff. They showed that CSE treatment induced apoptosis through mitochondrial mechanisms.(15, 16, 19, 22-32) In addition, two other studies proposed that CSE treatment could induces in changes lung cell lines that were similar to that of a cancerous state.(33, 34) However, it is well known that chronic cigarette smoke exposure and not acute exposures that induces carcinogenesis. These early studies revealed the pathways by which CSE may exert its effects, but did not investigate how chronic exposure could lead to carcinogenesis. To date, there has been no study of CSE exposure in oral keratinocytes even though cigarette smoke exposure is the primary risk factor for head and neck cancers. In addition, there has been no study comparing the chronic effects of CSE treatment versus the acute effects.
In our study, we treated a minimally transformed oral keratinocyte cell line with CSE to model cigarette smoking and compare acute versus chronic effects. We find that chronic CSE treatment selects for the baseline activation of several apoptotic events such as the depolarization of ΔΨm, the release of cytochrome c and the activation of caspase-3. In comparison, with the acute treatment period, the cells responded as expected by activating several apoptotic factors. Chronic CSE treatment selected for cells that are resistant to CSE-induced cell death, resistant to ΔΨm depolarization induced apoptosis and to a lesser degree other pathways. Interestingly, we also found that the vapor component of CSE was sufficient to select for cells that were identical to direct CSE treatment. Lastly, we found that chronic CSE treatment can provide the necessary selective pressure for propagating mitochondrial mutations.
Materials and Methods
Preparation of Cigarette Smoke Extract (CSE)
Cigarette smoke extract was prepared according to a modified Carp and Janoff method.(17) Research-grade cigarettes, 2R4F, from the Kentucky Tobacco Research and Development Center at the University of Kentucky were smoked to 0.25 cm above the filter. 100% CSE was prepared by bubbling smoke from one cigarette into 1 ml of PBS.(2) Each puff was 2seconds long at a rate 35ml/second. This extract was then filtered using a .22um filter from BD biosciences filter. This removed particles and microbiological contaminants of .22 micron and larger. This ensured that the cell cultures would remain free of contaminants. Each dilution was done by volume in media. Treatment concentrations were 1%, .1%, and .01%. To control for the vapor effect of the CSE, cells were also grown in the smoking incubator without direct CSE treatment.(23) Cells that were grown in a normal incubator that did not have any cell lines treated with CSE are labeled as control-N. Cells that were grown and passaged in the incubator in which cell lines were being treated with CSE, were not directly treated with CSE but were only exposed to CSE vapor are labeled as control-S, The CSE (100%) was stored in sterile Eppendorf tubes at -80 degree Celsius.
Drug treatments
Valinomycin is a potassium ionophore that facilitates the selective transport of K+ ions across the inner membrane of mitochondria(9) and induces apoptosis in various mammalian cell lines(6, 10-12) by disrupting the mitochondrial membrane potential.(13) Treatment was at 100nM and for 24hrs. Staurosporine is a non selective kinase inhibitor that was used to treat at 1uM and for 24hrs as well.
Cell culture and reagents
Immortalized human oral keratinocytes (OKF6/TERT1) were a generous gift from James Rheinwald at Brigham and Women's Hospital in Boston, MA. They retain the normal growth and differentiation characteristics of primary human oral keratinocytes. The cell line was expanded and passaged in keratinocyte serum-free medium (Gibco/Invitrogen; 10725-018). This medium was supplemented with BPE (25 ug/ml), epidermal growth factor (0.2 ng/ml), Calcium Chloride (0.4 mM) and 1% penicillin-streptomycin. This mixture was then filter-sterilized through a 0.2 um pore-size sterilization filter. The media for the cells were changed every 2-3 days. The cells were split when they reached >70% confluency. Both the cells treated with CSE and passaged cells were cultured in 37 deg C humidified air incubators with 5% CO2. All cell lines were grown in 35mm dishes.
Mitochondrial membrane potential analysis by flow cytometry
JC-1 is a mitochondrial dye (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine chloride) Stratagene (La Jolla, California) that stains mitochondria in living cells in a membrane potential-dependent fashion. In summary, after the cells were trypsinized and washed with PBS, the cells were labeled with the fluorescent dye JC-1 for 20 min at 37 deg C. The excess dye was then washed away with PBS and the remaining cells were suspended in the buffer solution. The JC-1 monomer is in equilibrium with J-aggregates which binds to the mitochondrial membrane. The monomer JC-1 fluoresces green (λ em = 527 nm), while the J-aggregates fluoresces red (λem = 590 nm). Therefore, cells with normal mitochondrial membrane potential fluoresce orange. The depolarization of mitochondria results in a decrease in the red component and a green fluorescence. J-aggregate fluorescence was recorded by flow cytometry as FL2 and monomer fluorescence as FL1. Necrotic fragments were electronically gated out by forward scatter channel (FSC) and side scatter channel (SSC). The ratio of FL1 (green) to FL2 (red) was regarded as semi-quantitative relative to the ΔΨm. All experiments were repeated at least 3 times and were run on a FACscalibur flow cytometer for 10,000 events. Representative data is shown. The data analysis was performed using Cell-Quest software (Becton Dickinson, USA). Depolarization was determined to be relative to the control cells that which were untreated OKF6 cells. The control cells were first analyzed by flow cytometry. After analysis the gates or thresholds of fluorescence were determined. The threshold was set so that >95% of cells were within the established gate as non-depolarized. Any shift from this threshold (ie outside the gate) was determined to be depolarized, in other words any decrease in fluorescence that fell below the established threshold was considered depolarized.
