

Abstract
Nuclear receptors are involved in a myriad of physiological processes, responding to ligands and binding to DNA at sequence-specific cis-regulatory elements. This binding occurs in the context of chromatin, a critical factor in regulating eukaryotic transcription. Recent high-throughput assays have examined nuclear receptor action genome-wide, advancing our understanding of receptor binding to regulatory elements. Here we discuss current knowledge of genome-wide response element occupancy by receptors, and the function of transcription factor networks in regulating nuclear receptor action. We highlight emerging roles for the epigenome, chromatin remodeling, histone modification, histone variants and long-range chromosomal interactions in nuclear receptor binding and receptor-dependent gene regulation. These mechanisms contribute importantly to the action of nuclear receptors in health and disease.
Nuclear receptor biology and epigenetic mechanisms
The nuclear receptor superfamily consists of transcription factors that regulate diverse physiological processes including metabolism, development, reproduction and immune responses. The ligand-induced superfamily of receptors responds to lipophilic molecules including fatty acids, vitamins and steroids such as glucocorticoids, estrogens, androgens and progesterone. Once ligand bound, nuclear receptors undergo a conformational change and drive target gene regulation through binding as sequence-specific transcription factors to DNA.
In eukaryotic cells, DNA is organized in the nucleus into higher order structures in the context of nucleoprotein complexes to form chromatin. While this organization facilitates compaction of substantial DNA quantities into the three-dimensional nuclear environment, the process was presumed to impede gene expression. However, the epigenome is now emerging as a critical regulator of expression profiles in both physiological and pathological states. The epigenomic landscape, consisting of posttranslational modifications of histone tails, histone variant localization, DNA methylation, chromatin accessibility and higher-order structure, is highly dynamic and thus permissive for complex regulation of eukaryotic gene expression.
Chromatin remodeling as a prerequisite for nuclear receptor binding to DNA
The nucleoprotein complex of DNA and nucleosomes occlude binding sites of DNA-binding proteins. To overcome this, sequence-specific DNA-binding factors including transcription factors and insulators recruit chromatin remodeling enzymes to mediate access to binding sites on DNA [1-3]. Nuclear receptors are a well characterized class of transcription factors capable of inducing chromatin remodeling events de novo to mediate DNA binding. For example, hormonal activation of the glucocorticoid receptor (GR) and the progesterone receptor (PR) have been long known to induce chromatin remodeling at hormone response elements (HRE) along the mouse mammary tumor virus (MMTV) long terminal repeat (LTR) [4,5]. For GR, this chromatin re-programming event results in remodeling of positioned nucleosomes to form accessible chromatin with increased susceptibility to cleavage by the nuclease DNaseI [5-7]. Indeed, this remodeling event at MMTV is dependent on GR forming a complex with the ATP-dependent brahma-related gene 1 (Brg1) subunit of the Swi/Snf (SWItch/Sucrose NonFermentable) chromatin remodeler [1]. Furthermore, a recent study shows that GR re-programs chromatin at binding sites around the loci of a number of GR responsive genes, with a subset of these sites requiring Brg1 [8]. It is currently unclear whether DNA-binding factors such as GR actively recruit or stabilize chromatin remodelers to target sites. Interestingly, chromatin remodelers contain multiple histone-binding motifs which might direct their targeting to genomic loci independent of DNA-binding factors, while association with binding factors might facilitate stabilization and activation of remodeling activity. The temporal order of factor recruitment to mediate transcriptional regulation is however currently unclear but is clearly promoter specific [9].
Nuclear receptors have also been shown to bind preset regions of accessible chromatin. Receptor binding to these pre-programmed sites of accessible chromatin has been observed for the estrogen receptor (ER) at the cyclin D1 gene [10], and for GR at a number of hormone responsive genes, including the tyrosine aminotransferase gene and chemokine genes [8,11]. The activity of nuclear receptors at pre-programmed sites contradicts their activity as pioneer factors in remodeling chromatin, and rather suggests that nuclear receptors act cooperatively through assisted loading or indirect DNA binding with other factors on chromatin to mediate transcriptional regulation. Understanding the repertoire of nuclear receptor interactions with transcription factors and co-factors will clearly be important for elucidating novel mechanisms in the pathophysiological processes of nuclear receptor action.
