

Abstract
Within the CHARMM polarizable force field based on the classical Drude oscillator, atomic polarizabilities are derived via fitting to ab initio calculated data on isolated gas phase molecules, with an empirical scaling factor applied to account for differences between the gas and condensed phases. In the development of polarizable models for the ethers, a polarizability scaling factor of 0.7 was previously applied. [Vorobyov et al. J Comput Chem 3:1120-1133, 2007] While the resulting force field models gave good agreement with a variety of experimental data, they systematically underestimated the liquid phase dielectric constants. Here, a new CHARMM polarizable model is developed for the ethers, employing a polarizability scaling factor of 0.85 and including atom-based Thole scale factors recently introduced into the CHARMM Drude polarizable force field. [Harder et al. J Phys Chem B 112:3509-3521, 2008] The new model offers a significant improvement in the reproduction of liquid phase dielectric constants, while maintaining the good agreement of the previous model with all other experimental and quantum mechanical data, highlighting the sensitivity of liquid phase properties to the choice of atomic polarizability parameters.
Keywords: Force field, CHARMM, polarizable, ethers, Thole, dielectric constant, heat of vaporization
Introduction
Parameters for a polarizable force field model of linear and cyclic ethers based on the Drude oscillator model [1] have previously been developed for use within the CHARMM force field. [2] While calculations performed using these parameters give results in satisfactory agreement with a wide range of reference data encompassing liquid phase thermodynamic properties; gas phase interactions with water and rare gas molecules, and free energies of solvation, the final force field systematically underestimates the liquid phase dielectric constants of the ether molecules.
A key parameter in the CHARMM polarizable force field is the atomic polarizability, which is obtained through fitting to a series of ab initio calculated electrostatic potentials for an isolated gas phase molecule, perturbed by a point charge. [3] It is well known that the polarizability of a molecule is dependent on its environment, [4, 5] and during the development of the Drude polarizable force field it has become apparent that gas phase polarizability values are often too large compared to the values required to obtain accurate condensed phase properties, [3, 6] with the liquid phase dielectric constant particularly sensitive to the polarizability. [7] The underlying physical reason for this difference in gas and condensed phase polarizabilities is still a point of debate. It was first explained in terms of the Pauli exclusion principle [6, 8]: with the accurate calculation of molecular polarizabilites by ab initio methods requiring the use of diffuse functions, which extend spatially beyond the van der Waals surface of the molecule, [7] it was suggested that in the condensed phase, when there are other molecules in close proximity, these orbitals will overlap with the similarly diffuse orbitals on neighboring molecules, causing “the Pauli exclusion principle to qualitatively raise the energies of these basis functions, thus reducing their mixing into the ground-state wave function.” [7] Elsewhere it has been attributed to “the inhomogeneities of the electric field within the volume of a water molecule embedded in liquid water.” [9] Other studies have observed the effect via quantum mechanical (QM) calculations on water [4, 8, 10] and on a lipid head group, [11] where the presence of up to eight water molecules resulted in a decrease of up to 20% in calculated polarizability, a result attributed to “charge stabilization” via the formation of H bonds, the authors proposing that electrons involved in H bonding are more tightly bound than those not involved in the formation of H bonds and therefore less able to respond to the presence of an external field.
In the development of the Drude-based CHARMM polarizable force field, this effect has been accounted for by scaling the gas phase atomic polarizabilites by an empirical scaling factor during the parametrization process. [3] In the original Drude oscillator model for the ether molecules, the values of the atomic polarizability, α, obtained from the charge fitting procedure were systematically scaled by a factor of 0.7: a value found to be appropriate in the development of the SWM4-NDP water model. [12] Since the development of the polarizable force field model for ethers, however, in a Drude polarizable model for N-containing heteroaromatics a polarizability scaling factor of 0.85 was found to be appropriate, [13] in good agreement with the work of Morita and Kato, [14] who showed that neutral species show a polarizability decrease of 13-18% on moving from the gas phase to aqueous solution. In another study of amides, no scaling (i.e. 1.0 scaling) was found to be necessary to give the correct dielectric constant, [15] with the same requirement for 1.0 scaling also observed in the parameterization of a Drude model for alkanes. [16] Given the systematic underestimation of the original ether dielectric constants and several studies showing that different scaling factors are appropriate for different classes of molecules, it was felt that the scaling factor used for the atomic polarizabilities in the Drude polarizable force field for ethers should be re-evaluated.
A second motivation for further optimization of the polarizable ether model was the inclusion of atom-based Thole scale factors in the polarizable Drude force field. Thole scale factors are parameters used to modify the electrostatic term in the force field to allow 1-2 and 1-3 screened dipole interactions via smearing of the charges associated with the dipole moments on the Drude particle and the real atom using a Slater distribution. [17] The Thole scaling factors dictate the extent of charge smearing and the standard Thole scaling procedure applies a single scale factor to all atom pairs. In the CHARMM Drude model a value of 2.6 was initially selected, based on the reproduction of the anisotropic molecular polarizability of benzene, and this is the value that was used in the previous work on ethers. [2] However, in subsequent studies on amides it was shown that the use of a single scale factor for all atom pairs resulted in a poor reproduction of the ab initio calculated orientation of the molecular polarizability tensor by the Drude model. Incorporating atom-based scaling terms into the Drude models overcame this limitation and, in combination with an atomic polarizability scaling factor of 1.0, allowed for reproduction of the dielectric constant of N-methylacetamide. [15] The use of atom-based Thole scaling has more recently been shown to facilitate reproduction of the polarizability tensor of N-containing heteroaromatics. [13]
Based on these advances, re-evaluation of the previously optimized ether force field was undertaken in the present study. This effort was primarily focused on the electrostatic parameters; however, given the interdependence of all parameters in a force field, it was necessary to optimize all remaining terms in the energy function. Accordingly, the objectives of this work are: (1) the development of new polarizable force field models for linear and cyclic ethers using a polarizability scaling factor of 0.85 in combination with atom-based Thole scaling factors, and (2) the assessment of the impact that the new parameter set has on calculated values of the dielectric constants.