Apoptosis analysis by flow cytometry
Apoptosis and cell death were analyzed by an Annexin V/7-AAD flow cytometric assay. The cells were washed twice with cold PBS and resuspended at 100ul Annexin V binding buffer. Cells were protected from light and incubated at room temperature for 15 min with 5 μl of FITC conjugated Annexin-V (BD Biosciences, USA) and 5μl of 7-AAD (BD Biosciences) and then analyzed by flow cytometry immediately. Flow cytometry analysis was performed using a FACScan flow cytometer (Becton Dickinson) by analyzing 10,000 events for cell viability. Cells were gated for analysis by a combination of forward scatter channel (FSC) and side scatter channel (SSC). All gating for the controls were exactly the same as the samples. The plots were divided into quadrants and the region in the lower left were considered viable and the other quadrants were classified as early apoptosis, late apoptosis and necrotic. The data is expressed as a simple percentage of these 10,000 events. Control cell lines were used to determine the thresholds. All experiments were run in triplicate. The data analysis was performed using Cell-Quest software (Becton Dickinson).
Cytochrome C release by Flow cytometry
Cytochrome C release was detected using flow cytometry and a CalbioChem Innocyte flow cytometric cytochrome c release kit (Darmastadt, Germany) which selectively permeabilized the plasma membrane without injuring the mitochondrial membrane. Cells were washed in PBS and centrifuged. We resuspended the cell pellets in 300 μl Permeabilization Buffer. Suspension was then incubated for 10 min on ice. Next, the cells were fixed by adding 300μl 8% paraformaldehyde in PBS directly to the tubes. After fixation, the cells were incubated for 20 min at room temperature. After incubation, the cells were washed by adding 1 ml 1× Wash Buffer 3 times and centrifuged. Then the cells were resuspended in 250 μl Blocking Buffer and incubated for 1hr at room temperature. Afterwards, 250 μl Anti-Cytochrome C WS was added and incubated for 1hr at room temperature. Then the cells were washed with the wash buffer. Next, 500 μl Anti-IgG FITC WS was added to the cells and incubate for 1 hr at room temperature in the dark. The cells were washed again with 1× Wash Buffer. Finally, the cells were resuspended in 500 μl Wash Buffer. Flow cytometry analysis was performed using a FACScan flow cytometer (Becton Dickinson) by analyzing 10,000 events. The data is expressed as a simple percentage of these 10,000 events as having released or retained mitochondrial cytochrome c. Control cell lines were used to determine the threshold of release or retention. Cells were gated for analysis by a combination of forward scatter channel (FSC) and side scatter channel (SSC). All gating for the controls were exactly the same as the samples. Each experiment was replicated independently. All experiments were run in triplicate. The data analysis was performed using Cell-Quest software (Becton Dickinson).
Caspase 3 activation by Flow Cytometry
Activated caspase-3 was detected using flow cytometry and a BD Pharmingen Caspase-3, Active Form, Apoptosis Kit (San Jose, California). In summary, the kit selectively permeabilized the cellular membrane to allow for a monoclonal antibody conjugated to FITC to bind specifically the activated form of caspase-3. Flow cytometry analysis was performed using a FACscalibur flow cytometer (Becton Dickinson) by analyzing 10,000 events for cell viability. The data is expressed as a simple percentage of these 10,000 events as having activated caspase-3. Control cell lines were used to determine the threshold of activation or inactivation of capase-3. Cells were gated for analysis by a combination of FSC and SSC. All experiments were repeated at least 3 times in triplicate. The data analysis was performed using Cell-Quest software (Becton Dickinson).
Mitochondrial DNA sequencing
All of the DNA samples were sequenced with MitoChip version 2.0, an oligonucleotide microarray. Briefly, the entire mtDNA sequence was amplified in three overlapping long PCR fragments, with each reaction containing 50 ng of genomic DNA. The amplified PCR products then were fragmented and labeled with GeneChip DNA labeling reagent and 3.4 μl of 30 units/μl terminal deoxynucleotidyl transferase (both from Affymetrix, Santa Clara, CA). Prehybridization, hybridization, washing, and scanning of the MitoChip were performed as described in the Affymetrix CustomSeq Resequencing protocol.(35) DNA sequences of the CSE treated cells and cells passaged in the normal incubator were compared to non-passaged “fresh” OKF6 cells. To verify, that the sequence of our “fresh” OKF6 cells, we also compared its sequence to the reference sequence (Supplementary Figure 1). Non-calls were excluded from the analysis.