Genome-scale analysis of transcription factor binding
Until recently, the study of nuclear receptor action was limited to promoters of well characterized target genes. A number of key technological advances allowed interrogation of nuclear receptor binding sites at a global scale. Chromatin immunoprecipitation (ChIP) hybridization to tiled oligonucleotide microarrays (ChIP-chip) allows global analysis of chromatin associated proteins including transcription factors and modified histones. However, the spatial resolution and genome coverage per chip remains relatively low and requires multiple arrays to encompass genomes of higher eukaryotes. More recently, ChIP coupled with ultra high-throughput sequencing (ChIP-Seq) has been developed, utilizing a sequencing approach of ChIP DNA that is mapped to annotated genomes to determine localization. This approach is advantageous in that it possesses inherent genome-wide capabilities while offering single base-pair resolution with relatively high cost-effective data generation [12]. The application of genome-scale studies to nuclear receptor biology by ChIP-chip, and more recently, ChIP-Seq have been critical in advancing our understanding of the genomic actions of nuclear receptors.
Nuclear receptor binding to the genome
Early chromosome-wide ChIP-chip studies for ER and the androgen receptor (AR) demonstrated that receptors can bind distal (up to 200 kb) to transcription start sites (TSS) [13,14]. Indeed, global and genome-scale analyses of receptor binding for ER, GR and the peroxisome proliferator-activated receptor γ (PPARγ) by ChIP-chip and ChIP-Seq showed that the majority (>60%) of receptor binding sites occur distally from promoters, in intergenic and intronic regions [15-19]. This surprising finding contradicts the promoter proximal action of nuclear receptors on classical hormone responsive genes, and posits that nuclear receptors frequently act as long-range enhancers rather than classic transcription factors. This topological distribution however is not unique to nuclear receptors. ChIP-Seq for the insulators CCCTC-binding factor (CTCF) and neuron-restrictive silencer factor (NRSF), and the signal transducer and activator of transcription protein 1 (STAT1) also show that >70% of binding sites are distributed in intergenic (>40%) and intronic (>20%) regions distal from promoters [12,20-22].
A major difficulty arising from the distal distribution of nuclear receptor binding sites is the assignment of binding events to target gene regulation. Genome-wide expression profiling shows that nuclear receptors upregulate and downregulate hundreds of genes [8,17,23,24]. Using bioinformatic approaches, analysis of global GR binding in A549 cells using a custom microarray tiled around GR regulated genes revealed that the majority (88%) of GR binding correlated with gene regulation [15]. Similarly, chromosome-wide ChIP-chip for ER in MCF-7 cells showed that 92% of ER regulated genes on these chromosomes had ER binding sites within 200 kb [13]. Genome-wide ER ChIP-chip and ChIP-Seq in MCF-7 cells indeed showed a correlation of binding events, occurring within 50 kb of TSS, to ER-regulated genes. Interestingly, binding was biased towards ER-upregulated genes (89%), compared to ER-downregulated genes (47%) [16,17]. Concurrently, ChIP-Seq for PPARγ demonstrated a bias towards genes upregulated during adipogenesis (53%), with highly induced genes associated with a greater number of binding sites [18]. However, relying on bioinformatics to correlate binding to gene regulation raises a number of critical issues. Primarily, such approaches assume that binding events located up- or downstream and/or occurring most proximal to regulated genes are involved in the receptor-mediated response. Furthermore, correlating binding sites to gene regulation may introduce bias by selecting for sites that are relatively promoter proximal, thereby not addressing distal binding sites in gene regulation. Despite the likelihood that relative to the TSS of regulated genes, proximal binding events are involved in the receptor-mediated response, discerning specific binding events to target gene regulation is problematic in vivo and particularly genome-wide. Incorporating data on RNA polymerase II loading and modification of histones such as H3 acetylation at binding sites and regulated genes might improve the correlation of binding to regulation [14,25]. Furthermore, novel approaches to targeted mutagenesis using engineered zinc finger nucleases can be utilized to examine the role of specific nuclear receptor cis elements [26]. The integration of nuclear receptor binding with mutagenic analysis and recruitment of cofactors has the potential to clarify our current understanding of genomic location of receptor binding to target gene regulation.