Methods
All ab initio calculations were performed using the program Gaussian03, [18] and all force field calculations were performed using version c35b1 of the program CHARMM. [19, 20, 21]
As a first step in the parametrization process, atomic charges and polarizabilities were obtained by restrained fitting to B3LYP/aug-cc-pVDZ perturbed electrostatic potential maps using MP2/6-31G(d) optimized geometries. This procedure was performed on the same molecular conformations used in the previous development of Drude polarizable models for ethers, [2] and the method employed was identical to that used in the previous work, except for two modifications. Firstly, a polarizability scaling factor of 0.85 was used in place of the original scaling factor of 0.7. The second modification was in the assignment of Thole scaling factors: in the previous work, a set value of 1.3 was used as the Thole scaling factor for every heavy atom, which corresponds to the value of 2.6 for each atom pair determined based on benzene (ie. a simple additive combining rule is used for the atom-based Thole scale factors). [15, 22] In the present study, atom specific Thole factors were calculated as part of the charge fitting procedure. As in the previous work, [2] initial values for the atomic charges were taken from the additive CHARMM model, and initial atomic polarizabilities were taken from the work of Miller, [23] modified to take account of the heavy-atom only polarizability model used in this work. For the cyclic ethers tetrahydrofuran (THF) and tetrahydropyran (THP), such a scheme was sufficient to obtain usable charge and polarizability parameters. For the linear ethers, however, it was a requirement that parameters should be transferable across the series, and only three distinct C atom types were used: Ca; Cb, and Cg, as shown in Figure 1. Charge fitting was initially performed on dimethyl ether (DME) to obtain charge and polarizability parameters for O and Ca. Charge fitting was then also performed on diethyl ether (DEE), with the O parameters restrained to those obtained during the fitting of DME. This yielded the required parameters for Cb and Cg, which were then (along with the Ca and O values from DME) transferred to 1,2-dimethoxyethane (DMOE) and methyl ethyl ether (MEE).
Fig. 1.
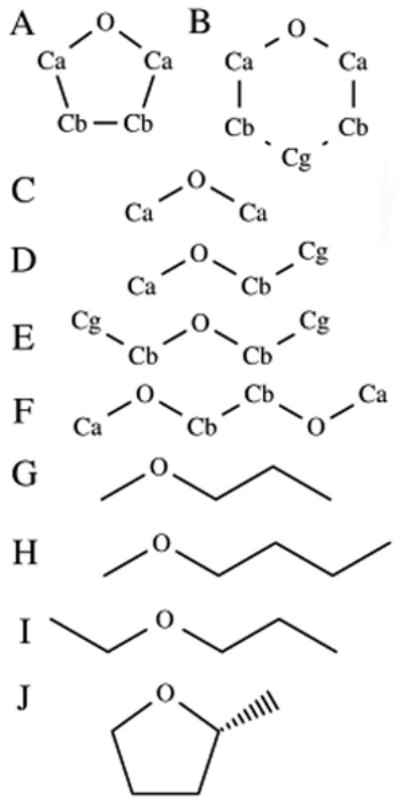
Ether model compounds, A-F, and test compounds, G-J: (A) tetrahydrofuran, THF; (B) tetrahydropyran, THP; (C) dimethyl ether, DME; (D) methyl ethyl ether, MEE; (E) diethyl ether, DEE; (F) 1,2-dimethoxyethane, DMOE; (G) methyl propyl ether, MPE; (H) methyl butyl ether, MBE; (I) ethyl propyl ether, EPE; (J) 2-(R)-methyl tetrahydrofuran, MTHF
Throughout this process, the Drude force constant is treated as being isotropic for all C atoms. For the O atoms, however, the Drude force constant is treated as a tensor, K(D), where the components Kxx(D), Kyy(D) and Kzz(D) “determine the stiffness of the atom-Drude bond in three orthogonal directions” [2] and hence provide an anisotropic representation of the polarizability. In the previous parametrization of the Drude oscillator model for ethers, the O atomic polarizability anisotropy was taken directly from that calculated previously for methanol, [22] with Kxx(D) = 1100, Kyy(D) = 800 and Kzz(D) = 1100 kcal/(molÅ2). The same approach was also adopted in this work, with the orientation of the anisotropy defined relative to the O lone pair atoms (see below) and a dummy atom connected to the O atom. To account for the anisotropy in the charge density around the O atom, two additional “lone pair” point charges are also included for each O atom within the ether molecules, a method that has been shown to give a better reproduction of the anisotropy of interactions between hydrogen bond acceptor atoms and water molecules when compared to models including only atom centered charges. [22, 24] While the charges on these lone pair sites were determined as part of the charge fitting procedure within this work, their positions were taken directly from the previous polarizable ether model, where they were “adjusted manually based on achieving as small as possible root-mean-square-error of empirical vs QM electrostatic potentials (ESP) and the qualitative reproduction of the local QM ESP around the oxygen atom.” [2] The final values used for the positions of the two lone pairs around each O atom, defined relative to the O atom and the two C atoms adjacent to the O atom (for convenience labeled C1 and C2) are: R(O-LP) = 0.35 Å, θ(LP-O-C) = 0.35°, φ (LP-O-C1-C2) = 90°, 270°.
With the electrostatic parameters in place, the next stage of the parametrization process was the development of Lennard-Jones parameters. In the previous work, two different O atom types were used for the ether molecules: one to describe the O atom in THF, and a second to describe the O atom in all other ethers (linear ethers and THP). Here, it was found that improved models of THP and the linear ethers could be obtained if the restriction that the O atom in THP should have the same Lennard-Jones parameters as O in the linear ethers was lifted, and a new CHARMM atom type, OD306A, was introduced for the O atom in THP. Initial Lennard-Jones parameters for all atoms were taken from the previous Drude force field ether model, and as a first assessment of the quality of Lennard-Jones parameters, interactions between the ether molecules and a single water molecule were considered. The method used was identical to that described in the previous work, [2] with the O Lennard-Jones parameters modified until a reasonable agreement with quantum mechanical data was obtained. Results from these calculations were used to guide the choice of a range of values for O Lennard-Jones parameters over which to scan in search of optimal agreement with experimental liquid phase data. For these scans, at each combination of the well depth, ε, and atomic radii, Rmin/2, a single liquid phase molecular dynamics simulation of 150 ps starting from a pre-equilibrated box of 128 ether molecules was performed, along with a single gas phase molecular dynamics simulation of 2.5 ns on an isolated molecule (simulations performed at the temperatures noted in Table 1). From the results of these simulations, the molecular volume, Vm, and heat of vaporization, ΔHvap, of the molecule were calculated and compared to known experimental values. The Lennard-Jones parameter combination that yielded the best agreement with the experimental values was then accepted as the final model, subject to the condition that calculated values of both Vm and ΔHvap must be within 2% of the experimental values.