Results
Acute versus Chronic CSE treatment of Oral Keratinocytes selects for resistance to depolarization induced apoptosis
Initially, the treatment of cells was performed at several concentrations varying from .001% to 10% CSE. CSE at .1% was found to be the highest concentration to which cells could be treated chronically. Cells treated at a higher concentration underwent apoptosis/necrosis within days of treatment (data not shown). After 1 month of CSE treatment (acute), cells showed baseline ΔΨm depolarization. CSE .1%, .01%, S-control, and N-control were 10.3%, 20.8%, 22.4%, and 21.8% depolarized respectively. After 6 months (chronic) of treatment the CSE .1%, .01%, and S-control cells were 80.8%, 80.4%, and 86.9% depolarized. In contrast, the N-control cells remained at 15.6% depolarization. In addition, CSE treated cells underwent decreased cell death over time. After 1 month of CSE treatment, the amount of non-viable cells of CSE-treated was 69.5% and 36.0% of cells analyzed for CSE .1% and CSE .01%, respectively (normalized to N-control). However after 6 months of CSE treatment, this difference was reduced to 0.68% and 0.25%. Therefore, chronic CSE treatment resulted in the selection of cells that had chronic depolarization of their ΔΨm, were resistant to CSE mediated cell death and were resistant to ΔΨm depolarization induced apoptosis. (Figure 1a,b)
Figure 1.
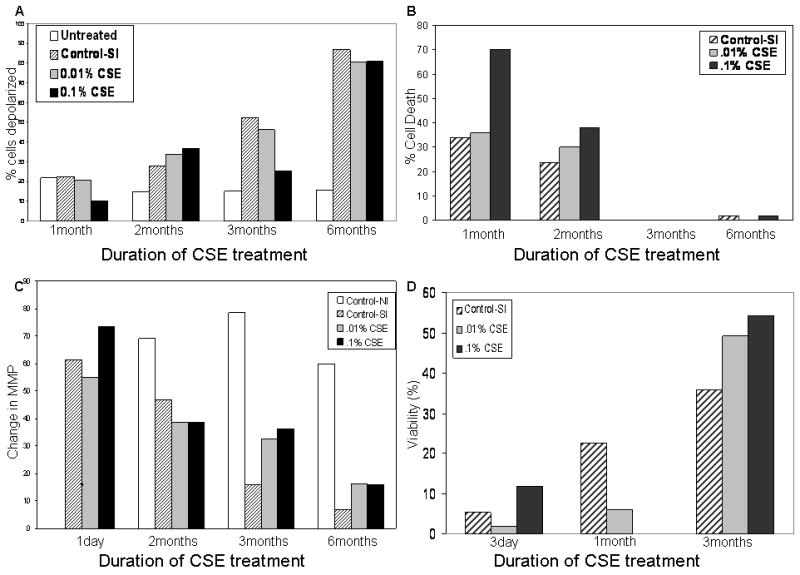
Acute versus Chronic CSE treatment of OKF6 induces apoptotic resistance.
a) CSE treatment of OKF6 results in chronic mitochondrial membrane potential depolarization. b) Chronic CSE treatment of OKF6 results in decreased cell death over time (Normalized to cells in normal incubator). c) Chronic CSE treated OKF6 are increasingly resistant to valinomycin induced depolarization. d) CSE treated OKF6 are increasingly resistant to mitochondrial membrane potential depolarization induced apoptosis. Values were normalized to normal incubator cells.
Chronic CSE treatment of Oral Keratinocytes selects for resistance to Valinomycin Treatment
Previously published data showed that HNSCC cell lines are resistant to valinomycin induced ΔΨm depolarization induced apoptosis. In order to determine whether these cell lines were truly resistant to ΔΨm depolarization induced cell death, the chronically treated cells were challenged with valinomycin treatment for 24hrs. Their ability to further depolarize their ΔΨm and percentage of cells that remained viable after treatment was assessed. The net ΔΨm decrease after valinomycin treatment of CSE .1%, CSE .01%, and S-control was 73.4%, 55.0%, and 61.2% respectively, after 1 day and then decreased to a change of 15.9%, 16.2%, and 6.7% after 6 months (percentages were normalized to N-control). In summary, chronic CSE treatment selected for oral keratinocytes that were resistant to valinomycin induced ΔΨm depolarization (Figure 1c).
However, to exclude the possibility that the cells were resistant to the depolarization of the ΔΨm but not resistant to cell death, cell viability via Annexin V staining was performed. CSE treatment was found to select for cells that were increasingly resistant to valinomycin treatment. Initially, CSE .1% and CSE .01% cells were 1.73% and 11.76% viable after valinomycin treatment at 3 days. At 3 months, CSE .1% and CSE .01% had increased their percentage of viable cells to 49.1% and 54.4% viable after valinomycin treatment. An interesting additional finding was that the S-control with had 5.2% viable cells after 3 days of CSE treatment and 24hrs of valinomycin treatment increased its viable component to 35.8% at 6 months. The S-control as mentioned above was exposed only to the vapor component of CSE. Therefore, CSE treated OKF6 appear to be resistant to valinomycin induced ΔΨm depolarization and apoptosis. Of note also, is that the S-control cells carried in an incubator containing CSE treated cell cultures, but not directly treated with CSE, showed a similar trend (Figure 1d).
