

Abstract
Mitochondrial uncoupling protein 2 (UCP2) is implicated in a wide range of pathophysiological processes, including immunity and diabetes mellitus, but its rapid degradation remains uncharacterized. Using pharmacological proteasome inhibitors, immunoprecipitation, dominant negative ubiqbiquitiuitin mutants, cellular fractionation and siRNA techniques, we demonstrate the involvement of the ubiquitin-proteasome system in the rapid degradation of UCP2. Importantly, we resolve the issue of whether intramitochondrial proteins can be degraded by the cytosolic proteasome by reconstituting a cell-free system that shows rapid proteasome-inhibitor-sensitive UCP2 degradation in isolated, energised mitochondria presented with an ATP regenerating system, ubiquitin and 26S proteasome fractions. These observations provide the first demonstration that a mitochondrial inner membrane protein is degraded by the cytosolic ubiquitin-proteasome system.
Keywords: Mitochondria, Mitochondrial inner membrane, Protein turnover, Ubiquitin-proteasome system, Uncoupling protein, UCP2
Introduction
The proteasome is a cytosolic multicatalytic protein degradation system involved in concerted degradation pathways in the cell, including those for the proteolysis of cytosolic, endoplasmic reticulum (Klausner and Sitia, 1990) and mitochondrial outer membrane proteins (Neutzner et al., 2007). This proteolytic pathway is largely, but not solely, mediated by the regulated recognition of proteins and the addition of polyubiquitin chains, which target proteins for proteasomal destruction (Chau et al., 1989; Murakami et al., 1992). One proteasomal pathway that has not convincingly been shown is the degradation of intramitochondrial proteins that are not directly in contact with the cytosol. To date, no mitochondrial protein export machinery has been identified, raising the question of how intramitochondrial proteins could be accessed by a cytosolic degradation machinery given the ostensible barrier of the outer membrane. Here, we identify uncoupling protein 2 (UCP2) as an example of a mitochondrial inner membrane protein that is degraded by this unusual pathway.
UCP2 regulates the bioenergetics of diverse mammalian tissues including the kidney, spleen, pancreas and central nervous system (Brand and Esteves, 2005; Mattiasson and Sullivan, 2006). UCP2 has a broad distribution and is implicated in a variety of processes, including regulation of reactive oxygen species production (Arsenijevic et al., 2000), food intake (Andrews et al., 2008), insulin secretion (Zhang et al., 2001) and immunity (Arsenijevic et al., 2000) as well as pathologies including atherosclerosis (Blanc et al., 2003), cancer (Derdak et al., 2008), diabetes (Zhang et al., 2001) and neuronal injury (Sullivan et al., 2003). UCP2 levels vary dynamically in response to nutrients and this is achieved by varied expression rates against a background of a very short UCP2 protein half-life of ~1 hour (Rousset et al., 2007; Giardina et al., 2008; Azzu et al., 2008). This rapid turnover is not a general result of mitochondrial inner membrane proteolysis or whole mitochondrial turnover by autophagy, since the adenine nucleotide translocase (ANT) — a related carrier also integral to the mitochondrial inner membrane − is not degraded in the same time period. In contrast to the situation in cells, UCP2 is stable in isolated mitochondria, suggesting that extramitochondrial factors may be involved in the UCP2 degradation pathway (Azzu et al., 2008).
Results
Proteasome inhibitors block UCP2 degradation
To explore the possibility that an extramitochondrial proteolytic pathway is involved in UCP2 degradation, we pharmacologically inhibited the cytosolic 26S proteasome, which is potentially involved in degradation of short-lived proteins.
Fig. 1 (controls) shows the rapid degradation of UCP2 in INS-1E cells following inhibition of protein synthesis by addition of cycloheximide at time zero. Two independent proteasome inhibitor cocktails (PIC)-1 and PIC-2, each containing peptide aldehyde inhibitors and covalent modifiers, blocked UCP2 degradation (Fig. 1A), supporting the hypothesis that the proteasome is involved in UCP2 degradation. Use of proteasome inhibitors did not cause noticeable cell death, and in the absence of cycloheximide resulted in increased UCP2 levels (not shown). To further characterise proteasomal inhibition, we explored the effects of the individual components of the two proteasome inhibitor cocktails, PIC-1 (Fig. 1B) and PIC-2 (Fig. 1C). Additionally, we examined the effects of a vinyl sulfone, a different class of proteasome inhibitor to those found in the cocktails (Fig. 1D). None of the individual inhibitors blocked UCP2 degradation as effectively as the cocktails. However, each of the inhibitors did significantly inhibit UCP2 degradation, and these inhibitors included members of all three classes of inhibitor tested − the peptide aldehydes, the covalent modifiers and the vinyl sulfone. Notably, the effects of the separate inhibitors were additive, approximating very closely to the extent of inhibition by the cocktail. We can speculate that this is because UCP2 can be degraded by all the catalytic activities of the proteasome, and whilst each of the inhibitors partially blocks proteasome activity, together they can inhibit more completely.
Fig. 1.

Proteasome inhibitors block UCP2 degradation in cells. INS-1E cells were preincubated with proteasome inhibitors for 2 hours then treated with 10 μg/ml cycloheximide. Samples were taken at the times shown, separated by SDS-PAGE (1×105 cells/lane) and immunoblotted for UCP2. (A) UCP2 degradation in cells treated with proteasome inhibitor cocktail-1 [PIC-1; containing 10 μM MG132, 10 μM lactacystin (Lact.) and 30 μM PI-1] or proteasome inhibitor cocktail-2 [PIC-2; containing 30 μM ALLN, 5 μM clastolactacystin β-lactone (Clasto.) and 5 μM epoxomicin (Epoxo.)] (n=6). (B-D) UCP2 degradation in cells treated with: (B) 10 μM MG132, 10 μM lactacystin or 30 μM PI-1 (n=3); (C) 30 μM ALLN, 5 μM clastolactacystin β-lactone or 5 μM epoxomicin (n=3); or (D) 5 μM adamantane-acetyl-6-aminohexanoyl-3-leucinyl-3-vinyl-methylsulfone (AdaAhx3L3VS; Ada.; n=3). Values are means ± s.e.m., corrected for loading (β-actin). Statistical significance was determined by repeated measures ANOVA (comparison of matching non-zero time points) with Dunnett's post-hoc testing (*P<0.05, **P<0.01, ***P<0.001). Typical UCP2 immunoblots (molecular mass ~30 kDa) are shown in supplementary material Fig. S1.
Although MG132 and lactacystin may also inhibit mitochondrial Lon protease, epoxomicin does not (Granot et al., 2003), and no other cellular targets of epoxomicin are known (Meng et al., 1999). The observation that all classes of proteasome inhibitor slowed UCP2 degradation reinforces the assertion that the proteasome is involved in the UCP2 degradation pathway [contrary to suggestions made by other authors (Giardina et al., 2008; Rousset et al., 2007)] and diminishes the possibility that cross-reactivity of these inhibitors with some unidentified mitochondrial protease is responsible for inhibition of UCP2 degradation.