Profiling nuclear receptor response elements genome-wide
While nuclear receptors regulate hundreds of genes, genome-wide receptor binding analysis reveals that the number of binding sites is an order of magnitude greater than genes regulated by hormone. ChIP-chip and ChIP-Seq respectively identified >3600 and >10,000 ER binding sites in MCF-7 cells, [16,17]. ChIP-chip and ChIp-Seq analyses for PPARγ in differentiated 3T3-L1 cells identified >5200 and >6900 binding sites respectively, at 10 and 6 days following differentiation. Aside from treatment, the differences between number of binding sites identified between ChIP-chip and ChIP-Seq may be due to the higher sensitivity of sequencing, but may also relate to the algorithm used to infer binding. Nevertheless, the discordance between the number of binding sites and the number of regulated genes not only further complicates bioinformatic assignment of binding to regulation, but also suggests that gene regulation and binding by nuclear receptors is highly complex.
Although there are more binding sites than regulated genes, receptor binding is known to occur in clusters both proximal and distal to functionally regulate target genes [13,16]. However, many binding sites do not occur in clusters and are greatly distal to TSS, positing that receptors might also bind at non-functional regulatory elements. Evolutionary conservation analysis of ChIP sites suggests that regulatory elements for GR, PPARγ and ER are highly localized around the center of the peak [15-17,19], suggesting a conserved regulatory role. Furthermore, luciferase reporter assays using response elements identified through genome-wide approaches show functionality independent of distance [15,19]. These assays do not confirm that response elements are active in vivo, but together with conservation analysis, suggest their functional significance.
Although binding sites occur in abundance distal to promoters on linear DNA, chromatin is organized into higher-order structures to form chromosomes that are non-randomly positioned in the three-dimensional nucleus (Figure 1a) [27]. This posits that distal elements acting as enhancers might be spatially organized in the nucleus to facilitate gene regulation by single or multiple response elements, proximal or distal, in cis- or in trans- (Figure 1b) [28, 29]. Enhancers may therefore associate physically with promoters of hormonally responsive genes or facilitate recruitment of co-factors and basal transcriptional machinery to transcriptionally active nuclear compartments (Figure 1c) [30]. Interestingly, AR recruits p160 co-activator complex and p300 with RNA polymerase II to the enhancer -4.2 kb of the prostate-specific antigen (PSA) gene [31], while ER bound enhancers recruit coactivator-associated arginine methyltransferase 1 (CARM1) in a hormone-dependent manner genome-wide [32], consistent with the role of enhancers in recruitment of co-factors.
Figure 1.
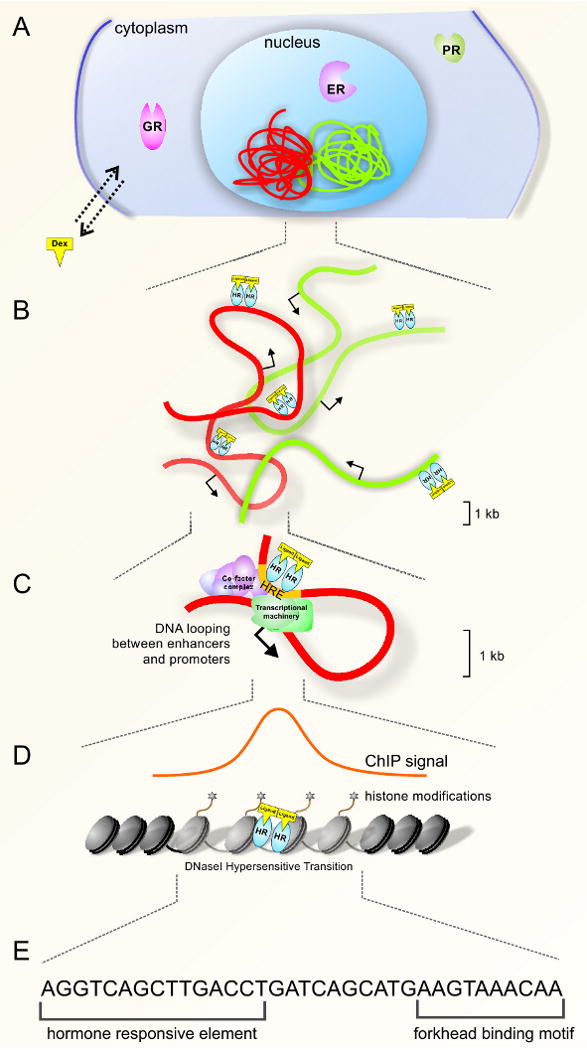
Epigenomic mechanism of nuclear receptor action. (a) Chromatin is organized into higher-order structures as chromosomes (red and green structures) which are non-randomly positioned in the nucleus. Nuclear receptors reside in the cytoplasm or nucleus in the unliganded state. Upon lipophilic ligand binding to receptors, the receptor undergoes a conformational change to mediate DNA binding and transcriptional regulation. (b) Hormone receptors (HR), such as estrogen receptor (ER), glucocorticoid receptor (GR) and progesterone