Chronic CSE treatment of Oral Keratinocytes selects for cells that are resistant to Staurosporine induced cell death
After discovering that chronically CSE treated cells selected for resistance to valinomycin, the possibility that these cells may resist alternate apoptosis inducing mechanisms was explored. To test this, the CSE treated cells were challenged with staurosporine (a non-specific protein kinase inhibitor) and analyzed by flow cytometry for signs of apoptosis via annexin staining. After treatment with staurosporine it was found that the S-control, CSE .01% and CSE .1% were only 19.2%, 11.2%, and 34.5% viable after treatment, respectively. However, only 4.9% of the N-control cells were found to be viable after staurosporine treatment. This suggests that chronically treated cells, though predominantly vulnerable to staurosporine treatment, also exhibited resistance (CSE dose dependant) to stuaurosporine treatment relative to the control. This finding suggests an overall loss of the apoptotic response after selection with CSE. (Figure 2)
Figure 2.
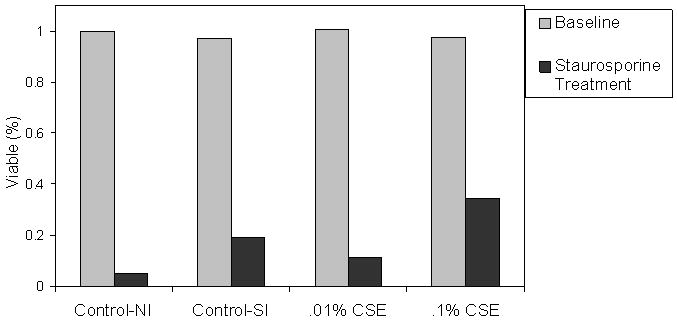
Chronic CSE treatment induces mild resistance to apoptosis induced by staurosporine a nonselective protein kinase inhibitor. Staurosporine induced apoptosis occurs via pathways that are different from valinomycin.
Chronic activation of the apoptotic pathway in CSE treated Oral Keratinocytes
Once it was established that chronic CSE treatment of cells could select for resistance to ΔΨm depolarization induced apoptosis the events that occur later in apoptosis were examined. Two major steps in the apoptotic pathway are the release of cytochrome-c from mitochondria and the subsequent downstream caspase-3 activation.
First, the status of cytochrome-c in chronically treated cells and in response to valinomycin treatment was evaluated. At baseline, the N-control cells release 23.9% of their cytochrome-c and cells treated with CSE .1% chronically release 59.4% of cytochrome-c. In response to valinomycin treatment, the N-control cells increased their release to 46.8% of their cytochrome-c and CSE .1% treated cells released 85.2% of cytochrome-c. Therefore, in addition to being chronically depolarized, CSE treated cells chronically released cytochrome-c. Treated cells were still sensitive to valinomycin and responded by releasing even more cytochrome c but did not undergo apoptosis as shown in the previous figure (Figure 3a).
Figure 3.
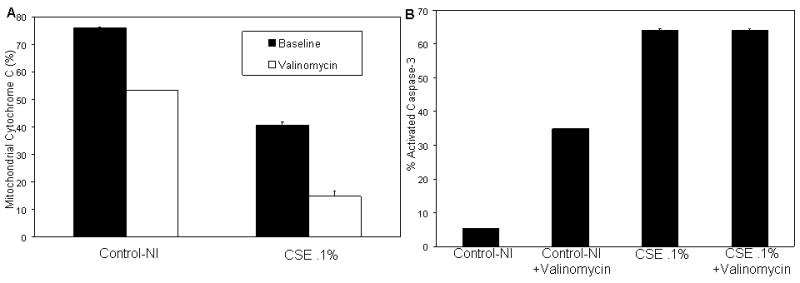
Chonic CSE treatment induces chronic activation of apoptotic mediators.
a) Mitochondrial cytochrome c at baseline and in response to valinomycin treatment shows that at baseline, chronically treated cells retain less cytochrome c. b) Caspase 3 activation at baseline and in response to valinomycin treatment. CSE treated cells show virtually no response to valinomycin treatment with regard to caspase-3 activation.
The activation of caspase-3 in response to CSE selective pressure was also assayed. CSE treated cells were found to have chronic activation of Caspase-3 (64.1%), but did not respond to valinomycin treatment (64.1%). In contrast, N-control cells had a low baseline activation of caspase-3 (5.32%) and a robust activation of caspase-3 (34.8%) upon valinomycin administration (Figure 3b).
Chronic CSE treatment induces mitochondrial mutations
The mitochondrial genome is uniquely sensitive to DNA damaging agents, and CSE is a complex mixture of genotoxic and other agents. We hypothesized that chronic mitochondrial depolarization and intrinsic pathway related resistance to apoptosis may be associated with mitochondrial genomic injury. We have previously demonstrated that mitochondrial mutations are common in HNSCC and single mutations may induce increased ROS production, HIF-1α stabilization, and proliferation.(35) Therefore, we assessed whether chronically CSE exposure would select for cells that contained mitochondrial mutations.
Cells that were treated with CSE at .1% for 7months and OKF6 cells that were passaged for 7 months were sequenced by array based sequencing.(35) Their sequences were compared to the sequence fresh OKF6 cells that were not passaged and chronically passaged, non-CSE treated cells. Analysis of sequencing data showed that CSE treatment selected for cells that contained 124 mutations compared to a baseline rate of 31 mutations in passaged cells. Of the 124 mutations in CSE treated cells, 70 mutations were amino acid changing mutations in the coding region of the mitochondrial genome. Of the 31 mutations in the passaged cells, none of the mutations overlapped with CSE treated mutations, and 8 were amino acid changing mutations in the coding region of the mitochondrial genome (Figure 4).