Ubiquitin is involved in UCP2 degradation
Since proteasomal degradation is not exclusively mediated by conjugation of polyubiquitin destruction tags to proteins (Murakami et al., 1992), we explored whether UCP2 is ubiquitylated in cells and whether such ubiquitylation affects UCP2 degradation.
Using immunoprecipitation (IP) of UCP2 from cell lysates followed by immunodetection of ubiquitin (Ub), we looked for species representing polyubiquitylated UCP2. We would expect to observe such species in cells treated with a scrambled short interfering (si)RNA (Scr) but less so in those knocked down for UCP2 (KD) and they should be absent from cell lysates pulled down with non-antigen-specific (normal) immunoglobulin. Fig. 2A, lane 1 shows strong signals from species with a range of molecular masses (>30 kDa) that disappear in a UCP2-specific fashion (cf. Fig. 2A, lanes 2-4).
Fig. 2.

Ubiquitin involvement in UCP2 degradation. (A) INS-1E cells were treated with 200 nM scrambled (Scr) or UCP2 (KD) siRNA for 48 hours. Cells were lysed with IP buffer and lysates were incubated for 1 hour with protein-A-conjugated beads and anti-UCP2 or anti-Ub antibodies, or non-antigen-specific (normal) immunoglobulin (N.Ig) at 4°C then pelleted and denatured by boiling in gel loading buffer. Proteins were separated by SDS-PAGE and immunoblotted for Ub, UCP2 or ANT. Non-specific: bands not dependent on specific interactions with UCP2 or ubiquitin. (B) INS-1E cells were transfected with 200 nM scrambled siRNA (Scr) or siRNAs targeted against UCP2 (KD). Scr/UCP2-KD cells used in the immunoprecipitation experiments were immunoblotted for UCP2, ANT and β-actin. Cell loading: 1×105 cells/lane. (C,D) Purified HA-tagged WT-ubiquitin, K48R-ubiquitin and lysine KO-ubiquitin plasmids were transfected at 0.5 μg/ml DNA for 20-24 hours. INS-1E cells were then treated with 10 μg/ml cycloheximide (CHX), harvested at the time points shown and resuspended in gel loading buffer. Proteins were separated using SDS-PAGE (1×105 cells/lane) and immunoblotted for HA (C) or UCP2 (D). Values in D are means ± s.e.m. (n=3), corrected for loading (β-actin). Statistical significance was determined by repeated measures ANOVA (comparison of matching non-zero time points) with Dunnett's post-hoc testing (*P<0.05, **P<0.01, ***P<0.001). Typical UCP2 immunoblots (molecular mass ~30 kDa) are shown.
The reciprocal immunoprecipitation of ubiquitin from cell lysates followed by immunodetection of UCP2 is depicted in Fig. 2A, lanes 5-8, and similarly shows that in the absence of UCP2 in cells (lane 6) or with the use of non-specific antibody for pull-down (lanes 7 and 8), signals representing polyubiquitylated UCP2 are less prominent or absent, and only non-specific band are seen.
To test whether ubiquitylation is specific for UCP2 or more general for mitochondrial carriers, we stripped the nitrocellulose membrane containing lanes 5-8 and reblotted with anti-ANT antibody (Fig. 2A, lanes 9-12). As expected, this resulted in the pull-down of non-specific bands which are also seen in normal immunoglobulin pull-downs (Fig. 2A, lanes 2, 4, 7 and 8), including the heavy and light chain immunoglobulins at ~50 kDa and ~23 kDa, respectively. However, there was no sign of ubiquitylated ANT in these experiments. Therefore, despite its similarity to and much higher abundance than UCP2, ANT is not pulled down by the anti-Ub antibody, and is therefore not measurably ubiquitylated.
To test whether polyubiquitylation of UCP2 is relevant to its degradation (and to probe the nature of the ubiquitin linkages), we transfected INS-1E cells with expression plasmids encoding wild-type (WT) Ub and two dominant negative mutants: a K48R-Ub mutant that is specifically unable to form K48-linked polyubiquitin chains and a lysine knockout (KO) Ub mutant, in which all lysines are mutated to arginines. The K48R mutant should still be able to form all types of polyubiquitin extensions and linkages, except those involving K48, whereas the lysine-knockout mutant should be unable to form linkages through any lysines, such as those through K29 and K33. If they compete with endogenous ubiquitin and modify UCP2 in a manner that prevents formation of polyubiquitin destruction tags, these mutants should extend UCP2 half-life. Since the WT and mutant Ub are HA-tagged, we used anti-HA antibodies to confirm successful expression of the appropriate Ub from the transfected plasmids (Fig. 2C).
Addition of cycloheximide to transfected cells followed by UCP2 immunoblotting analysis revealed that both of the mutant Ub plasmids (K48R-Ub and KO-Ub) delayed UCP2 decay significantly (Fig. 2D), with K48R-Ub extending UCP2 half-life to ~150% of the control value, and KO-Ub to ~175% of control. Although the WT-Ub induced a trend towards faster UCP2 degradation (and shorter UCP2 half-life), the kinetics of degradation were not significantly different from mock-transfected cells. The inhibition caused by these dominant negative mutants was incomplete, possibly because of the presence of endogenous Ub. The effects of K48R-Ub and KO-Ub were not significantly different from each other, suggesting that Ub linkage via K48 residues is the primary chain extension that targets UCP2 to the proteasome, although other linkages cannot be excluded.
Regulation of UCP2 degradation
In addition to inhibition of the ubiquitin-proteasome system, compounds that influence mitochondrial bioenergetic status also affected UCP2 turnover (Fig. 3). All tested compounds that lowered mitochondrial protonmotive force, Δp (Fig. S3C), slowed UCP2 turnover. Such compounds include respiratory inhibitors, which prevent Δp generation (Fig. 3A), and compounds that dissipate Δp, namely the chemical uncoupler carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) and the redox cycling quinone menadione (Fig. 3B), a known inducer of the mitochondrial permeability transition pore (Toninello et al., 2004). Dissipation of the chemical component of Δp (ΔpH) by nigericin had no effect on UCP2 degradation (Fig. 3B), suggesting that ΔpH was not required for UCP2 degradation. Cycloheximide treatment had no significant effect on mitochondrial membrane potential compared with untreated cells (not shown).
Fig. 3.