Table 1.
Molecular Volumes and Heats of Vaporizations (Vm in Å3, ΔHvap in kcal/mol). a Experimental data from [32]. b Experimental data from [33]. c Experimental data from [34]. d Experimental data from [35]
| T, K | exper | D0.7 | % err | D0.85 | % err | |
|---|---|---|---|---|---|---|
| Tetrahydrofuran (THF) | ||||||
| Vm | 298.15 | 135.6a | 134.0 ± 0.8 | -1.2 | 134.8 ± 0.4 | -0.6 |
| ΔHvap | 298.15 | 7.65a | 7.80 ± 0.06 | 2.0 | 7.69 ± 0.03 | 0.9 |
| Tetrahydropyran (THP) | ||||||
| Vm | 298.15 | 162.3a | 165.3 ± 0.3 | 1.8 | 163.8 ± 0.8 | 0.9 |
| ΔHvap | 298.15 | 8.26a | 8.27 ± 0.02 | 0.1 | 8.41 ± 0.04 | 1.8 |
| Diethyl Ether (DEE) | ||||||
| Vm | 298.15 | 173.9b | 171.8 ± 1.0 | -1.2 | 176.9 ± 1.2 | 1.9 |
| ΔHvap | 298.15 | 6.48a | 6.80 ± 0.10 | 4.9 | 6.46 ± 0.06 | -0.3 |
| Dimethoxyethane (DMOE) | ||||||
| Vm | 298.15 | 173.6b | 176.6 ± 0.8 | 1.7 | 178.1 ± 0.9 | 2.6 |
| ΔHvap | 298.15 | 8.79a | 8.79 ± 0.10 | 0.0 | 8.67 ± 0.07 | -1.4 |
| Dimethyl Ether (DME) | ||||||
| Vm | 248.34 | 104.9c | 106.6 ± 0.9 | 1.6 | 104.2 ± 0.8 | -0.7 |
| ΔHvap | 248.34 | 5.14a | 4.94 ± 0.05 | -3.9 | 5.18 ± 0.02 | 0.8 |
| Methyl Ethyl Ether (MEE) | ||||||
| Vm | 273.20 | 137.5d | 137.5 ± 0.6 | 0.0 | 140.2 ± 0.8 | 2.0 |
| ΔHvap | 280.60 | 5.90a | 5.72 ± 0.06 | -3.1 | 5.85 ± 0.04 | -0.8 |
For THF and THP this approach, in which only the O Lennard-Jones parameters were varied, was sufficient to yield models giving good agreement with experimental data. However, for the linear ethers such an approach was unsuccessful. While varying only the O Lennard-Jones parameters was sufficient to give a model that accurately reproduced experimental liquid phase properties of any one molecule, it was unable to yield a set of parameters giving acceptable results across the whole series of linear ethers. Accordingly, optimization of the O Lennard-Jones parameters was followed by optimization of the Lennard-Jones parameters for C atoms adjacent to the O atoms. This was again achieved by running scans over ranges of ε and Rmin/2 parameter values, employing the same criteria for acceptance as outlined above. In this way, parameter sets were obtained giving good agreement with experimental data for all four linear ethers considered. This method also required the introduction of two new CHARMM atom types: CD32E, for an sp3 hybridized C atom bonded to two H atoms and adjacent to an ether O atom, and CD33E, for an sp3 hybridized C atom bonded to three H atoms and adjacent to an ether O atom.
While an assessment of Vm and ΔHvap for each molecule had been obtained in the previous step, longer simulations were required to calculate more precise values of these thermodynamic properties. For each molecule, ten liquid phase molecular dynamics simulations were performed using the parameters obtained in the steps above, each simulation of 150 ps. For each molecule, all ten liquid phase simulations were commenced from an identical pre-equilibrated box of 128 ether molecules, with a random number seed used to assign different initial velocities in each case. For each simulation, the first 50 ps was treated as equilibration, with the remaining 100 ps used for analysis. Volumes and energies were averaged over all ten simulations, and the gas phase contribution to the heat of vaporization was calculated from a single simulation of 2.5 ns, with 0.5 ns used for equilibration and 2.0 ns for analysis. All simulations were performed at the temperatures reported in Table 1.
Once a set of parameters had been obtained giving good agreement with experimental values of Vm and ΔHvap, longer simulations were performed for the calculation of dielectric constants, ε. For each molecule, four simulations were run for 5 ns each, with the final 4.5 ns of each simulation used for analysis. Values of ε were calculated as described before, [4] with the high-frequency optical dielectric constant εinf estimated using the Clausius-Mossotti equation, and averaged over all four simulations, with errors calculated as the standard deviations over the four simulations.
Free energies of aqueous solvation were calculated via the free energy perturbation method [25] using the staged protocol of Deng and Roux [26] in a manner analogous to that described in the previous work, [2] with only minor differences in the simulation protocol. Specifically, the simulation time was extended to 10 ps of equilibration and 100 ps of production for a given value of the coupling and/or staging parameter and a long range correction [27] was included to account for errors introduced by the truncation of Lennard-Jones interactions. For every calculated value of the free energy of hydration, the long range correction was calculated from a single simulation of a single solute molecule in a box of 250 SWM4-NDP [12] water molecules run for 50 ps of molecular dynamics in the NVT ensemble, during which coordinates were saved every 1 ps. Following completion of the MD simulation, coordinates were extracted from the final 30 ps of the CHARMM trajectory file and energies were calculated for each set of coordinates using two different non-bonded interaction cutoff schemes. In the first scheme, nonbond pair lists were maintained to 16 Å with a real space cutoff of 12 Å used for both electrostatic and van der Waals terms, with the latter truncated via an atom-based force switch algorithm. In the second scheme, the only differences were that nonbond pair lists were maintained to 36 Å, and a real space cutoff of 32 Å was used. The difference in the van der Waals interaction energy calculated using the two non-bonded interaction cutoff schemes, averaged over all sets of coordinates, was taken as the long range correction.