receptor (PR), bind sequences on DNA, predominantly at distal regulatory elements. Arrows indicate direction of transcription. (c) Nuclear receptor binding to distal regulatory elements may involve long-range interactions including looping to facilitate recruitment of enhancers to promoter regions. (d) Analysis of receptor binding by chromatin immunoprecipitation (ChIP) coupled with tiled microarrays (ChIP-chip) or sequencing (ChIP-Seq) permits genome-wide resolution of receptor binding. Binding sites are associated with modifications of histone tails and chromatin accessibility, monitored by transitions in sensitivity to nucleases such as DNaseI. (e) Motif analysis of nuclear receptor binding sites identifies nuclear receptor response elements, but also motifs for transcription factors that may play important roles in nuclear receptor function.
Long-range interactions between regulatory elements can be detected using chromosome conformation capture (3C) [33] and its high-throughput counterparts, 3C-chip (4C) and 3C carbon-copy (5C). Indeed, ER and GR-mediated transcriptional regulation have been shown to involve cis-interactions of promoter-enhancer regions. For example, in a hormone-dependent manner, ER facilitates physical interactions between ER-bound enhancer regions and the promoters of human trefoil factor 1 (TFF-1), nuclear receptor interacting protein (NRIP-1) and B-cell CLL/lymphoma 2 (bcl-2) in MCF-7 cells to mediate transcription [13,34]. In murine mammary epithelial cells and hepatocyte cells, GR mediates transcriptional regulation of the Ciz1 and Lcn2 genes through binding a single response element upstream of Lcn2. 3C detected that the Lcn2 and Ciz1 promoters interact through a 30 kb loop, present prior to and after hormone, facilitating the hormone response [35]. Interestingly, the loop between Lcn2 and Ciz1 is cell-specific, i.e. absent in murine pituitary corticotrophs where the genes are inert to hormone regulation despite the presence of functional GR [35]. This demonstrates that the hormonal response is highly cell-specific, and chromatin loops are likely to be programmed during lineage commitment.
The anatomical distribution of nuclear receptors indicates that some receptors have ubiquitous or widespread expression such as GR and ER, while others such as the pregnane X receptor (PXR) have restricted distribution [36]. Despite ubiquitous expression of a subset of nuclear receptors, the ligand-mediated transcriptional response by these receptors is highly cell-specific with a strikingly limited number of genes regulated between cell lines. For GR, cell-specific binding has been associated with cell-specific hormone regulation in A549 and U2OS cells using low coverage ChIP-chip, tiled 100 kb around TSS of 548 hormone responsive genes [15]. Similarly, ER binding in MCF-7 and U2OS cells on chromosomes 1 and 6 show less than 15% overlap, despite similarities in the distribution profile, that is, binding events are nevertheless distal relative to promoters [37]. It can be reasonably assumed that cell lines derived from the same species do not have dissimilar genetic content. Taking this into account, genome-scanning for consensus sequence motifs reveals that only a small fraction (<5%) of motifs are bound by receptor in vivo in a given cell line. Therefore, the selective binding to a subset of motifs in a given cell, and cell-specific profiles of nuclear receptor occupancy mediating transcriptional regulation, begs the question of how binding is orchestrated.