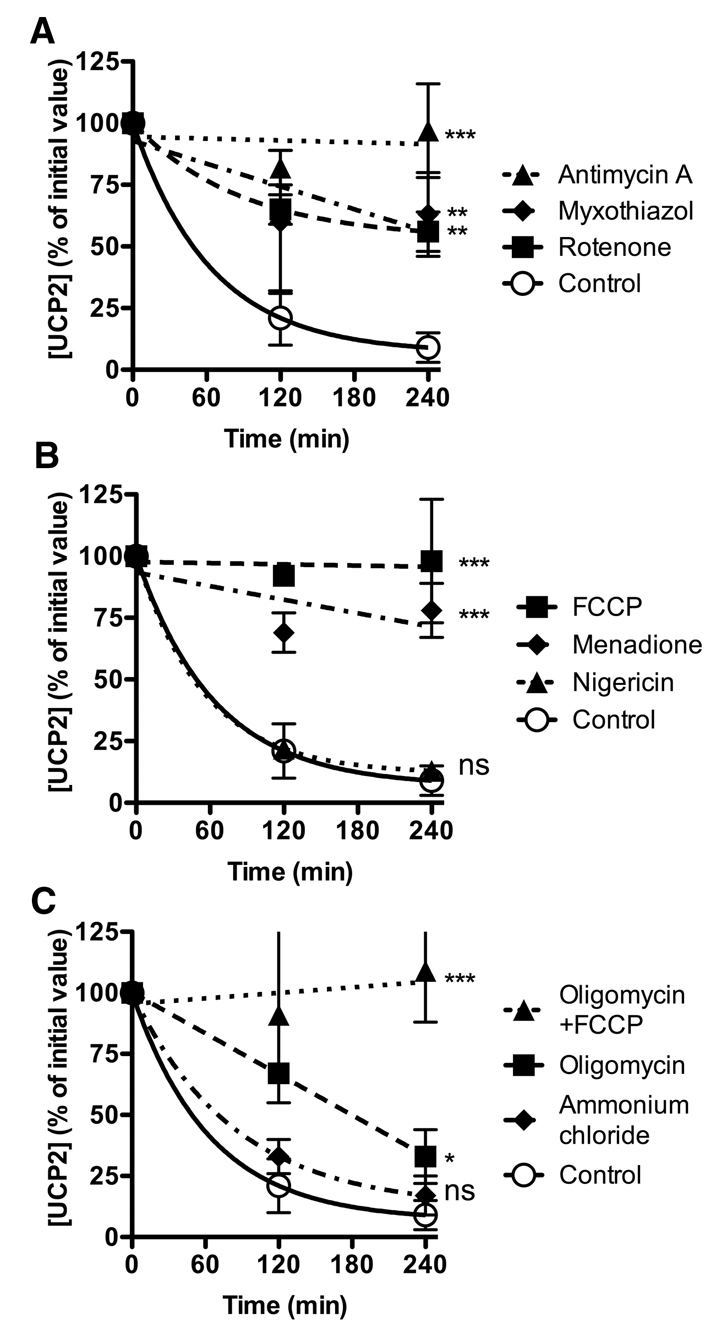
Regulation of UCP2 degradation by mitochondrial bioenergetics. INS-1E cells were treated with the effectors described below for 30 minutes, then treated with 10 μg/ml cycloheximide. Samples were taken at the times shown, separated by SDS-PAGE (1×105 cells/lane), immunoblotted for UCP2 and quantified by densitometry. (A) Inhibition of respiratory chain using 10 μM rotenone (inhibits complex I), 10 μM antimycin A or 10 μM myxothiazol (inhibit Qo and Qi site of complex III, respectively). (B) Treatment with 20 μM FCCP (dissipates Δp), 0.5 μM nigericin (dissipates ΔpH) or 75 μM menadione. (C) Treatment with 1 μg/ml oligomycin (inhibits Fo/F1 ATP synthase) with or without 20 μM FCCP, or lysosomal inhibitor 10 mM NH4Cl. Values are means ± s.e.m. (n=3), corrected for loading (β-actin). Statistical significance was determined by repeated measures ANOVA (comparison of matching non-zero time points) with Dunnett's post-hoc testing (*P<0.05, **P<0.01, ***P<0.001). Typical UCP2 immunoblots (molecular mass ~30 kDa) are shown in supplementary material Fig. S2.
Since dissipation of Δp or its electrical component (membrane potential, ΔΨm) also results in ATP depletion, we tested the effects of the Fo/F1 ATP synthase inhibitor oligomycin, which halts mitochondrial ATP synthesis and lowers cellular ATP but does not depolarise mitochondria (supplementary material Fig. S3C,D). Oligomycin partially inhibited UCP2 degradation (Fig. 3C), suggesting that ATP is needed, as would be expected for ubiquitin-proteasome system function. However, preincubation of cells with both oligomycin and FCCP led to much stronger inhibition of UCP2 degradation (Fig. 3C), suggesting that as well as ATP, Δp maintenance is also important for rapid UCP2 degradation (Fig. 3C). We can speculate that ΔΨm may be required for UCP2 unfolding [as it is for other proteins (Huang et al., 2002; Prakash and Matouschek, 2004)] and extraction from the membrane.
In contrast to ΔΨm and ATP, effects on reactive oxygen species (ROS) did not correlate with inhibition of UCP2 degradation (Fig. 3, supplementary material Fig. S3A,B). Some compounds that inhibited UCP2 degradation raised ROS (rotenone, antimycin A) whereas others had no great effect (oligomycin) or slightly lowered ROS (menadione, FCCP). It has been reported that respiratory inhibitors may stabilise UCP2 in the inner membrane (that is, by decreasing turnover) and it was suggested that this may be through increasing ROS (Giardina et al., 2008). Our results show that respiratory inhibitors affect UCP2 degradation through their effects on ATP and ΔΨm and not through their effects on ROS.
Next we examined whether FK506-binding protein 8 (FKBP8), a protein that has been implicated in tethering the 26S proteasome to mitochondria (Nakagawa et al., 2007), might be important in facilitating proteasome binding to the mitochondrial outer membrane and hence important in UCP2 degradation. Fig. 4A shows that knockdown of FKBP8 by siRNA statistically significantly slowed UCP2 turnover in cells, indicating that FKBP8 may be involved in the UCP2 degradation pathway and that the proteasome may access UCP2 at the interface between the cytosol and the mitochondrial outer membrane.
Fig. 4.