Results and Discussion
The results given below are those obtained using the final set of optimized parameters (final parameter values are listed in Table S1). The new model obtained using a polarizability scaling factor of 0.85 and atom-based Thole scaling factors is termed D0.85; the previous model, obtained using a polarizability scaling factor of 0.7 with a single Thole scaling factor for all atom pairs, is termed D0.7: all results presented for the D0.7 model are taken from [2]. Allowing the Thole scaling factors to vary, along with the atomic charges and polarizabilities, during the electrostatic parameter fitting procedure resulted in significant differences between the Thole scaling factors used in the D0.7 and D0.85 models (Table S1). In the D0.7 model, all atoms were assigned a Thole scaling factor of 1.3, as noted above. In the D0.85 model, the final Thole scaling factors ranged from 0.316 for C in THP to 2.046 for O in THP. In general, Thole scaling factors for C atoms decreased from the default value of 1.3 (final values ranged from 0.316 to 1.103), while Thole scaling factors for O atoms increased from the default value of 1.3 (final values ranged from 1.312 to 2.046). This pattern is consistent with earlier work on liquid amides, [15] where fitted Thole scaling factors were consistently larger for O atoms than for C atoms.
Intramolecular Properties
While this work is focused on an examination of the nonbonded properties of the ether models, it is important to bear in mind that, within both the polarizable and additive versions of the CHARMM force field, both the nonbonded and bonded (ie bond, angle, dihedral) parameters are developed iteratively to ensure the optimum reproduction of both the bonded and nonbonded target data. [28] Therefore, when adjusting nonbonded parameters it is essential to examine what effect these adjustments have on the internal properties of the force field models, if necessary adjusting the bonded parameters before returning to examine the effect that these subsequent changes have on the properties used in the parametrization of the nonbonded parameters. This iterative process should then be repeated until optimal agreement with the target data is obtained. In this case, following the development of the nonbonded parameters, a series of tests were performed to evaluate the impact of these new parameters on the internal properties of the molecules. For each molecule, minimum energy conformations obtained from ab initio calculation at the MP2/6-31G(d) level were optimized within CHARMM, and their geometric parameters compared to those obtained from the ab initio structures as well as experimental data where available (Tables S4-S9). For the global energy minima of THF, THP and DEE, vibrational spectra were also calculated within CHARMM and compared to the corresponding ab initio calculated vibrational spectra (Tables S10-S12). As a final test, potential energy surfaces were calculated for rotations about conformationally important dihedral angles in THF, THP, DEE and DMOE, and compared to equivalent potential energy surfaces calculated at the MP2/cc-pVTZ//MP2/6-31G(d) level of theory (Figures S1-S11).
Results from these tests show that the changes in the nonbonded parameters have had little effect on the geometric properties of the molecules (Tables S4-S9). Bond lengths are reproduced well by both models, with only the Cb-Cb bond of DMOE (Table S9) showing a deviation of greater than 0.02 Å from the reference QM data. For bond angles, the D0.85 model generally performs similarly to the D0.7 model, with an RMSD of 0.92° across all molecules and conformations and a maximum deviation of magnitude 3.8°, compared to an RMSD of 0.93° and a maximum deviation of magnitude 3.8 ° for the D0.7 model; both models provide excellent agreement with experimental data. For dihedral angles, both models again perform well, with the D0.7 model giving an RMSD of 2.56° over all molecules and conformations, the D0.85 model giving an RMSD of 2.45° over all molecules and conformations and each model giving only one deviation from the reference data greater than 5°: 6.8° and 6.9° for the Ca-O-Ca-Cb dihedral of the twist25 conformation of THP with the D0.7 and D0.85 models respectively (Table S5). Overall, the results obtained using the D0.85 model are very close to those obtained using the D0.7 model, with both being in good agreement with the reference quantum mechanical data.
In the development of the original Drude force field for ethers, “sacrifices were made in the vibrational data to allow for better reproduction of the relative energies of different conformations of the target molecules.” [2] For this reason, neither the D0.85 nor the D0.7 model gives perfect agreement with the quantum mechanical results. However, the agreement between the vibrational spectra calculated using the D0.85 and D0.7 models is good, (Tables S10-S12) indicating that the intermolecular properties of the molecules have not been adversely affected by the new nonbonded parameters. Dihedral angle rotational profiles calculated using both the D0.7 and D0.85 models give good agreement with the results from quantum mechanical reference calculations, (Figures S1-S11) supporting the conclusion that the new nonbonded parameters have not adversely affected the overall models. The final conclusion from these three sets of calculations is that no further reparameterization of the bonded parameters is required.
Intermolecular Properties
In general, the D0.85 model gives good agreement with experimental and quantum mechanical reference data (Tables 1-4), usually at a level comparable to that obtained with the D0.7 model for the ethers. The largest difference occurs in the calculated values of the dielectric constant (Table 2). While the D0.7 model systematically underestimates the dielectric constants of the ether molecules (with errors ranging from -1.60 to -0.17, an average error of -0.72 and an RMSD of 0.87), the errors in the dielectric constant values obtained using the D0.85 model range from -0.30 to 0.81, with an average error of 0.01 and an RMSD of 0.50, suggesting that the D0.85 model yields atomic polarziabilities that are appropriate for condensed phase simulations.
Table 4.