Figure 4.
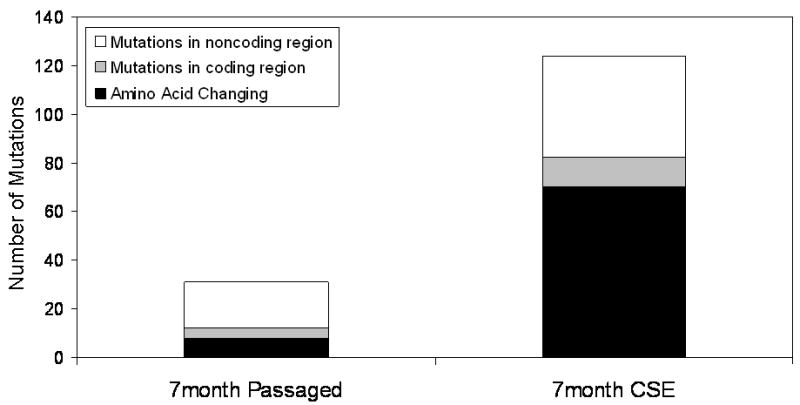
Chronic CSE treatment induces amino acid changing mitochondrial DNA mutations. Chronic CSE treatment induces a greater number of all types of mutations, but as shown, its shows the greatest difference in the amino acid changing mutations.
We then assessed whether these mutations occurred early or late relative to the functional resistance to apoptosis. We examined the total number of mutations accrued by the CSE treated cell lines over time (2 months, 3 months and 7 months at .1% CSE). We then subdivided these mutations into whether the mutation was located in the coding or non coding region of the mitochondrial genome. Sequencing of cells treated for two months of CSE showed a total of only 2 mutations and both were in the coding region of the genome. Analysis of cells that were CSE treated for seven months showed 82 coding mutations and 42 non coding mutations for a total of 124 mutations (Figure 5). This suggests that the functional resistance to apoptosis precedes mitochondrial mutation i.e. the genotoxic effects of CSE.
Figure 5.
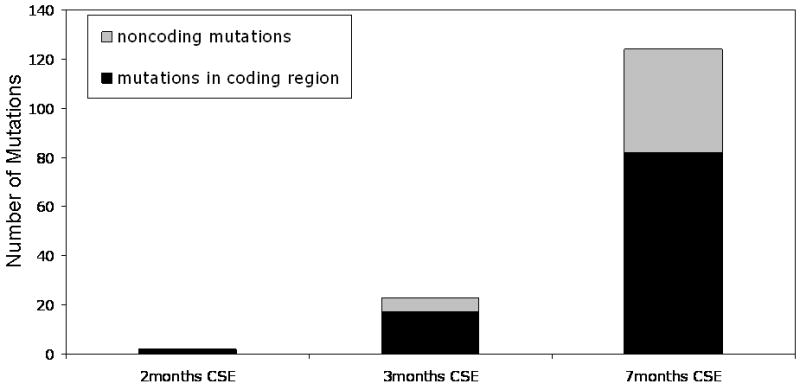
CSE treatment of cells acute versus chronic. The number of total mutations and mutations in the coding region of the genome induced by .1% CSE over time increased.
Discussion
There have been previous studies studying the effect of CSE on cell cultures. These studies generally employed high doses of CSE and exposed the cells for very short periods of time. These early investigators demonstrated that CSE treatment induced apoptosis through the intrinsic pathway.(15, 16, 19, 22-32) In addition, two studies demonstrated that cigarette smoke extract could induces in changes in lung cell lines that were similar to that of a cancerous state. Specifically, they observed that CSE treatment of alveolar epithelial cells could induce a pro-inflammatory/activated state that could predispose cells to malignant transformation.(33, 34) Several groups also examined the role of caspase-3 activation in response to CSE treatment of various pulmonary cell lines, but had conflicting findings. It is difficult to compare these studies since each study examined a different cell type (neutrophils, macrophages, lung epithelial cells etc.) (15, 29, 30) However, all these studies confirmed that cell death in response to CSE treatment is mediated predominantly through the intrinsic pathway and only examined the acute response to cigarette smoke exposure. Our findings concur with these previous studies. However, we further postulate and prove that it is the chronic treatment of cells that best models in vivo cigarette smoke exposure as compared to acute exposure cell line models. We submit that chronic CSE exposure provides the environment necessary for the selection of cellular clones that contain the functional molecular changes that set the stage for the progression to malignancy.
However, the critical carcinogenic effects of tobacco smoke occur in the context of chronic, rather than acute, exposure in vivo, and ongoing selection pressures may result in the development of genetic and phenotypic characteristics that require a model of chronic exposure. Our findings suggest that intrinsic pathway apoptotic factors become activated in response to CSE exposure, and select for a chronically active state during long term administration. This chronic low level activation of cellular apoptotic pathways selects for cells that are resistant to normal apoptotic signaling. This growing functional resistance to apoptosis also prevents cells from responding to apoptotic inducing agents that act through the intrinsic pathway. An interesting finding of our study was that the CSE treated cells were also resistant, in a dose dependent manner, to staurosporine treatment, and marked by the presence of chronically activated caspase 3. This suggests that CSE acts on cells beyond simply mitochondrial or intrinsic pathways and that there are likely effects of CSE that have not been recognized. There are several areas of ongoing investigations including the evaluation of status the death receptor mediated pathway and apoptotic events down stream of caspase-3 activation.