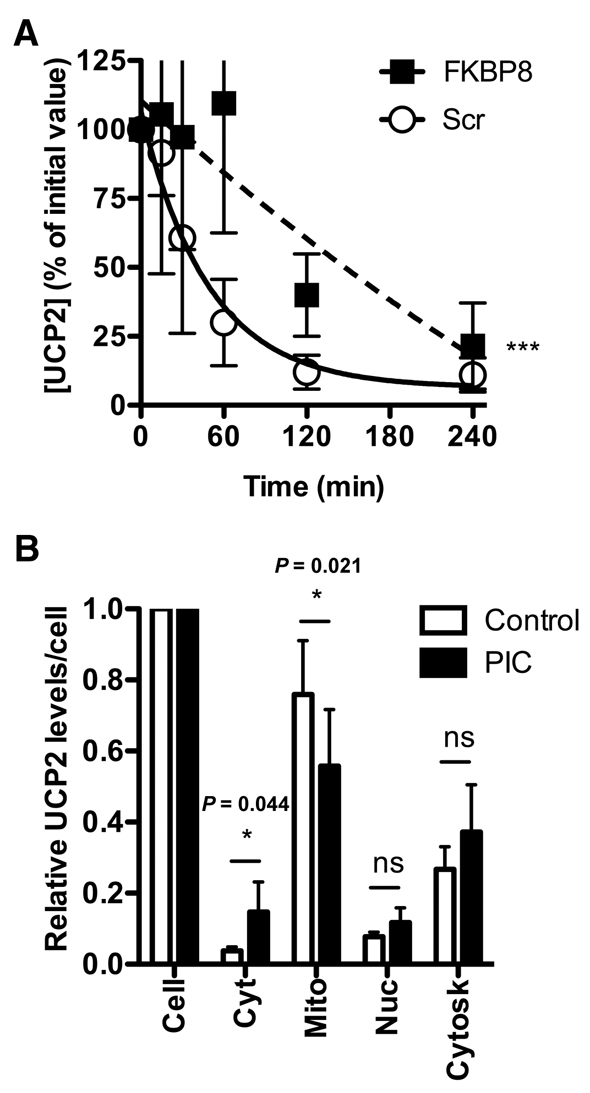
UCP2 attachment and retrotranslocation. (A) INS-1E cells were treated with 200 nM scrambled siRNA or siRNAs targeted against FKBP8 for 48 hours. UCP2 degradation was measured as in Fig. 1. Proteins (1×105 cells/lane) were separated by SDS-PAGE and immunoblotted for UCP2 and β-actin. Values are means ± s.e.m. (n=4), corrected for loading (β-actin). All statistical significances were determined by repeated measures ANOVA (comparison of matching non-zero time points) with Dunnett's post-hoc testing (*P<0.05, **P<0.01, ***P<0.001). (B) INS-1E cells were treated with PIC-1 for 2 hours and fractionated using the Q Proteome cell compartment kit. Proteins were separated by SDS-PAGE and immunoblotted for UCP2. Values are normalised to the cell numbers used to generate the fractions and show means ± s.e.m. (n=3). Statistical significance was determined by Student's t-test. Significant P values are shown.
Fig. 4B shows the results of an experiment designed to test whether UCP2 can be exported from the inner membrane to the cytosol. Cells were incubated with or without proteasome inhibitor cocktail for 2 hours, and then fractionated. In the presence of PIC, a significantly greater proportion of the UCP2 was recovered in the cytosolic and nuclear fractions. This is unlikely to be preimported UCP2 as similar experiments in the presence of cycloheximide also showed cytosolic increases in UCP2 following proteasome inhibition (not shown). This observation suggests that when the proteasome is inhibited, some UCP2 is exported from the inner membrane to the cytosol and de-ubiquitylated, but not further degraded. This reaction may be carried out by the proteasome caps, whose de-ubiquitylation activity remains active in the presence of proteasome inhibitors (Verma et al., 2002), or by de-ubiquitylating enzymes. Similar observations obtained with a polytopic endoplasmic reticulum protein have been interpreted in the same way (Oberdorf et al., 2006).
Reconstitution of UCP2 degradation in vitro
To verify that UCP2 embedded in the mitochondrial inner membrane can be degraded by the cytosolic proteasome, we reconstituted an in vitro system in which components of the ubiquitin-proteasome system were added to isolated INS-1E mitochondria. We have previously reported that UCP2 is very stable in isolated mitochondria in a standard incubation medium (Azzu et al., 2008). Fig. 5A shows that UCP2 remains stable in succinate-energised mitochondria (which maintain high Δp) supplied with an ATP-regenerating system (ATP plus phosphocreatine plus creatine kinase). By contrast, when we added highly purified commercial fractions of 26S proteasome and ubiquitin plus conjugation enzymes, UCP2 was degraded in vitro with very similar kinetics to its degradation in intact cells. The addition of the proteasomal inhibitor cocktail PIC-1 resulted in strong and statistically significant inhibition of UCP2 degradation in vitro, mimicking its effect in cells and strongly suggesting that the reconstituted pathway is similar to the normal cellular pathway.
Fig. 5.
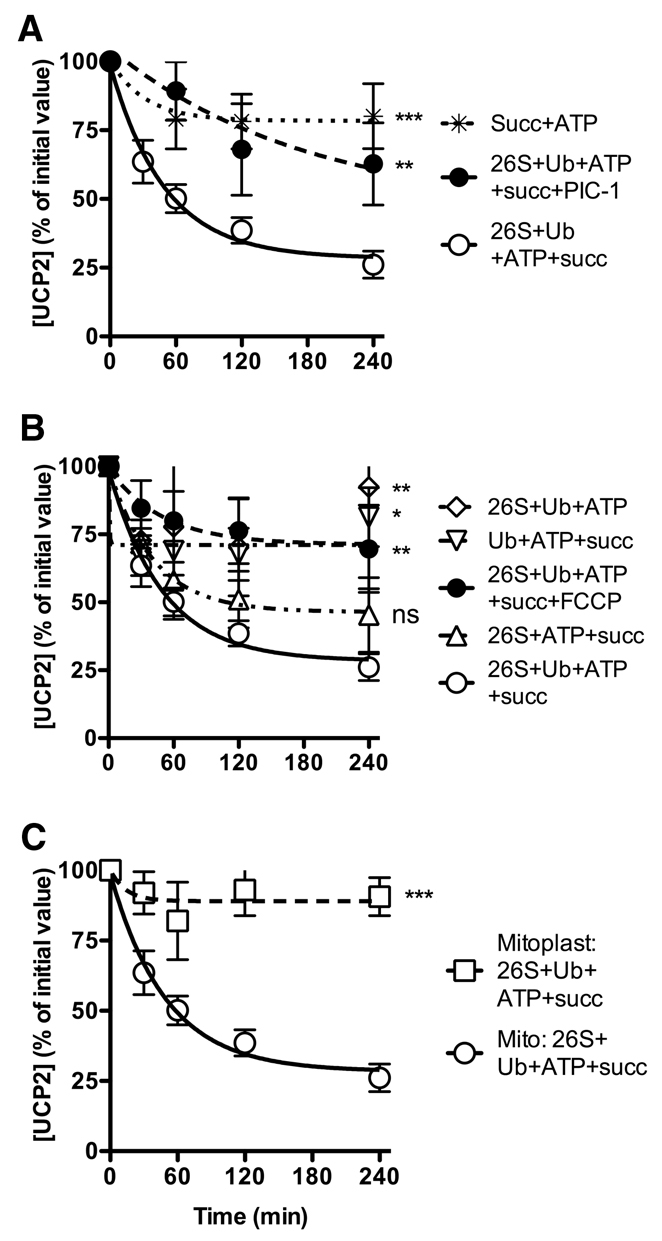
Reconstitution of UCP2 degradation in vitro. Isolated INS-1E mitochondria (A,B) or mitoplasts (C) (240 μg per 260 μl) in sucrose-HEPES buffer (pH 7.4) were incubated at 37°C together with (as indicated) an ATP regeneration system (0.5 mM ATP, 10 mM phosphocreatine and 0.5 μg creatine kinase), ubiquitin mix (70 μg ubiquitin, 1.4 μg fraction 1, 1.4 μg fraction 2), 3.5 μg 26S proteasome fraction, 20 mM succinate, 50 μM PIC-1, and 20 μM FCCP. Aliquots were removed at the time points shown. Proteins (25 μg/lane) were separated by SDS-PAGE and immunoblotted for UCP2. Values are means ± s.e.m. (n=5), corrected for loading (Coomassie-Blue-stained membranes). All statistical significances were determined by repeated measures ANOVA (comparison of matching non-zero time points) with Dunnett's post-hoc testing (*P<0.05, **P<0.01, ***P<0.001). Typical UCP2 immunoblots (molecular mass ~30 kDa) are shown in supplementary material Fig. S4.