Gas-Phase Relative Conformational Energies and Dipole Moments. Relative energies are in kcal/mol, dipole moments in Debye. QM results are from [2] and were calculated at the MP2/cc-pVTZ//MP2/6-31G(d) level of theory. Experimental data from [32]
| Relative Energies | Dipole Moments | ||||||
|---|---|---|---|---|---|---|---|
| QM | D0.7 | D0.85 | exper | QM | D0.7 | D0.85 | |
| Tetrahydrofuran (THF) | |||||||
| C2 | 0.00 | 0.00 | 0.00 | 1.75 | 1.78 | 1.69 | 1.68 |
| Cs | 0.15 | 0.15 | 0.12 | 1.75 | 1.56 | 1.78 | 1.78 |
| C2v | 4.47 | 2.84 | 3.09 | 1.77 | 1.70 | 1.72 | |
| Tetrahydropyran (THP) | |||||||
| chair | 0.00 | 0.00 | 0.00 | 1.58 | 1.44 | 1.58 | 1.61 |
| twist25 | 5.67 | 5.22 | 5.58 | 1.43 | 1.61 | 1.62 | |
| twist14 | 6.74 | 6.48 | 6.70 | 1.63 | 1.67 | 1.74 | |
| boat25 | 6.76 | 6.42 | 6.75 | 1.33 | 1.62 | 1.70 | |
| boat14 | 7.48 | 6.41 | 6.89 | 1.62 | 1.64 | 1.65 | |
| Diethyl Ether (DEE) | |||||||
| tt | 0.00 | 0.00 | 0.00 | 1.15 | 1.11 | 1.20 | 1.35 |
| gt | 1.36 | 1.52 | 1.48 | 1.21 | 1.23 | 1.32 | |
| gg | 2.66 | 3.24 | 3.17 | 1.19 | 1.21 | 1.27 | |
| Methyl Ethyl Ether (MEE) | |||||||
| t | 0.00 | 0.00 | 0.00 | 1.17 | 1.19 | 1.25 | 1.35 |
| g | 1.38 | 1.53 | 1.45 | 1.30 | 1.27 | 1.30 | |
| Dimethyl Ether (DME) | |||||||
| s | 0.00 | 0.00 | 0.00 | 1.30 | 1.29 | 1.30 | 1.33 |
| Dimethoxyethane (DMOE) | |||||||
| ttt | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| gg't | 0.21 | 0.37 | 0.44 | 1.64 | 1.45 | 1.58 | |
| tgt | 0.26 | 0.03 | 0.24 | 1.35 | 1.67 | 1.76 | |
| gtt | 1.41 | 1.19 | 1.20 | 1.64 | 1.66 | 1.70 | |
| tgg | 1.50 | 1.29 | 1.53 | 2.40 | 2.46 | 2.55 | |
| ggg | 1.51 | 2.35 | 2.61 | 1.20 | 1.86 | 1.97 | |
| ggg' | 1.68 | 1.62 | 1.69 | 1.91 | 1.84 | 1.91 | |
| gtg' | 2.84 | 2.41 | 2.45 | 0.00 | 0.00 | 0.00 | |
| gtg | 2.91 | 2.30 | 2.31 | 2.19 | 2.12 | 2.16 | |
Table 2.
Dielectric Constants of Neat Liquids. Experimental data from [32]
| Molecule | T/K | exper | D0.7 | Error | D0.85 | error |
|---|---|---|---|---|---|---|
| THF | 298.15 | 7.43 | 6.80 ± 0.78 | -0.63 | 7.13 ± 0.06 | -0.30 |
| THP | 298.15 | 5.54 | 5.03 ± 0.20 | -0.51 | 5.39 ± 0.10 | -0.15 |
| DEE | 298.15 | 4.24 | 3.53 ± 0.34 | -0.71 | 4.55 ± 0.05 | 0.31 |
| DMOE | 298.15 | 7.22 | 5.61 ± 0.82 | -1.60 | 6.58 ± 0.14 | -0.64 |
| DME | 248.34 | 6.53 | 6.36 ± 0.18 | -0.17 | 7.34 ± 0.12 | 0.81 |
| RMSD | 0.87 | RMSD | 0.50 |
In the reproduction of other properties, the D0.7 and D0.85 models generally perform similarly, though there are some small differences. The D0.85 model performs slightly better than the D0.7 model in the reproduction of experimental values of Vm and ΔHvap (Table 1). The D0.85 model yields only one value that falls outside the target region (experimental datum ± 2%), with a Vm error of 2.6% for DMOE. The D0.7 model gives three values that do not fall within 2% of the experimental data: DEE, DME and MEE show errors in ΔHvap of 4.9%, -3.9% and 3.1% respectively. Notable is the fact that these errors all occur in the linear ethers, for which transferability of parameters was a key consideration. The improvement shown by the D0.85 model is likely due to the increased flexibility in the parametrization process, where the O parameters were not required to be identical to those in THP, and the Lennard-Jones parameters on C atoms adjacent to O atoms were also optimized. In the development of the D0.7 model, O parameters were required to be the same in both THP and the linear ethers, and only O Lennard-Jones parameters were optimized.
The D0.7 model performs noticeably better than the D0.85 model in the reproduction of experimental free energies of solvation for THF and THP (Table 3). In the development of the D0.85 model, accurate reproduction of this value was not considered a fundamental requirement (though approximate reproduction was), with priority placed on the reproduction of liquid phase properties instead. The reason that the accurate reproduction of free energies of solvation was not considered a requirement in this work was that, in the development of Drude polarizable force field parameters for alkanes, [16] alcohols [29] and N-containing aromatic heterocycles, [13] it was found that a force field giving a good representation of liquid phase properties yields free energies of hydration that are too favorable, as is also observed in this work (Table 3); the same result was also seen in the development of alkane parameters [30] for a CHARMM fluctuating charge [31] polarizable force field. For alcohols and N-containing aromatic heterocycles, accurate reproduction of the free energies of hydration with the CHARMM Drude polarizable force field has required the introduction of atom-pair specific Lennard-Jones parameters between the water O atom and the relevant solute atoms using the NBFIX keyword of CHARMM. [13] This method could also be used to obtain accurate free energies of hydration for the ethers. Current efforts towards a systematic approach to the development of NBFIX parameters within the CHARMM Drude polarizable force field are ongoing and will be published elsewhere.
Table 3.