Binding sequence motifs reveal clues to receptor biology
Nuclear receptors interact with a number of sequence specific transcription factors to up- or downregulate transcription. Known interactions involve activator protein-1 (AP-1), forkhead proteins, CCAAT/enhancer-binding proteins (C/EBP), octamer transcription factors (Oct) and nuclear factor-kappa B (NF-kB). Many of these interactions are cell- and gene-specific as inferred by promoter studies of well characterized genes. Datasets from genome-wide ChIP analysis, enrichment for sequence motifs by de novo discovery, or comparison to databases of known motif weight matrices (Transfac or Jaspar), can potentially reveal biologically important or novel interactions of DNA-binding factors with nuclear receptors (Figure 1e). Indeed, using de novo motif discovery 500bp around GR bound elements from ChIP-chip data, enrichment of the motifs for AP-1, ETS, Sp1, C/EBP and forkhead proteins were found in addition to the GR response elements (GRE) [15]. This is in agreement with reports that GR acts at composite elements, consisting of GRE and non-receptor DNA binding factor motifs [38], or tether to mediate transcriptional interference [39]. Genome-wide approaches can therefore identify prevalent and novel nuclear receptors interactions with other sequence specific DNA-binding factors.
During adipogenesis using an in vitro model in the 3T3-L1 preadipocyte cell line, motif analysis reveals that PPARγ binding sites are enriched with C/EBP and DR1, the response element to which PPARγ:RXR heterodimers bind [18,19]. In agreement with this, genome-scale analysis of C/EBP binding in adipogenesis showed that more than 60% of PPARγ binding occurs in close proximity to C/EBPα, biased toward genes upregulated during adipogenesis. C/EBPα and C/EBPβ are required for PPARγ-dependent gene expression, and interestingly, C/EBPβ expression precedes C/EBPα and PPARγ expression in adipogenesis and might act as a pioneer protein to facilitate PPARγ binding. The co-operativity between PPARγ and C/EBP is therefore an important mechanism for adipogenesis, forming a transcriptional network for spatio-temporal control of gene expression [19].
In MCF-7 cells, ER binding is associated with the forkhead motif, as well as AP-1, Oct, C/EBP and SP-1 motifs [13,16,17]. Prior to hormone binding ER, FOXA1 was found at over 50% of ER sites, proposed to act as a pioneer factor to facilitate recruitment of ER to chromatin upon hormone binding[13,40]. In agreement with this hypothesis, knock-down of FOXA1 decreased ER binding and ER-induced transcriptional responses [13]. Interestingly, in U2OS cells where only 10% of estrogen responsive genes overlap with MCF-7 cells, FOXA1 is not expressed and the forkhead motif was not enriched at ER bound elements. This suggests that cell-specific nuclear receptor-transcription factor interactions play a role in cell-specific binding of nuclear receptors [37].
Nuclease accessibility identifies regulatory regions
Protein interactions with nuclear receptors may direct cell-specific binding to mediate specific transcriptional profiles. However, transcription factors such as FOXA1, like nuclear receptors, only occupy a small fraction of consensus motifs; therefore, this cannot fully explain a cell-specific binding mechanism. It was recently demonstrated that GR binding sites identified by global ChIP-chip analysis are invariably associated with chromatin accessibility, as monitored by hypersensitivity to DNaseI coupled with high-throughput qPCR (Figure 1d) [8, 41]. DNaseI hypersensitive sites (DHS) identify regulatory elements such as promoters, enhancers, insulators, silencers and locus control regions [42-44]. Indeed, GR binding correlated with receptor-dependent re-programming of chromatin to mediate formation of accessible chromatin regions. Interestingly, GR was also found to occupy a large number of pre-programmed DHS sites accessible prior to and following hormone treatment [8]. Both re-programmed and preprogrammed DHS were correlated with cell-specific GR binding. Compared to a mammary cell line, GR binding at genes inert to GR-mediated regulation in a pituitary corticotroph cell line was ablated, and re-programmed and pre-programmed DHS were absent. This strongly suggests that cells undergo epigenomic programming during lineage commitment to encode cell-specific hormonal responses.
DHS coupled with chip (DNase-chip) and sequencing (DNase-Seq) have provided global and genome-wide analysis of functional regulatory elements in the genome [45-47]. In agreement with identification of regulatory elements by DHS, evolutionary analysis reveals that DHS are highly conserved and thus likely to be important cis-regulatory sites [45,47]. In human primary CD4+ T cells, genome-wide DHS profiling found that regulatory regions cover <3% of the genome, while only 16% of DHS are located in promoter regions [47]. Furthermore, DHS at TSS correlate with expression levels, suggesting that chromatin accessibility is an important determinant of cellular phenotype. Considering the small fraction of nuclear receptor binding events relative to the high abundance of consensus motifs in the genome, and that GR binding is invariably linked to DHS at a limited number of loci, we proposed that chromatin accessibility is a major determinant of nuclear receptor binding, critically contributing to cell-specific binding [8]. Indeed, genome-wide DNaseI ‘footprinting’ by deep-sequencing in yeast revealed that motif-predicted Reb1 binding sites in DHS were protected from nuclease cleavage [48]. This supports the notion that motifs scans do not reveal factor binding in vivo considering Reb1 motifs are abundant throughout the yeast genome. Thus transcription factors most likely bind to accessible chromatin in a sequence-specific manner at functional regulatory elements, imparting DNaseI protection.
