

The m.3243 A>G point mutation in the mitochondrial-encoded MTTL1 gene is the single most common cause of mitochondrial disease.1 The range of clinical phenotypes is highly variable, from isolated diabetes and deafness to the devastating mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome.
Mitochondria contain multiple copies of mtDNA, and both wild-type and mutated mtDNA molecules can coexist in the same cell, a condition referred to as heteroplasmy. The expression of different clinical phenotypes is presumed to relate to different heteroplasmy levels within affected tissues.2 Being able to predict which patients are likely to develop complications of mitochondrial disease is essential for optimal clinical management. An important question therefore is which of the tissues which are suitable for analysis best reflect the level of mutation in clinically affected tissues, and hence are likely to best predict clinical outcome.
Although the m.3243A>G mutation is readily measurable in blood leukocytes, the level of mutation declines over time.3 Thus even in severely affected patients presenting with MELAS, the level of m.3243A>G mutation may be very low, or even escape detection. Muscle biopsy is the current gold standard for the measurement of heteroplasmy to predict the incidence of specific clinical features. However, although a high heteroplasmy level of the m.3243A>G mutation in muscle increases the probability of developing some of the clinical features seen in this mutation, for example epilepsy and stroke-like episodes, for other clinical features such as myopathy, external ophthalmoplegia, and deafness, this is not the case.4 Furthermore, no published data exist as to the stability of mutation load in muscle over time.
In recent years, several laboratories have investigated screening for the m.3243A>G mutation using a range of noninvasive tissues, including hair follicles, fibroblasts, buccal mucosa, and urinary epithelium.5,6 Of these, urinary epithelium shows the most consistent mutation load within individual patients. For example, in a small cohort of 12 patients studied over a 5-year period, we find that there is negligible change in the mutation load of m.3243A>G in urine (J. Blackwood, unpublished data), possibly reflecting replicative differences between urinary epithelium and blood leukocytes, where the mutation is lost. The main advantage of using urine is the ease of obtaining tissue; a 30 mL sample of urine provides enough DNA for accurate detection and quantitation of the m.3243A>G mutation, avoiding the need for patients to undergo a painful and expensive muscle biopsy. We therefore wanted to investigate whether urine was a useful tissue for assessing disease severity.
We studied 24 adult patients harboring the m.3243A>G mutation recruited via a specialist mitochondrial clinic. In all patients, the initial molecular diagnosis had been made using skeletal muscle DNA obtained at muscle biopsy. In addition, an early morning sample of urine and EDTA-blood sample was obtained and the m.3243A>G percentage heteroplasmy level determined. Skeletal muscle and urine samples were obtained from all 24 patients, with blood samples from 21. The time between the initial diagnosis using muscle DNA and subsequent blood and urine samples was 0–6 years. Clinical features were assessed using the Newcastle Mitochondrial Disease Adult Scale (NMDAS), a validated scale in which 29 clinical features are rated according to a numerical scale to give an overall value indicating disease severity.7
We examined the correlation between mutation load in these three tissues and disease severity as indicated by the total score on the NMDAS (table). We found a very weak correlation between both blood and muscle m.3243A>G mutation load and total NMDAS score (rho = 0.205, p = 0.372 for blood, rho = 0.191, p = 0.372 for muscle). The highest correlation with clinical score was with m.3243A>G mutation load in urinary epithelium (rho = 0.451, p = 0.027). When we extended the analysis to include all our patients in whom m.3243A>G mutation load in urinary epithelium had been obtained (n = 58), we found a similar correlation (rho = 0.327, p = 0.012).