As shown in Fig. 5B, omission of the proteasome fraction, the ATP-regenerating system, or succinate (to maintain Δp) greatly inhibited UCP2 degradation in vitro, showing that each is required, as they are in cells. As in cells, addition of FCCP to dissipate Δp also strongly inhibited degradation. Interestingly, although absence of the Ub fraction tended to inhibit degradation, the effect was not statistically significant, suggesting that crude mitochondrial isolates obtained by differential centrifugation may have retained a sufficient Ub pool to allow substantial UCP2 degradation. Consistent with this conclusion, blotting a mitochondrial preparation with anti-Ub antibody gave a strong signal (not shown), which might represent cytosolic ubiquitin contamination.
Partial removal of the mitochondrial outer membrane by preparation of crude mitoplasts abolished UCP2 degradation (Fig. 5C), suggesting that components of the outer membrane or of the intermembrane space − perhaps a proteasome anchor site or a ubiquitin ligase − are required for UCP2 degradation. However, since the mitoplasts were frozen and thawed, and were not coupled, inadequate ΔΨm may be a sufficient explanation.
Discussion
Although the role of the ubiquitin-proteasome system in many cellular processes is well known, its interaction with mitochondria is only just surfacing. However, our finding that UCP2 is rapidly degraded in cells, but is stable in isolated mitochondria (Azzu et al., 2008) led us to consider extramitochondrial factors involved in UCP2 degradation. In the present study, we show that two proteasome inhibitor cocktails are remarkably effective at halting UCP2 degradation in cells, with immunoprecipitation studies indicating that UCP2 is likely to be ubiquitylated.
However, there are currently some hurdles in the analysis of ubiquitin-proteasome system-mitochondrial interactions. Firstly, the existence of mitochondrial proteases that partially resemble the cytosolic 26S proteasome in structure or function (Bayot et al., 2008) and reports indicating the cross reactivity of some proteasome inhibitors with mitochondrial proteases (Granot et al., 2003) weaken conclusions regarding ubiquitin-proteasome system-mediated degradation of intramitochondrial proteins (Margineantu et al., 2007; Sutovsky et al., 2003). Secondly, the prokaryotic origins of mitochondria and the recent discovery of a prokaryotic destruction tag named prokaryotic ubiquitin-like protein (Pup) (Pearce et al., 2008), raises the possibility that observations of proteasome-inhibitor sensitivity can be attributed to cross-reactivity. Thirdly, although mitochondrially encoded proteins have been reported to be found at extramitochondrial locations (Soltys and Gupta, 1999) and a peptide export machinery has been described for mitochondria (Young et al., 2001), the lack of a known protein retrotranslocation machinery from mitochondria to the cytosol makes it difficult to envision how intramitochondrial proteins can become substrates of the cytosolic proteasome.
Since UCP2 is stable in isolated mitochondria (Azzu et al., 2008), we used a reductionist approach to provide evidence that added components of the ubiquitin-proteasome system can induce the degradation of mitochondrially embedded UCP2. We find that efficient UCP2 degradation minimally requires mitochondrial substrate energisation, ATP and purified proteasomal fractions. Additionally, this cell-free system provides important insights into the mechanism by which UCP2 is retrotranslocated from mitochondria. If UCP2 was simply exported from intact mitochondria [say by mitochondrial unfoldases or exportases (Soltys and Gupta, 1999; Young et al., 2001)], the apparent UCP2 half-life should have remained short in vitro in the absence of the 26S proteasome, where mitochondria were pelleted at each assessed time point and the supernatant, which would contain exported UCP2, was discarded. However, the 26S proteasome fraction was required for efficient loss of UCP2 signal, suggesting that the proteasome may be directly involved in extracting UCP2 from mitochondria. Interestingly, preincubation of the proteasome fraction at room temperature prevented UCP2 degradation (not shown), presumably by allowing the dissociation of the proteasome caps from the catalytic subunits, again suggesting that functional 26S proteasomes are required for efficient UCP2 degradation. UCP2 might be directly retrotranslocated (perhaps via contact sites between the inner and outer membranes) by the caps of the proteasome, which can perform this process (Mayer et al., 1998), or retrotranslocation may occur in conjunction with other unfoldases and/or exportases in mitochondria (Soltys and Gupta, 1999).
Although there are reports of mitochondrial protein ubiquitylation (Abu-Farha et al., 2005; Jeon et al., 2007; Margineantu et al., 2007; Radke et al., 2008; Sutovsky et al., 2003), this is the first compelling evidence that a mitochondrial inner membrane protein is degraded by the cytosolic 26S proteasome via the ubiquitin-mediated pathway.
In a process analogous to ER-associated degradation (ERAD), Neutzner et al. proposed a mitochondrial outer membrane-associated degradation (OMMAD) (Neutzner et al., 2007). We now extend this model to involve mitochondrial inner membrane proteins, tentatively suggesting the mechanistic working model shown in Fig. 6. In this hypothetical model, UCP2 is ubiquitylated (inside or outside the mitochondrion) by an unidentified putative E3 ligase and unfolded from the mitochondrial inner membrane by processes which may be ATP or ΔΨm dependent. At the mitochondrial outer membrane, the proteasome, perhaps tethered by FKBP8, recognises polyubiquitylated UCP2 and participates in its extraction from mitochondria in an ATP-dependent fashion, whereby the protein is subsequently degraded by the peptidase activity of the proteasome core. Despite this model's hypothetical nature, we suggest that it provides a useful platform for further investigation,
Fig. 6.