These data imply that prolonged cigarette smoking may provide the selective pressure for a state of chronic resistance to apoptosis in exposed mucosal tissues during long term exposure. This finding expands the role of cigarette smoking as a factor that allows evasion of apoptosis while concurrently inducing genetic damage, allowing genetic alterations to propagate through continued cellular division. This may provide support for a paradigm in which development of clonal populations with apoptosis resistance are a critical factor for tobacco induced carcinogenesis. Supporting this theory is a recent study by Tan et al that found both an increase in the amount of mitochondrial DNA content and somatic mitochondrial DNA mutations in buccal mucosal cells from smokers.(36)
Cigarette smoking exposes the individual to an entire array of chemicals, some of which are well known carcinogens. Many of these carcinogens are well studied. However, our intent in this study was to model in vivo conditions in cell. More specifically, we were interested in how the components of cigarette smoke act in conjunction with one another, if at all, to induce cellular changes. Our next step is to determine the components of the CSE and their respective additive or synergistic effects on apoptotic dysfunction and DNA damage.
A surprising and interesting finding of this study was that the vapor component of CSE alone was sufficient to cause the same effect as direct CSE treatment. An entire incubator was dedicated to growing cells treated with CSE. However, cells grown in the same incubator but without direct CSE treatment displayed similar effects as the treated cells. It is likely that it is the aromatic component that is most essential to produce the functional apoptotic resistance we observed. This implies that the carcinogenic risk conferred by second hand smoke exposure may be due to exposure of these same volatile components of cigarette smoke, and that this may also mediate induction of apoptosis resistance. Therefore, further characterization of this component of CSE may have significant implications for second hand smoke exposure studies. Once characterized the vapor component may then be further isolated by other methods, such as Cambridge filters. This would then allow us to determine which components are essential to produce the effects of CSE. The components of the vapor component are an active area of research in our lab.
We have previously investigated the functional effects of mitochondrial mutations found in primary HNSCC, noting that these mutations induce increased proliferation and an aerobic, glycolytic phenotype. It is well known that CSE exhibits mitochondrial toxicity, however, we were able to demonstrate increased mitochondrial mutation in CSE exposed cells with apoptosis resistance. These mutations occurred in respiratory chain enzymes, and we have also noted stabilization of HIF-1α in cells chronically exposed to CSE (data not shown). These pathways are promising avenues of investigation as possible mediators of CSE induced apoptosis resistance.
We therefore cannot rule out the possibility that CSE may affect other cellular pathways in our system including epigenetic mechanisms. One of these additional pathways was proposed by a study that correlated pirin over-expression in smokers with over-expression in normal lung cells after CSE treatment.(22) There is also some evidence that there is a close relationship between CSE treatment and epigenetic regulation of genes. A recent study found that CSE treatment of lung cancer cells caused the hypomethylation of an oncogene by the downregulation of DNA methyltransferase 3B, an enzyme important in epigenetic regulation.(37) Several other studies have found that CSE treatment in cell lines can induce histone modification, chromatin remodeling and activation of the NFKB cell proliferation pathway in lung cells.(33, 34, 38) Continued investigation into the basis for CSE mediated apoptosis resistance in in vitro and in vivo/human systems will likely further define the mechanisms by which this phenotype is produced.
The assertion of paper is loosely based upon the events of apoptosis following as described: ΔΨm, cytochrome c release, caspase-3 activation. It should be noted that all of these events are still actively researched and debated. The apoptotic cascade of events that we selected to base our study upon was supported by Lakhani et al.(8). We do acknowledge that our model is a simplification of a very complex signaling pathway. For instance, ΔΨm disruption and its role in apoptosis is still under investigation (8, 39-41). However, we believe that the dominant paradigm today is the one proposed by Lakhani et al and we selected three events in apoptosis that we felt were well studied (8, 39-41). Regardless of the order in which these events are believed to occur and whether one event is sufficient to produce apoptosis, we found that all three events are activated in our cell lines after CSE selection.
An interesting finding of our study was that activated caspase-3, known to be an effector caspase (as opposed to the initiator caspases), was not sufficient to induce complete apoptosis in cell lines (8, 39, 40). Caspase-3 is an inactive proenzyme that undergoes proteolytic processing to form the active enzyme. This protein then activates caspases 6, 7, and 9. It is possible that CSE exposure alters the environment and therefore the enzymatic properties of the active caspase-3, disrupting its ability to activate its downstream targets but this has not been proven. The status of the other downstream caspases is still under investigation. The more likely explanation is that the redundancy of the apoptotic pathway prevented cellular death. In other words, it is possible that although the activation of these apoptotic events (ΔΨm, cytochrome c release, caspase-3 activation) was offset by the activation or de-activation of other events that was not studied.