Global microarray analysis of DHS in multiple cell lines mapping 1% of the human genome defined by the ENCODE consortium, showed that the majority of ubiquitous DHS are located at promoters, while cell-specific DHS localize at enhancer regions [49]. The perception that cell-specific DHS are enhancers, and nuclear receptors predominantly bind distal to promoters with enhancer-like behavior, further supports the concept that nuclear receptors are associated with accessible chromatin and drive cell-specific binding. Enhancers have thus emerged as important regulatory elements in regulating cell-specific gene expression [50].
Interestingly, the majority of ubiquitous distal regulatory elements were found to be CTCF bound, supporting the cell-type invariant binding profiles of CTCF [51]. CTCF has also been proposed to partition the genome into functional blocks and mediate long-range interactions in cis and in trans [51-53]. This functionality may facilitate the enhancer-like function of nuclear receptors through the proximity of distal elements with promoters. Indeed, computational analysis of CTCF binding with ER action showed CTCF demarcates ER-mediated gene expression into co-regulated blocks [54]. However, as CTCF is largely invariable across cell-types, CTCF is unlikely to mediate cell-specific receptor binding and gene regulation but might divide the genome into functional units, contributing to clustering of nuclear receptor binding sites.
Global analysis of cell-specific enhancers, associated with cell-specific DHS, demonstrated a correlation with post-translational modifications of histone tails including the active marks H3K4me1 and H3K4me2 [49,50]. Recent genome-wide studies of histone modifications in T cells indeed show that distal sites are marked by H3K4me1, H3K4me2, H3K4me3 but also repressive marks H3K9me1, H3K9me2, H3K9me3 and H3K27me3 and the histone variant H2A.Z [20,47]. During adipogenesis, global ChIP-chip analysis found H3K9ac enriched at PPARγ sites and interestingly, the mark increased in >60% of PPARγ bound elements during differentiation, implicating a lineage specific epigenomic mark associated with PPARγ [19]. In the case of ER, FOXA1 was shown to be important for ER recruitment in MCF-7 cells and binds in a cell-specific manner compared to FOXA1 binding in LNCaP cells. These cell-specific FOXA1 binding sites at enhancers were associated with enrichment for H3K4me2, while inactive enhancers are marked by H3K9me2 prior to hormone binding its receptor [32,37,55]. Histone marks may therefore represent a mechanism for cell-specific binding of nuclear receptor to chromatin, in association with cell-specific DNA-binding transcription factors and chromatin accessibility through programming during lineage commitment (Figure 1d).
Concluding remarks
Recent high-throughput advances in assays to interrogate genome biology have led to important discoveries in nuclear receptor action. ChIP coupled with microarray or sequencing allows localization studies for genome-wide nuclear receptor binding sites. Surprisingly, these studies show that the majority of nuclear receptor binding sites are distal to promoters, a characteristic likely to be true for most transcription factors. The spatial organization of receptor response elements in the nucleus may involve the amalgamation of multiple binding sites, distal and proximal, to specialized nuclear compartments or looping mechanisms to bring cis- and trans- enhancer elements in proximity to promoters.