Solvation free energies in Aqueous Solution. All values in kcal/mol. Experimental data from [36, 37]
| exper | D0.7 | diff | D0.85 | diff | |
|---|---|---|---|---|---|
| THF | -3.47 | -3.78 ± 0.15 | -0.31 | -4.80 ± 0.08 | -1.33 |
| THP | -3.12 | -3.20 ± 0.83 | -0.08 | -5.34 ± 0.27 | -2.22 |
| DEE | -1.76 | -1.60 ± 0.11 | 0.16 | -2.77 ± 0.10 | -1.01 |
| DMOE | -4.84 | -3.78 ± 0.59 | 1.06 | -5.61 ± 0.54 | -0.77 |
| DME | -1.92 | -1.25 ± 0.22 | 0.67 | -1.97 ± 0.13 | -0.05 |
| MEE | -2.10 | -1.38 ± 0.16 | 0.72 | -2.27 ± 0.25 | -0.17 |
In the reproduction of gas phase interactions with water molecules (Table S3), the D0.85 model does a better job of reproducing interaction energies in all cases except THF, but systematically gives a shorter minimum interaction distance than does the D0.7 model, which already gave an interaction distance shorter than that obtained from quantum mechanical calculation: this underestimation of gas phase water interaction distances has previously been shown to be a requirement for the accurate reproduction of condensed-phase properties, [28] and could also be improved by the inclusion of appropriate NBFIX parameters. The major contribution to the improvement shown by the D0.85 model in reproducing the interaction energies comes from the linear ethers, again suggesting that by removing some of the constraints imposed during the paramtrization of the D0.7 model, an improved model has been obtained. Despite this, however, all of the linear ethers still perform worse than either THF or THP, providing evidence for the compromise in accuracy necessary to achieve parameter transferability.
Another area in which the D0.7 model outperforms the D0.85 model is in the reproduction of gas phase dipole moments (Table 4), where the D0.85 generally provides an overestimate. In the calculation of the relative energies of the minimum energy conformations (Table 4), however, the D0.85 model generally gives a better reproduction of the ab initio calculated values.
Test Compounds
To test the transferability of the obtained parameters, simulations were performed on another four ether molecules: methyl propyl ether (MPE); methyl butyl ether (MBE); ethyl propyl ether (EPE), and 2-(R)-methyl tetrahydrofuran (MTHF) (Figure 1). For the linear ethers, all parameters used in these simulations were taken directly from those developed in the steps described above, with only minor modifications to the C charges as required to maintain charge neutrality in the molecules. For example, to construct a model for methyl propyl ether (MPE) (structure G, Figure 1), an additional CH2 group was inserted between the existing CH2 and CH3 groups of methyl ethyl ether (MEE) (structure D, Figure 1). The H atoms in this new CH2 group were assigned the standard Hb charge of 0.060 (Table S1) with the C atom charge then determined so as to ensure a total molecular charge of 0: in this case resulting in a C atom charge of -0.120, rather than the standard Cb charge of -0.004 (Table S1). For MTHF, the situation was more complicated. In the development of the original Drude force field model for ethers, all C atoms within MTHF were treated as being equivalent, (Figure 2) an approach that is not consistent with standard CHARMM atom typing, which would require the C atom in the 2 position of MTHF (bonded to the methyl group: Cd in Figure 2 and henceforth termed C1) to have a different atom type to the other ring C atoms. In this case it was decided that new parameters should be included to represent more accurately the atom types present in the model. Accordingly, two new atom types were introduced, CD315B to represent C1 and HDA1R5 to represent H1, the H atom bonded to C1. New parameters introduced for angle and dihedral terms were taken from the previous work on ethers, [2] with Lennard-Jones parameters taken directly from the equivalent alkane atoms. [16] All bonding parameters were identical to those used in the previous work, with electrostatic parameters taken directly from those developed in the steps above for THF C atoms. The resulting parameters are detailed in Table S2 of the supplementary material. Following construction of the models, for each molecule 10 liquid phase molecular dynamics simulations were performed at 298.15 K, following the protocol outlined above, from which averages and standard deviations of the Vm and ΔHvap were obtained. Reasonable reproduction of experimental values for these quantities was considered to signify acceptable levels of transferability for the derived parameters, indicating that they are applicable across a range of ether molecules.
Fig. 2.
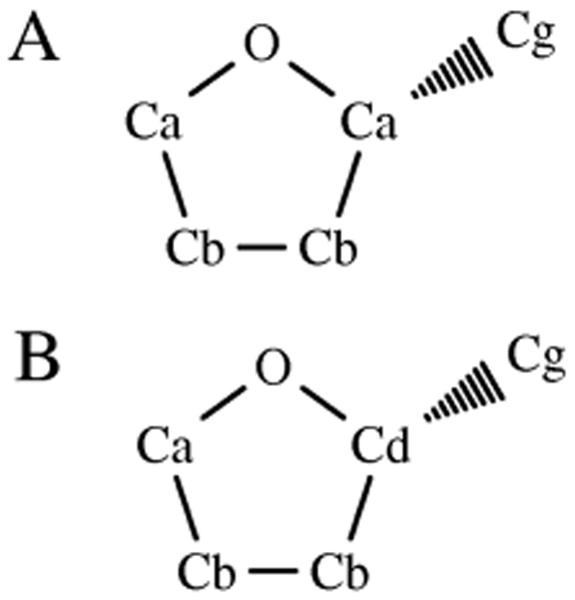
Atom typing in 2-(R)-methyltetrahydrofuran: (A) Original atom typing used in previous work [2]; (B) Revised atom typing used in this work
The results of the calculations of liquid phase properties are shown in Table 5. For the linear ethers, the values of Vm and ΔHvap calculated with the D0.85 model are comparable to those obtained with the original ether model and agreement with experimental data is generally good. The largest improvement occurs for MTHF, which now shows deviations from the experimental values within the target of 2% for the D0.85 model: it had been significantly outside the target range using the D0.7 model. Most of this improvement can be attributed to the change in the parameters for atoms C1 and H1. Simulations performed using the original methodology (ie C1, H1 atom types the same as other C and H atoms in the THF ring) but the newly calculated electrostatic parameters gave ΔHvap = 8.74 ± 0.03 kcal/mol (% err = 7.5 %) and Vm = 163.19 ± 0.5 Å (% err = -2.1%), providing a vivid illustration of the importance of accurate atom type selection in the construction of new compounds for inclusion within a force field.
Table 5.