Supplementary Material
Mitochondrial DNA sequencing
Acknowledgments
Financial Support:
The following work was supported by a Clinical Innovator Award from the Flight Attendant Medical Research Institute, (Dr. Califano), SPORE (5P50CA096784-05), and NIH T32DC000027-20 (Ian Smith) and NIH T32DC000027-19 (Steven Chang)
References
- 1.Hashibe M, Brennan P, Benhamou S, Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova E, Wunsch-Filho V, Franceschi S, Hayes RB, Herrero R, Koifman S, La Vecchia C, Lazarus P, Levi F, Mates D, Matos E, Menezes A, Muscat J, Eluf-Neto J, Olshan AF, Rudnai P, Schwartz SM, Smith E, Sturgis EM, Szeszenia-Dabrowska N, Talamini R, Wei Q, Winn DM, Zaridze D, Zatonski W, Zhang ZF, Berthiller J, Boffetta P. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007;99:777–789. doi: 10.1093/jnci/djk179. [DOI] [PubMed] [Google Scholar]
- 2.Chortyk OT, Bock FG. Tumor-promoting activity of certain extracts of tobacco. J Natl Cancer Inst. 1976;56:1041–1045. doi: 10.1093/jnci/56.5.1041. [DOI] [PubMed] [Google Scholar]
- 3.Bock FG, Moore GE, Crouch SK. Tumor-Promoting Activity of Extracts of Unburned Tobacco. Science. 1964;145:831–833. doi: 10.1126/science.145.3634.831. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 5.Krammer PH. CD95(APO-1/Fas)-mediated apoptosis: live and let die. Adv Immunol. 1999;71:163–210. doi: 10.1016/s0065-2776(08)60402-2. [DOI] [PubMed] [Google Scholar]
- 6.Gottlieb RA. Programmed cell death. Drug News Perspect. 2000;13:471–476. [PubMed] [Google Scholar]
- 7.Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–781. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- 8.Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: and key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duax WL, Griffin JF, Langs DA, Smith GD, Grochulski P, Pletnev V, Ivanov V. Molecular structure and mechanisms of action of cyclic and linear ion transport antibiotics. Biopolymers. 1996;40:141–155. doi: 10.1002/(SICI)1097-0282(1996)40:1%3C141::AID-BIP6%3E3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 10.Inai Y, Yabuki M, Kanno T, Akiyama J, Yasuda T, Utsumi K. Valinomycin induces apoptosis of ascites hepatoma cells (AH-130) in relation to mitochondrial membrane potential. Cell Struct Funct. 1997;22:555–563. doi: 10.1247/csf.22.555. [DOI] [PubMed] [Google Scholar]
- 11.Krick S, Platoshyn O, McDaniel SS, Rubin LJ, Yuan JX. Augmented K(+) currents and mitochondrial membrane depolarization in pulmonary artery myocyte apoptosis. Am J Physiol Lung Cell Mol Physiol. 2001;281:L887–894. doi: 10.1152/ajplung.2001.281.4.L887. [DOI] [PubMed] [Google Scholar]
- 12.Penning LC, Denecker G, Vercammen D, Declercq W, Schipper RG, Vandenabeele P. A role for potassium in TNF-induced apoptosis and gene-induction in human and rodent tumour cell lines. Cytokine. 2000;12:747–750. doi: 10.1006/cyto.1999.0626. [DOI] [PubMed] [Google Scholar]
- 13.Paananen A, Mikkola R, Sareneva T, Matikainen S, Hess M, Andersson M, Julkunen I, Salkinoja-Salonen MS, Timonen T. Inhibition of human natural killer cell activity by cereulide, an emetic toxin from Bacillus cereus. Clin Exp Immunol. 2002;129:420–428. doi: 10.1046/j.1365-2249.2002.01898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao M, Mydlarz WK, Zhou S, Califano J. Head and neck cancer cell lines are resistant to mitochondrial-depolarization-induced apoptosis. ORL J Otorhinolaryngol Relat Spec. 2008;70:257–263. doi: 10.1159/000133280. [DOI] [PubMed] [Google Scholar]
- 15.Aoshiba K, Tamaoki J, Nagai A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1392–1401. doi: 10.1152/ajplung.2001.281.6.L1392. [DOI] [PubMed] [Google Scholar]
- 16.Carnevali S, Petruzzelli S, Longoni B, Vanacore R, Barale R, Cipollini M, Scatena F, Paggiaro P, Celi A, Giuntini C. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;284:L955–963. doi: 10.1152/ajplung.00466.2001. [DOI] [PubMed] [Google Scholar]
- 17.Carp H, Janoff A. Possible mechanisms of emphysema in smokers. In vitro suppression of serum elastase-inhibitory capacity by fresh cigarette smoke and its prevention by antioxidants. Am Rev Respir Dis. 1978;118:617–621. doi: 10.1164/arrd.1978.118.3.617. [DOI] [PubMed] [Google Scholar]
- 18.Kim H, Liu X, Kobayashi T, Conner H, Kohyama T, Wen FQ, Fang Q, Abe S, Bitterman P, Rennard SI. Reversible cigarette smoke extract-induced DNA damage in human lung fibroblasts. Am J Respir Cell Mol Biol. 2004;31:483–490. doi: 10.1165/rcmb.2002-0300OC. [DOI] [PubMed] [Google Scholar]
- 19.Ishii T, Matsuse T, Igarashi H, Masuda M, Teramoto S, Ouchi Y. Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1189–1195. doi: 10.1152/ajplung.2001.280.6.L1189. [DOI] [PubMed] [Google Scholar]