Model of UCP2 degradation via the ubiquitin-proteasome system. At the mitochondrial outer membrane (MOM), the proteasome − tethered by FKBP8 − recognises UCP2 that has been polyubiquitylated by an unidentified putative E3 ligase. The proteasome cap participates in the unfolding and extraction of UCP2 from the mitochondrial inner membrane (MIM) by processes that may be ATP- or ΔΨm-dependent. The cap also catalyses de-ubiquitylation, and the ubiquitin (Ub) is recycled. UCP2 is subsequently degraded by the peptidase activity of the proteasome core.
On a more functional note, we suggest that regulation of UCP2 degradation by the ubiquitin-proteasome system serves to regulate it in line with other proteins. In the pancreatic β-cell, the proteasome is known to regulate proteins involved in the insulin secretion pathway such as ATP-sensitive potassium channels (Yan et al., 2005), voltage-dependent calcium channels (Kawaguchi et al., 2006) and proinsulin levels (Kitiphongspattana et al., 2005). In all these cases, proteasome inhibition leads to decreased glucose-stimulated insulin secretion, suggesting that proteasome dysfunction may be one of the molecular mechanisms of the β-cell impairment seen in type 2 diabetes mellitus. Increased UCP2 levels seen in type 2 diabetes mellitus (Sasahara et al., 2004) may be a direct result of proteasomal dysfunction and decreased degradation as well as increased UCP2 expression.
Materials and Methods
Cell growth and UCP2 degradation
INS-1E cells were grown and maintained as described previously (Merglen et al., 2004) in RPMI 1640 medium containing 11 mM glucose. UCP2 degradation kinetics were determined as described by Azzu et al. (Azzu et al., 2008). Briefly, INS-1E cells were seeded at 3×105 cells/well in 24-well plates (Falcon) and incubated overnight. Cells were then treated with 10 μg/ml cycloheximide at time zero to arrest mRNA translation. At various time points they were harvested by trypsinisation in RPMI containing protease inhibitors and pelleted by centrifugation (800 g, 2 minutes).
Proteasome inhibition
INS-1E cells were treated with proteasome inhibitors for 2 hours before conducting UCP2 turnover experiments. Proteasome inhibitor cocktail-1 (PIC-1) included 30 μM MG132, 10 μM lactacystin and 10 μM PI-1 [Z-Ile-Glu(Ot-Bu)-Ala-Leucinal]. PIC-2 included 30 μM ALLN (N-acetyl-Leu-Leu-Nle-CHO; a calpain inhibitor), 5 μM clastolactacystin β-lactone and 5 μM epoxomicin. AdaAhx3L3VS was used at 5 μM. Proteasome inhibitors were used at the concentrations indicated and the cocktails were used at the additive concentration of their components. All proteasome inhibitors were from Calbiochem.
Bioenergetic modulation of UCP2 degradation in cells
INS-1E cells were incubated with 10 μM rotenone, 10 μM antimycin A, 10 μM myxothiazol, 75 μM menadione, 20 μM FCCP, 0.5 μM nigericin, 1 μg/ml oligomycin, or 10 mM NH4Cl for 30 minutes before measuring UCP2 degradation.
Measurement of reactive oxygen species, mitochondrial membrane potential and ATP levels in INS-1E cells
Measurements were made in 96-well plates seeded overnight with 40,000 INS-1E cells/well. Cells were incubated in Krebs-Ringer-Hepes buffer (135 mM NaCl, 3.6 mM KCl, 10 mM Hepes, 1.5 mM CaCl2·H2O, 0.5 mM MgCl2, 0.5 mM NaH2PO4, 2 mM glucose, 5% v/v FCS, 2 mM glutamine, pH 7.4) 30 minutes prior to experiments. Reactive oxygen species (ROS) production was measured using 5 μM mitoSOX (Invitrogen) as a mitochondrial ROS indicator, at λex/λem of 510 nm/580 nm. Mitochondrial membrane potential was measured using 25 nM tetramethyl rhodamine methyl ester (TMRM), at λex/λem of 555 nm/588 nm. Cellular ATP levels were determined using the ATPlite luminescence luciferase assay kit (Perkin Elmer) according to the manufacturer's instructions. All data were collected using a Spectramax Gemini XPS plate reader (Molecular Devices) and recorded using SoftMax Pro software.
Immunoblotting
Typically, 1×105 cells or 25 μg mitochondrial protein in gel loading buffer (10% (w/v) SDS, 250 mM Tris-HCl (pH 6.8), 5 mM EDTA, 50% (v/v) glycerol, 5% (v/v) β-mercaptoethanol, 0.05% (w/v) Bromophenol Blue) were separated by 12.5% SDS-PAGE, transferred onto a Protran nitrocellulose membrane (Whatman) using the semi-dry method (20 V for 30 minutes), and probed with 0.2 μg/ml anti-human UCP2 goat polyclonal IgG antibody (Santa Cruz Biotechnology, cat. no. sc-6525), 0.2 μg/ml anti-human adenine nucleotide translocase (ANT) goat polyclonal IgG antibody (Santa Cruz Biotechnology, cat. no. sc-9300), 0.2 μg/ml anti-human β-actin rabbit polyclonal IgG antibody (Abcam, UK, cat. no. ab8227), or 0.2 μg/ml anti-HA (Abcam, UK, cat. no. ab9110). The secondary antibodies were Immunopure (Pierce) peroxidase-conjugated, either 0.08 μg/ml goat anti-rabbit (cat. no. 31463) IgG or 0.04 μg/ml rabbit anti-goat IgG (cat. no. 31433). Where required, membranes were stripped using Restore PLUS western blot stripping buffer (Pierce), and reblotted according to the manufacturer's instructions.
Immunoblots were developed using a Lumigen® ECL Plus Western Blotting Detection system (Amersham Biosciences). Protein was quantified by analysing band intensities using ImageJ software (http://rsb.info.nih.gov/ij/) as described by Affourtit and Brand (Affourtit and Brand, 2008). Additional loading controls were conducted as follows: for cell experiments, UCP2 signals were corrected using β-actin signals, and for mitochondrial experiments, UCP2 signals were corrected for total protein using the entire density of each lane in Coomassie-stained membranes (with Gelcode Blue Stain Reagent, Pierce).
Ubiquitin plasmid amplification and isolation
E. coli containing HA-tagged wild-type (WT), knockout (KO) or K48R-ubiquitin pRK5 plasmids (Addgene cat. nos 17608, 17603, 17604, respectively) were grown overnight at 37°C in Luria-Bertani medium with 100 μg/ml ampicillin. Plasmids were isolated using the EndoFree Plasmid Maxi Kit (Qiagen) according to the manufacturer's instructions. A NanoDrop 1000 spectrophotometer was used to determine DNA concentration (A260) and plasmid purity (where A260/A280 of >1.8 indicated little or no protein contamination). All values obtained were >1.8.