Table Comparison of Newcastle Mitochondrial Disease Adult Scale clinical score with m.3243A>G heteroplasmy level in blood, skeletal muscle, and urinary epithelium
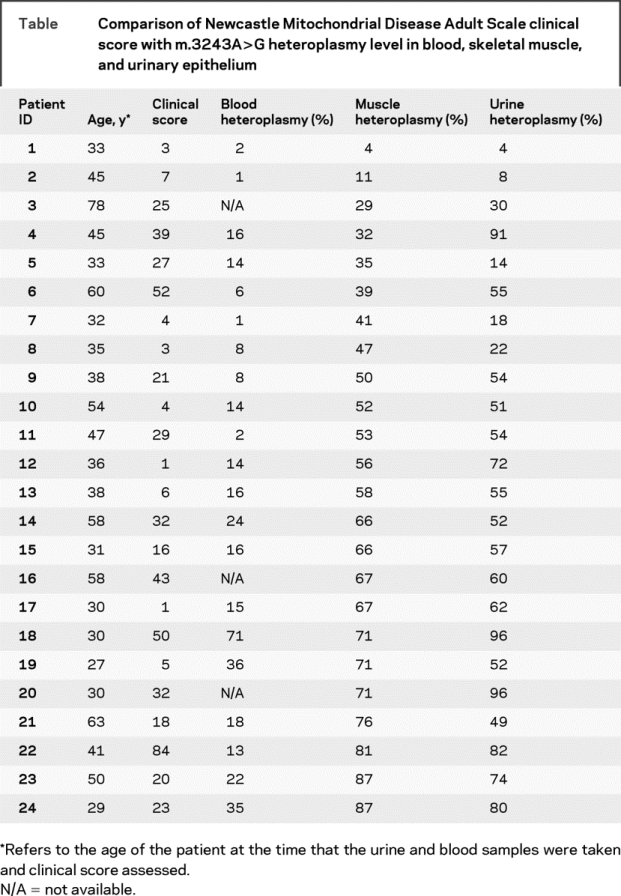
Far from being a compromise in terms of ease of obtaining tissue over diagnostic accuracy, screening of m.3243A>G mutation load in urinary epithelium is a better predictor of outcome than the present gold standard of skeletal muscle tissue. We believe that urinary epithelium completely replaces muscle biopsy as the most suitable tissue for the initial diagnosis in patients suspected of harboring the m.3243A>G mutation. We have adopted this approach in our practice, and believe that if this approach were to become widespread, then countless patients would avoid the need to undergo an unnecessary muscle biopsy.
Funded by a project grant from SPARKS and by the Wellcome Trust. R.G.W. is funded by EUmitocombat.
Disclosure: The authors report no disclosures.
Received August 12, 2008. Accepted in final form September 15, 2008.
Address correspondence and reprint requests to Dr. Roger G. Whittaker, Mitochondrial Research Group, The Medical School, Newcastle University, Framlington Place, Newcastle upon Tyne, NE2 4HH, UK; r.whittaker@ncl.ac.uk
&NA;
- 1.Schaefer AM, McFarland R, Blakely EL, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol 2008;63:35–39. [DOI] [PubMed] [Google Scholar]
- 2.Macmillan C, Lach B, Shoubridge EA. Variable distribution of mutant mitochondrial DNAs (tRNA(Leu[3243])) in tissues of symptomatic relatives with MELAS: the role of mitotic segregation. Neurology 1993;43:1586–1590. [DOI] [PubMed] [Google Scholar]
- 3.Rahman S, Poulton J, Marchington D, et al. Decrease of 3243 A>G mtDNA mutation from blood in MELAS syndrome: a longitudinal study. Am J Hum Genet 2001;68:238–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chinnery PF, Howell N, Lightowlers RN, et al. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain 1997;120:1713–1721. [DOI] [PubMed] [Google Scholar]
- 5.Shanske S, Pancrudo J, Kaufmann P, et al. Varying loads of the mitochondrial DNA A3243G mutation in different tissues: implications for diagnosis. Am J Med Genet A 2004;130A:134–137. [DOI] [PubMed] [Google Scholar]
- 6.McDonnell MT, Schaefer AM, Blakely EL, et al. Noninvasive diagnosis of the 3243A>G mitochondrial DNA mutation using urinary epithelial cells. Eur J Hum Genet 2004;12:778–781. [DOI] [PubMed] [Google Scholar]
- 7.Schaefer AM, Phoenix C, Elson JL, et al. Mitochondrial disease in adults: a scale to monitor progression and treatment. Neurology 2006;66:1932–1934. [DOI] [PubMed] [Google Scholar]