Pure solvent properties of test compounds (MPE = methyl propyl ether, MBE = methyl butyl ether, EPE = ethyl propyl ether, MTHF = 2-methyl tetrahydrofuran). (Vm in Å3, ΔHvap in kcal/mol). a Experimental data from [38]. b Experimental data from [33]. c Experimental data from [32]. d Experimental data from [39]
| exper | D0.7 | % err | D0.85 | % err | |
|---|---|---|---|---|---|
| Molecular Volumes | |||||
| MPE | 171.3a | 172.8 ± 1.1 | 0.9 | 174.5 ± 0.9 | 1.9 |
| MBE | 198.0a | 199.0 ± 0.7 | 0.5 | 201.0 ± 0.9 | 1.5 |
| EPE | 200.3a | 199.6 ± 0.9 | -0.3 | 204.4 ± 0.8 | 2.0 |
| MTHF | 166.7b | 162.5 ± 0.4 | -2.5 | 167.5 ± 0.6 | 0.6 |
| Heats of Vaporization | |||||
| MPE | 6.60c | 6.60 ± 0.12 | 0.0 | 6.46 ± 0.03 | -2.1 |
| MBE | 7.74c | 7.75 ± 0.12 | 0.1 | 7.57 ± 0.04 | 2.2 |
| EPE | 7.51c | 7.80 ± 0.12 | 3.8 | 7.33 ± 0.04 | -2.4 |
| MTHF | 8.13d | 8.82 ± 0.05 | 8.5 | 8.08 ± 0.03 | -0.6 |
Polarizability Scaling Factor 0.9
As a final test of the impact of polarizability scaling on the calculated dielectric constant, a series of simulations were performed using parameter sets in which atomic polarizabilities were scaled by 0.9 rather than 0.85. In this case no additional charge fitting was performed. Rather, the models obtained for THF and THP via the procedures described above had their atomic polarizabilities rescaled to values of 0.9 rather than 0.85. The charges within the corresponding models were unaltered, which would be expected to have a negative impact on the dipole moments of the model compounds [3] (since the charge on the Drude particle depends on the atomic polarizability), and all Lennard-Jones parameters were also unaltered. With the new models in place, simulations were performed as described above to evaluate liquid phase properties (Table 6). For THP, the new parameters resulted in a value of ΔHvap with an error outside of the 2% range considered acceptable, while for THF, the new parameters resulted in Vm and ΔHvap values within the acceptable range. For both molecules, further calculations were performed to evaluate the dielectric constants, which were, in both cases, in good agreement with the experimental values: comparison of the values calculated using the D0.9 model with the equivalent values obtained using the D0.7 and D0.85 models provides a good illustration of the sensitivity of the calculated dielectric constant to the magnitude of the atomic polarizabilities, and supports the assumption that increasing the atomic polarizability increases the calculated dielectric constant.
Table 6.
Liquid phase properties calculated using a Drude polarizable force field with polarizability scaling factors of 0.7, 0.85 and 0.9 (Vm in Å3, ΔHvap in kcal/mol). Experimental data from [32]
| T/K | exper | D0.7 | % err | D0.85 | % err | D0.9 | % err | |
|---|---|---|---|---|---|---|---|---|
| Tetrahydrofuran (THF) | ||||||||
| Vm | 298.15 | 135.6 | 134.0 ± 0.8 | -1.2 | 134.8 ± 0.4 | -0.6 | 134.7 ± 0.6 | -0.6 |
| ΔHvap | 298.15 | 7.65 | 7.80 ± 0.06 | 2.0 | 7.69 ± 0.03 | 0.9 | 7.58 ± 0.03 | -0.9 |
| ε | 298.15 | 7.42 | 6.80 ± 0.78 | -8.4 | 7.13 ± 0.06 | -3.9 | 7.49 ± 0.13 | 0.9 |
| Tetrahydropyran (THP) | ||||||||
| Vm | 298.15 | 162.3 | 165.3 ± 0.3 | 1.8 | 163.8 ± 0.8 | 0.9 | 163.8 ± 0.8 | 0.9 |
| ΔHvap | 298.15 | 8.26 | 8.27 ± 0.02 | 0.1 | 8.41 ± 0.04 | 1.8 | 8.45 ± 0.04 | 2.3 |
| ε | 298.15 | 5.54 | 5.03 ± 0.20 | -9.2 | 5.39 ± 0.10 | -2.7 | 5.48 ± 0.09 | -1.1 |
Summary
A Drude polarizable force field model for linear and cyclic ethers has been parameterized based on the molecules THF, THP, DEE, DMOE, DME and MEE. Where a previous Drude polarizable force field model for the ethers was developed by scaling the atomic polarizabilities by a factor of 0.7 in the initial charge fitting procedure and using a uniform Thole scaling factor for all atom pairs, here the atomic polarizabilites were scaled by a factor of 0.85 and atom-based Thole scaling factors were employed. The newly developed parameters, which included additional optimization of the LJ parameters, have been used to calculate a variety of properties of the ether molecules, and have generally been found to give good agreement with experimental and quantum mechanical reference data. While the D0.7 model for ethers systematically underestimated the dielectric constants of these molecules, the D0.85 model corrects for this underestimation. Multiple tests were performed with the D0.85 model to evaluate the intramolecular properties of the ether molecules, giving results very similar to those obtained with the previously published D0.7 model, indicating that no further modification of the bonded parameters is required.
The D0.85 model has also been used to calculate liquid phase properties for a series of ether molecules not considered in the parametrization process: MPE; MBE; EPE, and MTHF. In these cases the new model yields an acceptable reproduction of experimental properties, suggesting that the new model is broadly transferable across the ethers.
A subsequent test with force field models of THF and THP in which atomic polarizabilities were scaled by a factor of 0.90 yielded dielectric constants even higher than those obtained with the D0.85 model which, when taken with the previous results, illustrates the sensitivity of calculated values of the liquid phase dielectric constant to the magnitude of the atomic polarizability.
Supplementary Material
Acknowledgments
The authors would like to thank the National Institutes of Health (NIH) for financial support (GM51501, GM07855) and Dr. Igor V. Vorobyov for helpful discussions and access to input scripts and results from previous simulations.