Transfection experiments
Scr/UCP2 KD
2.5 μg/ml Lipofectamine 2000 (Invitrogen), UCP2 knockdown (Ambion ID 199050) or scrambled siRNA (negative control 1, Ambion ID 4636) at 200 nM was used to transfect INS-1E cells seeded overnight at 1×107 cells/10 cm2 dish. Cells were washed with PBS and harvested 48 hours post-transfection. An aliquot was used to create a cell sample. The remaining cells were lysed using 1 ml immunoprecipitation (IP) buffer containing 150 mM NaCl, 10 mM Tris, 1 mM EGTA, 1 mM EDTA, 5 mM N-ethylmaleimide, 1% (v/v) Triton X-100 and 1× protease inhibitor, pH 7.4.
FKBP8 KD
200 nM FKBP8 knockdown (Ambion ID s145125-7) or scrambled siRNA was used to transfect INS-1E cells seeded overnight at 3×105 cells/well in 24 well plates. After 48 hours, UCP2 turnover experiments were conducted.
Ubiquitin plasmids
Purified ubiquitin (Ub) plasmids were transfected at 0.5 μg/ml DNA per 3×105 cells (seeded overnight in 24-well plates) using Lipofectamine 2000 according to the manufacturer's instruction. After 20-24 hours, UCP2 turnover experiments were performed.
Immunoprecipitation
Lysates from scrambled siRNA-treated or UCP2-KD cells were cleared by incubation with protein-A-conjugated beads for 1 hour at 4°C, followed by pelleting of the beads and transfer of the 1 ml cell lysate into a fresh tube. Cleared cell lysates were incubated with various antibodies for 1 hour at 4°C, then incubated with protein-A-conjugated beads for a further hour at 4°C. Antibodies used were 0.5 μl of anti-Ub serum (Abcam ab19247), 0.05 μg polyclonal rabbit anti-UCP2 (Calbiochem, cat. no. 144157) or 0.05 μg rabbit normal IgG (Calbiochem, cat. no. NI01). The rabbit UCP2 antibody was chosen for this application for its specificity, and was validated as described for the goat anti-UCP2 antibody (Azzu et al., 2008).
Reconstituted in vitro UCP2 degradation assay
Mitochondria were isolated as described previously (Affourtit and Brand, 2006) but without protease inhibitors. Crude mitoplasts were prepared by centrifugation of frozen and thawed mitochondria at 10,000 g for 10 minutes. Mitochondria and mitoplasts were resuspended to 920 μg/ml in modified sucrose-Hepes buffer (2 mM dithiothreitol (DTT), 0.25 M sucrose, 20 mM Hepes, 2 mM EGTA, 10 mM KCl, 5 mM MgCl2 and 0.1% (w/v) defatted bovine serum albumin, pH 7.4). For each assay, 240 μg mitochondria and/or mitoplasts were incubated with an ATP regeneration system (1 mM ATP, 10 mM phosphocreatine and 20 IU/ml creatine kinase) and ubiquitin mix (70 μg ubiquitin, 1.4 μg fraction 1, 1.4 μg fraction 2 from Calbiochem, cat. no. 662096) for 30 minutes. Time zero was taken to be at the addition of 3.5 μg mammalian 26S proteasome fraction (Biomol International cat. no. PW8950), 20 mM succinate, 0.3 μM FCCP or 50 μM PIC-1 as indicated. Mitochondria were incubated at 37°C and aliquots were removed at the time points indicated. A sample of 40 μg mitochondrial protein was removed at each time point, pelleted and resuspended in gel loading buffer.
Cellular fractionation
INS-1E cells were treated with PIC-1 for 2 hours and then fractionated using the Q proteome cell compartment kit (Qiagen) according to the manufacturer's instructions.
Chemicals
All chemicals were obtained from Sigma or BDH unless otherwise indicated.
Data analysis
Data are presented as means ± s.e.m. of independent experiments. Each experiment represents independent cell cultures, transfections, immunoprecipitations, in vitro assays and western blots. Where appropriate, data were fitted with non-linear regression curves using Prism 5 software. Statistical significance was determined by repeated measures ANOVA (comparison of matching non-zero time points where required) with Dunnett's post-hoc testing or by Student's t-test as appropriate. Values of P<0.05 were considered significant. *P<0.05, **P<0.01, ***P<0.001.
Supplementary Material
Acknowledgments
Support from the Medical Research Council (UK), the School of Clinical Medicine, University of Cambridge, and National Institutes of Health grants P01 AG025901, PL1 AG032118 and P30 AG025708 is gratefully acknowledged. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/4/578/DC1
References
- Abu-Farha M., Niles J., Willmore W. G. (2005). Erythroid-specific 5-aminolevulinate synthase protein is stabilized by low oxygen and proteasomal inhibition. Biochem. Cell Biol. 83, 620-630 [DOI] [PubMed] [Google Scholar]
- Affourtit C., Brand M. D. (2006). Stronger control of ATP/ADP by proton leak in pancreatic beta-cells than skeletal muscle mitochondria. Biochem. J. 393, 151-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affourtit C., Brand M. D. (2008). Uncoupling protein-2 contributes significantly to high mitochondrial proton leak in INS-1E insulinoma cells and attenuates glucose-stimulated insulin secretion. Biochem. J. 409, 199-204 [DOI] [PubMed] [Google Scholar]
- Andrews Z., Liu Z., Walllingford N., Erion D., Borok E., Friedman J., Tschöp M., Shanabrough M., Cline G., Shulman G., et al. (2008). UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature 454, 846-851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic D., Onuma H., Pecqueur C., Raimbault S., Manning B. S., Miroux B., Couplan E., Alves-Guerra M. C., Goubern M., Surwit R., et al. (2000). Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 26, 435-439 [DOI] [PubMed] [Google Scholar]
- Azzu V., Affourtit C., Breen E. P., Parker N., Brand M. D. (2008). Dynamic regulation of uncoupling protein 2 content in INS-1E insulinoma cells. Biochim. Biophys. Acta 1777, 1378-1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayot A., Basse N., Lee I., Gareil M., Pirotte B., Bulteau A. L., Friguet B., Reboud-Ravaux M. (2008). Towards the control of intracellular protein turnover: mitochondrial Lon protease inhibitors versus proteasome inhibitors. Biochimie 90, 260-269 [DOI] [PubMed] [Google Scholar]
- Blanc J., Alves-Guerra M. C., Esposito B., Rousset S., Gourdy P., Ricquier D., Tedgui A., Miroux B., Mallat Z. (2003). Protective role of uncoupling protein 2 in atherosclerosis. Circulation 107, 388-390 [DOI] [PubMed] [Google Scholar]
- Brand M. D., Esteves T. C. (2005). Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2, 85-93 [DOI] [PubMed] [Google Scholar]
- Chau V., Tobias J. W., Bachmair A., Marriott D., Ecker D. J., Gonda D. K., Varshavsky A. (1989). A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243, 1576-1583 [DOI] [PubMed] [Google Scholar]