One of the critical questions in molecular biology relates to the development of cell identity. For nuclear receptor biology, this relates to the programming of cell-specific nuclear receptor-mediated gene regulation despite ubiquitous receptor expression. Functional regulatory elements are known to be nuclease hypersensitive, displaying cell-specific topologies. Importantly, nuclear receptors bind to accessible chromatin; thus cell-specific binding events are likely to be driven by cell-specific remodeled chromatin. Evidence has also emerged for the role of cell-specific transcription factors in programming hormone responses, as suggested for ER and FOXA1 in MCF-7 cells, for example. The current studies on global and genome-wide nuclear receptor binding events have clearly made important discoveries in nuclear receptor action. However, we do not yet have a full understanding of the regulatory mechanisms which direct nuclear receptor binding. For instance, FOXA1 acting as a pioneer protein in ER binding, is itself a cell-specific protein. The question arises how a pioneer factor is driven to specific regulatory elements on chromatin; this might require cell-specific histone modifications and cofactor recruitment to response elements for direct binding. Our temporal understanding of the coordinated control of nuclear receptor binding remains unclear, whether cofactors such as chromatin remodelers are pre-bound on chromatin independent of transcription factors, marking active regulatory elements, or require active recruitment to target sites. Nuclear organization plays important roles in gene expression, and a greater understanding of the organization of genome-wide regulatory elements in the cell nucleus might reveal important mechanisms contributing to cell-specific binding and the control of gene expression.
Furthermore, an integrative analysis of genome-scale data with multiple datasets including the proteome, transcriptome and metabolome will allow correlations and identification of novel pathways in physiology and pathology and uncover interactions with other transcription factors, the chromatin landscape and signaling pathways. Indeed, recent genome-wide association studies (GWAS) in breast cancer identified novel susceptibility loci associated with estrogen receptor signaling [56,57]. GWAS-identified loci are often in non-coding regions, potentially harboring enhancer elements critical in disease signaling [58-59]. Integrating such data with genome-scale cis binding and epigenetic maps will be important for novel analysis into pathophysiological mechanisms of disease.
As with new technologies, the relatively high costs of sequencing and lack of consensus of appropriate analytical sequencing tools hinders investigators who wish to pursue genome-wide studies. Nevertheless, with current technology rapidly progressing and cost drastically dropping over the foreseeable future, the increased feasibility for more labs to engage in ultra high-throughput investigations to examine action of nuclear receptors genome-wide will undoubtedly make important contributions to receptor biology.
Acknowledgments
This research was supported, in part, by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. S.C.B was supported, in part, by The Needham Cooper Postgraduate Medicine Scholarship held by Stafford L. Lightman and the Faculty of Medicine, University of Bristol. The authors report no conflicts of interest. The authors would like to thank Karen J. Meaburn and Ofir Hakim for helpful comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fryer CJ, Archer TK. Chromatin remodeling by the glucocorticoid receptor requires the BRG1 complex. Nature. 1998;393:88–91. doi: 10.1038/30032. [DOI] [PubMed] [Google Scholar]
- 2.Gargiulo G, et al. NA-Seq: a discovery tool for the analysis of chromatin structure and dynamics during differentiation. Dev Cell. 2009;16:466–481. doi: 10.1016/j.devcel.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Ishihara K, et al. CTCF-dependent chromatin insulator is linked to epigenetic remodeling. Mol Cell. 2006;23:733–742. doi: 10.1016/j.molcel.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Truss M, et al. Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J. 1995;14:1737–1751. doi: 10.1002/j.1460-2075.1995.tb07163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richard-Foy H, Hager GL. Sequence specific positioning of nucleosomes over the steroid-inducible MMTV promoter. EMBO J. 1987;6:2321–2328. doi: 10.1002/j.1460-2075.1987.tb02507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaret KS, Yamamoto KR. Reversible and persistent changes in chromatin structure accompany activation of a glucocorticoid-dependent enhancer element. Cell. 1984;38:29–38. doi: 10.1016/0092-8674(84)90523-3. [DOI] [PubMed] [Google Scholar]
- 7.Belikov S, et al. Hormone activation induces nucleosome positioning in vivo. EMBO J. 2000;19:1023–1033. doi: 10.1093/emboj/19.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.John S, et al. Interaction of the glucocorticoid receptor with the global chromatin landscape. Mol Cell. 2008;29:611–624. doi: 10.1016/j.molcel.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Cosma MP, et al. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell. 1999;97:299–311. doi: 10.1016/s0092-8674(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 10.Eeckhoute J, et al. A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev. 2006;20:2513–2526. doi: 10.1101/gad.1446006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grange T, et al. Two remote glucocorticoid responsive units interact cooperatively to promote glucocorticoid induction of rat tyrosine aminotransferase gene expression. Nucleic Acids Res. 1989;17:8695–8709. doi: 10.1093/nar/17.21.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 13.Carroll JS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Wang Q, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.So AY, et al. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. 2007;3:e94. doi: 10.1371/journal.pgen.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll JS, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 17.Welboren WJ, et al. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009;28:1418–1428. doi: 10.1038/emboj.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen R, et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lefterova MI, et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008;22:2941–2952. doi: 10.1101/gad.1709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Johnson DS, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 22.Jothi R, et al. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John S, et al. Kinetic complexity of the global response to glucocorticoid receptor action. Endocrinology. 2009;150:1766–1774. doi: 10.1210/en.2008-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DePrimo SE, et al. Transcriptional programs activated by exposure of human prostate cancer cells to androgen. Genome Biol. 2002;3:1–12. doi: 10.1186/gb-2002-3-7-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia L, et al. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS ONE. 2008;3:e3645. doi: 10.1371/journal.pone.0003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geurts AM, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325:433. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takizawa T, et al. The meaning of gene positioning. Cell. 2008;135:9–13. doi: 10.1016/j.cell.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Q, et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci U S A. 2008;105:19199–19204. doi: 10.1073/pnas.0810634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lomvardas S, et al. Interchromosomal interactions and olfactory receptor choice. Cell. 2006;126:403–413. doi: 10.1016/j.cell.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 30.Osborne CS, et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 31.Louie MC, et al. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor-p160 coactivator complex. Proc Natl Acad Sci U S A. 2003;100:2226–2230. doi: 10.1073/pnas.0437824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lupien M, et al. Coactivator function defines the active estrogen receptor alpha cistrome. Mol Cell Biol. 2009;29:3413–3423. doi: 10.1128/MCB.00020-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dekker J, et al. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 34.Perillo B, et al. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 35.Hakim O, et al. Glucocorticoid receptor activation of the Ciz1-Lcn2 locus by long range interactions. J Biol Chem. 2009;284:6048–6052. doi: 10.1074/jbc.C800212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bookout AL, et al. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krum SA, et al. Unique ERalpha cistromes control cell type-specific gene regulation. Mol Endocrinol. 2008;22:2393–2406. doi: 10.1210/me.2008-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diamond MI, et al. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science. 1990;249:1266–1272. doi: 10.1126/science.2119054. [DOI] [PubMed] [Google Scholar]
- 39.Schule R, et al. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell. 1990;62:1217–1226. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- 40.Laganiere J, et al. From the Cover: Location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc Natl Acad Sci U S A. 2005;102:11651–11656. doi: 10.1073/pnas.0505575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dorschner MO, et al. High-throughput localization of functional elements by quantitative chromatin profiling. Nat Methods. 2004;1:219–225. doi: 10.1038/nmeth721. [DOI] [PubMed] [Google Scholar]
- 42.Hager GL. Footprints from deep sequencing. Nat Methods. 2009;6:252–253. doi: 10.1038/nmeth0409-254. [DOI] [PubMed] [Google Scholar]
- 43.Hager GL, et al. Chromatin dynamics and the evolution of alternate promoter states. Chromosome Res. 2006;14:107–116. doi: 10.1007/s10577-006-1030-0. [DOI] [PubMed] [Google Scholar]
- 44.Hager GL, et al. Dynamics of nuclear receptor movement and transcription. Biochim Biophys Acta. 2004;1677:46–51. doi: 10.1016/j.bbaexp.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 45.Sabo PJ, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- 46.Crawford GE, et al. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods. 2006;3:503–509. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyle AP, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hesselberth JR, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xi H, et al. Identification and characterization of cell type-specific and ubiquitous chromatin regulatory structures in the human genome. PLoS Genet. 2007;3:e136. doi: 10.1371/journal.pgen.0030136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim TH, et al. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ling JQ, et al. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312:269–272. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Z, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 54.Chan CS, Song JS. CCCTC-binding factor confines the distal action of estrogen receptor. Cancer Res. 2008;68:9041–9049. doi: 10.1158/0008-5472.CAN-08-2632. [DOI] [PubMed] [Google Scholar]
- 55.Lupien M, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomas G, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat Genet. 2009;41:579–584. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng W, et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet. 2009;41:324–328. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pomerantz MM, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tuupanen S, et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet. 2009;41:885–890. doi: 10.1038/ng.406. [DOI] [PubMed] [Google Scholar]