- 20.Fadeel B. Programmed cell clearance. Cell Mol Life Sci. 2003;60:2575–2585. doi: 10.1007/s00018-003-3145-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ethell DW, Buhler LA. Fas ligand-mediated apoptosis in degenerative disorders of the brain. J Clin Immunol. 2003;23:439–446. doi: 10.1023/b:joci.0000010420.96419.a8. [DOI] [PubMed] [Google Scholar]
- 22.Gelbman BD, Heguy A, O'Connor TP, Zabner J, Crystal RG. Upregulation of pirin expression by chronic cigarette smoking is associated with bronchial epithelial cell apoptosis. Respir Res. 2007;8:10. doi: 10.1186/1465-9921-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoshino Y, Mio T, Nagai S, Miki H, Ito I, Izumi T. Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell line. Am J Physiol Lung Cell Mol Physiol. 2001;281:L509–516. doi: 10.1152/ajplung.2001.281.2.L509. [DOI] [PubMed] [Google Scholar]
- 24.Jiao ZX, Ao QL, Xiong M. Cigarette smoke extract inhibits the proliferation of alveolar epithelial cells and induces apoptosis. Sheng Li Xue Bao. 2006;58:244–254. [PubMed] [Google Scholar]
- 25.Kode A, Yang SR, Rahman I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir Res. 2006;7:132. doi: 10.1186/1465-9921-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan MY, Ho CY, Lee TC, Yang AH. Cigarette smoke extract induces cytotoxicity on human nasal epithelial cells. Am J Rhinol. 2007;21:218–223. doi: 10.2500/ajr.2007.21.2966. [DOI] [PubMed] [Google Scholar]
- 27.Ramage L, Jones AC, Whelan CJ. Induction of apoptosis with tobacco smoke and related products in A549 lung epithelial cells in vitro. J Inflamm (Lond) 2006;3:3. doi: 10.1186/1476-9255-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, Karlsson JM, Watkins SC, Kim HP, Wang X, Lee JS, Postma DS, Kauffman HF, Choi AM. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol. 2007;36:409–417. doi: 10.1165/rcmb.2006-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Stringer KA, Tobias M, O'Neill HC, Franklin CC. Cigarette smoke extract-induced suppression of caspase-3-like activity impairs human neutrophil phagocytosis. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1572–1579. doi: 10.1152/ajplung.00325.2006. [DOI] [PubMed] [Google Scholar]
- 30.Wang HY, Shin VY, Leung SY, Yuen ST, Cho CH. Involvement of bcl-2 and caspase-3 in apoptosis induced by cigarette smoke extract in the gastric epithelial cell. Toxicol Pathol. 2003;31:220–226. doi: 10.1080/01926230390183715. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Wilcken DE, Wang XL. Cigarette smoke activates caspase-3 to induce apoptosis of human umbilical venous endothelial cells. Mol Genet Metab. 2001;72:82–88. doi: 10.1006/mgme.2000.3115. [DOI] [PubMed] [Google Scholar]
- 32.Yang YM, Liu GT. Damaging effect of cigarette smoke extract on primary cultured human umbilical vein endothelial cells and its mechanism. Biomed Environ Sci. 2004;17:121–134. [PubMed] [Google Scholar]
- 33.Moodie FM, Marwick JA, Anderson CS, Szulakowski P, Biswas SK, Bauter MR, Kilty I, Rahman I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. Faseb J. 2004;18:1897–1899. doi: 10.1096/fj.04-1506fje. [DOI] [PubMed] [Google Scholar]
- 34.Martey CA, Pollock SJ, Turner CK, O'Reilly KM, Baglole CJ, Phipps RP, Sime PJ. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol. 2004;287:L981–991. doi: 10.1152/ajplung.00239.2003. [DOI] [PubMed] [Google Scholar]
- 35.Zhou S, Kassauei K, Cutler DJ, Kennedy GC, Sidransky D, Maitra A, Califano J. An oligonucleotide microarray for high-throughput sequencing of the mitochondrial genome. J Mol Diagn. 2006;8:476–482. doi: 10.2353/jmoldx.2006.060008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan D, Goerlitz DS, Dumitrescu RG, Han D, Seillier-Moiseiwitsch F, Spernak SM, Orden RA, Chen J, Goldman R, Shields PG. Associations between cigarette smoking and mitochondrial DNA abnormalities in buccal cells. Carcinogenesis. 2008;29:1170–1177. doi: 10.1093/carcin/bgn034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Zhou Y, Boggs SE, Belinsky SA, Liu J. Cigarette smoke induces demethylation of prometastatic oncogene synuclein-gamma in lung cancer cells by downregulation of DNMT3B. Oncogene. 2007;26:5900–5910. doi: 10.1038/sj.onc.1210400. [DOI] [PubMed] [Google Scholar]
- 38.Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46–57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- 39.Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 41.Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial membrane permeability transition and cell death. Biochim Biophys Acta. 2006;1757:1297–1300. doi: 10.1016/j.bbabio.2006.03.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mitochondrial DNA sequencing