References
- 1.Drude P. The Theory of Optics. Green, New York: 1902. [Google Scholar]
- 2.Vorobyov I, Anisimov VM, Greene S, Venable RM, Moser A, Pastor RW, MacKerell AD., Jr J Chem Theory Comput. 2007;3:1120–1133. doi: 10.1021/ct600350s. [DOI] [PubMed] [Google Scholar]
- 3.Anisimov VM, Lamoureux G, Vorobyov IV, Huang N, Roux B, MacKerell AD., Jr J Chem Theory Comput. 2005;1:153–168. doi: 10.1021/ct049930p. [DOI] [PubMed] [Google Scholar]
- 4.Tu Y, Laaksonen A. Chem Phys Lett. 2000;329:283–288. [Google Scholar]
- 5.in het Panhuis M, Popelier PLA, Munn RW, Ángyán JG. J Chem Phys. 2001;114:7951–7961. [Google Scholar]
- 6.Lamoureux G, MacKerell AD, Jr, Roux B. J Chem Phys. 2003;119:5185–5197. doi: 10.1063/5.0019987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaminski GA, Stern HA, Berne BJ, Friesner RA. J Phys Chem A. 2004;108:621–627. [Google Scholar]
- 8.Giese TJ, York DM. J Chem Phys. 2004;120:9903–9906. doi: 10.1063/1.1756583. [DOI] [PubMed] [Google Scholar]
- 9.Schropp B, Tavan P. J Phys Chem B. 2008;112:6233–6240. doi: 10.1021/jp0757356. [DOI] [PubMed] [Google Scholar]
- 10.Morita A. J Comput Chem. 2002;23:1466–1471. doi: 10.1002/jcc.10169. [DOI] [PubMed] [Google Scholar]
- 11.Botek E, Giribet C, de Azúa MR, Negri RM, Bernik D. J Phys Chem A. 2008;112:6992–6998. doi: 10.1021/jp711620m. [DOI] [PubMed] [Google Scholar]
- 12.Lamoureux G, Harder E, Vorobyov IV, Roux B, MacKerell AD., Jr Chem Phys Lett. 2006;418:245–249. [Google Scholar]
- 13.Lopes PEM, Lamoureux G, MacKerell AD., Jr J Comput Chem. 2009;30:1821–1838. doi: 10.1002/jcc.21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morita A, Kato S. J Chem Phys. 1999;110:11987–11998. [Google Scholar]
- 15.Harder E, Anisimov VM, Whitfield T, MacKerell AD, Jr, Roux B. J Phys Chem B. 2008;112:3509–3521. doi: 10.1021/jp709729d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vorobyov IV, Anisimov VM, MacKerell AD., Jr J Phys Chem B. 2005;109:18988–18999. doi: 10.1021/jp053182y. [DOI] [PubMed] [Google Scholar]
- 17.Noskov SY, Lamoureux G, Roux B. J Phys Chem B. 2005;109:6705–6713. doi: 10.1021/jp045438q. [DOI] [PubMed] [Google Scholar]
- 18.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Corss JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision D.01. Gaussian Inc; Wallingford, CT: 2004. [Google Scholar]
- 19.Brooks BR, Brooks CL, III, MacKerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacKerell AD, Jr, Brooks B, Brooks CL, III, Nilsson L, Roux B, Won Y, Karplus M. CHARMM: The Energy Function and Its Parameterization with an Overview of the program. In: Schleyer PvR, Allinger NL, Clark T, Gasteiger J, Kollman PA, Schaefer HF, III, Schreiner PR., editors. Encyclopedia of Computational Chemistry. Vol. 1. John Wiley and Sons; Chichester: 1998. p. p271. [Google Scholar]
- 21.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 22.Harder E, Anisimov VM, Vorobyov IV, Lopes PEM, Noskov SY, MacKerell AD, Jr, Roux B. J Chem Theory Comput. 2006;2:1587–1597. doi: 10.1021/ct600180x. [DOI] [PubMed] [Google Scholar]
- 23.Miller KJ. J Am Chem Soc. 1990;112:8533–8542. [Google Scholar]
- 24.Dixon RW, Kollman PA. J Comput Chem. 1997;18:1632–1646. [Google Scholar]
- 25.Kollman P. Chem Rev. 1993;93:2395–2417. [Google Scholar]
- 26.Deng Y, Roux B. J Phys Chem B. 2004;108:16567–16576. [Google Scholar]
- 27.Lagüe P, Pastor RW, Brooks BR. J Phys Chem B. 2004;108:363–368. [Google Scholar]
- 28.MacKerell AD., Jr J Comput Chem. 2004;25:1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 29.Anisimov VM, Vorobyov IV, Roux B, MacKerell AD., Jr J Chem Theory Comput. 2007;3:1927–1946. doi: 10.1021/ct700100a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis JE, Warren GL, Patel S. J Phys Chem B. 2008;112:8298–8310. doi: 10.1021/jp8003129. [DOI] [PubMed] [Google Scholar]
- 31.Rick SW, Berne BJ. J Am Chem Soc. 1996;118:672–679. [Google Scholar]
- 32.Lide DR, editor. CRC Handbook Chemistry and Physics. 84th. CRC Press; Boca Raton: 2003. [Google Scholar]
- 33.Poling BE, Prausnitz JM, O'Connell JP. The properties of gases and liquids. 5th. McGraw-Hill; New York: 2001. [Google Scholar]
- 34.Wu J, Liu Z, Bi S, Meng X. J Chem Eng Data. 2003;48:426–429. [Google Scholar]
- 35.Liu ZY, Chen ZC. Chem Eng J Biochem Eng J. 1995;59:127–132. [Google Scholar]
- 36.Kelly CP, Cramer CJ, Truhlar DG. J Chem Theory Comput. 2005;1:1133–1152. doi: 10.1021/ct050164b. [DOI] [PubMed] [Google Scholar]
- 37.Rizzo RC, Aynechi T, Case DA, Kuntz ID. J Chem Theory Comput. 2006;2:128–139. doi: 10.1021/ct050097l. [DOI] [PubMed] [Google Scholar]
- 38.Obama M, Oodera Y, Kohama N, Yanase T, Saito Y, Kusano K. J Chem Eng Data. 1985;30:1–5. [Google Scholar]
- 39.Chickos JS, Acree WE., Jr J Phys Chem Ref Data. 2003;32:519–878. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.