- Derdak Z., Mark N. M., Beldi G., Robson S. C., Wands J. R., Baffy G. (2008). The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 68, 2813-2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardina T. M., Steer J. H., Lo S. Z., Joyce D. A. (2008). Uncoupling protein-2 accumulates rapidly in the inner mitochondrial membrane during mitochondrial reactive oxygen stress in macrophages. Biochim. Biophys. Acta 1777, 118-129 [DOI] [PubMed] [Google Scholar]
- Granot Z., Geiss-Friedlander R., Melamed-Book N., Eimerl S., Timberg R., Weiss A. M., Hales K. H., Hales D. B., Stocco D. M., Orly J. (2003). Proteolysis of normal and mutated steroidogenic acute regulatory proteins in the mitochondria: the fate of unwanted proteins. Mol. Endocrinol. 17, 2461-2476 [DOI] [PubMed] [Google Scholar]
- Huang S., Ratliff K., Matouschek A. (2002). Protein unfolding by the mitochondrial membrane potential. Nat. Struct. Biol. 9, 301-307 [DOI] [PubMed] [Google Scholar]
- Jeon H. B., Choi E. S., Yoon J. H., Hwang J. H., Chang J. W., Lee E. K., Choi H. W., Park Z. Y., Yoo Y. J. (2007). A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun. 357, 731-736 [DOI] [PubMed] [Google Scholar]
- Kawaguchi M., Minami K., Nagashima K., Seino S. (2006). Essential role of ubiquitin-proteasome system in normal regulation of insulin secretion. J. Biol. Chem. 281, 13015-13020 [DOI] [PubMed] [Google Scholar]
- Kitiphongspattana K., Mathews C. E., Leiter E. H., Gaskins H. R. (2005). Proteasome inhibition alters glucose-stimulated (pro)insulin secretion and turnover in pancreatic β-cells. J. Biol. Chem. 280, 15727-15734 [DOI] [PubMed] [Google Scholar]
- Klausner R. D., Sitia R. (1990). Protein degradation in the endoplasmic reticulum. Cell 62, 611-614 [DOI] [PubMed] [Google Scholar]
- Margineantu D. H., Emerson C. B., Diaz D., Hockenbery D. M. (2007). Hsp90 inhibition decreases mitochondrial protein turnover. PLoS ONE 2, e1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiasson G., Sullivan P. G. (2006). The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid. Redox Signal. 8, 1-38 [DOI] [PubMed] [Google Scholar]
- Mayer T. U., Braun T., Jentsch S. (1998). Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO J. 17, 3251-3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L., Mohan R., Kwok B. H., Elofsson M., Sin N., Crews C. M. (1999). Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA 96, 10403-10408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merglen A., Theander S., Rubi B., Chaffard G., Wollheim C. B., Maechler P. (2004). Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145, 667-678 [DOI] [PubMed] [Google Scholar]
- Murakami Y., Matsufuji S., Kameji T., Hayashi S., Igarashi K., Tamura T., Tanaka K., Ichihara A. (1992). Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 360, 597-599 [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Shirane M., Iemura S., Natsume T., Nakayama K. (2007). Anchoring of the 26S proteasome to the organellar membrane by FKBP38. Genes Cells 12, 709-719 [DOI] [PubMed] [Google Scholar]
- Neutzner A., Youle R. J., Karbowski M. (2007). Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found Symp. 287, 4-14; discussion 14-20 [PubMed] [Google Scholar]
- Oberdorf J., Carlson E. J., Skach W. R. (2006). Uncoupling proteasome peptidase and ATPase activities results in cytosolic release of an ER polytopic protein. J. Cell Sci. 119, 303-313 [DOI] [PubMed] [Google Scholar]
- Pearce M. J., Mintseris J., Ferreyra J., Gygi S. P., Darwin K. H. (2008). Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322, 1104-1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S., Matouschek A. (2004). Protein unfolding in the cell. Trends Biochem. Sci. 29, 593-600 [DOI] [PubMed] [Google Scholar]
- Radke S., Chander H., Schäfer P., Meiss G., Krüger R., Schulz J. B., Germain D. (2008). Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 283, 12681-12685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset S., Mozo J., Dujardin G., Emre Y., Masscheleyn S., Ricquier D., Cassard-Doulcier A. M. (2007). UCP2 is a mitochondrial transporter with an unusual very short half-life. FEBS Lett. 581, 479-482 [DOI] [PubMed] [Google Scholar]
- Sasahara M., Nishi M., Kawashima H., Ueda K., Sakagashira S., Furuta H., Matsumoto E., Hanabusa T., Sasaki H., Nanjo K. (2004). Uncoupling protein 2 promoter polymorphism -866G/A affects its expression in beta-cells and modulates clinical profiles of Japanese type 2 diabetic patients. Diabetes 53, 482-485 [DOI] [PubMed] [Google Scholar]
- Soltys B. J., Gupta R. S. (1999). Mitochondrial-matrix proteins at unexpected locations: are they exported? Trends Biochem. Sci. 24, 174-177 [DOI] [PubMed] [Google Scholar]
- Sullivan P. G., Dubé C., Dorenbos K., Steward O., Baram T. Z. (2003). Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann. Neurol. 53, 711-717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutovsky P., McCauley T. C., Sutovsky M., Day B. N. (2003). Early degradation of paternal mitochondria in domestic pig (Sus scrofa) is prevented by selective proteasomal inhibitors lactacystin and MG132. Biol. Reprod. 68, 1793-1800 [DOI] [PubMed] [Google Scholar]
- Toninello A., Salvi M., Schweizer M., Richter C. (2004). Menadione induces a low conductance state of the mitochondrial inner membrane sensitive to bongkrekic acid. Free Radic. Biol. Med. 37, 1073-1080 [DOI] [PubMed] [Google Scholar]
- Verma R., Aravind L., Oania R., McDonald W. H., Yates J. R., Koonin E. V., Deshaies R. J. (2002). Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611-615 [DOI] [PubMed] [Google Scholar]
- Yan F. F., Lin C. W., Cartier E. A., Shyng S. L. (2005). Role of ubiquitin-proteasome degradation pathway in biogenesis efficiency of β-cell ATP-sensitive potassium channels. Am. J. Physiol, Cell Physiol. 289, C1351-C1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L., Leonhard K., Tatsuta T., Trowsdale J., Langer T. (2001). Role of the ABC transporter Mdl1 in peptide export from mitochondria. Science 291, 2135-2138 [DOI] [PubMed] [Google Scholar]
- Zhang C. Y., Baffy G., Perret P., Krauss S., Peroni O., Grujic D., Hagen T., Vidal-Puig A. J., Boss O., Kim Y. B., et al. (2001). Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105, 745-755 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.