

Abstract
An important function of the RNAase-III enzyme Dicer is to process microRNA precursors into ~22-nucleotide non-coding small RNAs. But little is known about the role of Dicer in mammalian brain formation and neural stem cell (NSC) development. Here we show that Dicer plays a crucial role in controlling mouse cortical NSC development. We found that Dicer function is essential for expanding cortical neural progenitors and NSCs. We have identified a population of Dicer-deficient NSCs that can self-renew, and that display normal karyotype and heterochromatin protein expression levels but show enlarged nuclei. Dicer-deficient NSCs display abnormal differentiation and undergo cell death when mitogens are withdrawn. Dicer deletion affects the levels of many proteins, as revealed by a mass spectrometry proteomic approach. We have found that an increase of anti-survival and/or pro-apoptosis proteins and a decrease of pro-survival and/or anti-apoptosis proteins contribute to the cell death of Dicer-deficient NSCs, implying a general role for Dicer in protecting cells from apoptosis. Our results demonstrate important functions for Dicer in regulating NSC development by maintaining proper signaling pathways related to cell survival and differentiation.
Keywords: Neural stem cells, Dicer, MicroRNAs, Epigenetics, Apoptosis, Differentiation
Introduction
The precise regulation of proliferation, survival, migration and differentiation of neural stem cells (NSCs) and neural progenitors is crucial for proper formation of the mammalian cerebral cortex during embryonic, postnatal and adult stages (Kokovay et al., 2008; Merkle and Alvarez-Buylla, 2006; Perrier and Studer, 2003; Zhao et al., 2008). NSCs are able to self-renew and give rise to multiple cell types such as neurons and glia. In the embryonic dorsolateral cortical wall, NSCs are transformed into neuroepithelial cells and radial glial cells residing in the ventricular zone (VZ), and later on intermediate progenitors in the subventricular zone (SVZ) through both asymmetric and symmetric divisions (Anthony et al., 2004; Chenn and McConnell, 1995; Guillemot, 2005; Hevner et al., 2006; Kriegstein, 2005; Molyneaux et al., 2007; Noctor et al., 2004). Cortical neural progenitors give rise to distinct neurons under an intrinsic timing control mediated by a complex gene expression program (Shen et al., 2006). Revealing the regulation of gene expression profiles that are crucial for NSCs expansion and lineage commitment over the course of development can provide the molecular basis for developing effective methods for NSC-based therapies.
Recently, a class of ~22-nucleotide endogenous non-coding small RNAs, called microRNAs (miRNAs), has been identified (Lee et al., 1993; Wightman et al., 1993). The RNAase-III enzyme Dicer is the essential molecule that processes miRNA precursors into mature miRNAs (Carmell and Hannon, 2004). The mature miRNA recognizes the 3′-untranslated region (3′-UTR) of its target gene and silences protein translation. Dicer plays an important role in stem-cell development. In Drosophila, the lack of Dicer-1 severely affects proliferation of germline stem cells (Hatfield et al., 2005; Jin and Xie, 2007; Park et al., 2007). Dicer-deficient embryonic stem (ES) cells have proliferation defects and fail to differentiate properly (Kanellopoulou et al., 2005; Murchison et al., 2005). However, little is known about the role of Dicer in mammalian brain formation and NSC development (Cheng et al., 2005; Kosik, 2006).
We here show that Dicer plays an essential role in expanding cortical neural progenitors and NSCs. We have identified a population of Dicer-deficient cortical NSCs that can survive with mitogens in a culture medium, retain properties of self-renewal, and display normal karyotype and heterochromatin protein expression levels. However, Dicer-deficient NSCs have abnormal differentiation and undergo cell death when mitogens are withdrawn. Because Dicer deletion blocks miRNA biogenesis and alters miRNA-mediated protein translational silencing, we measured protein levels in NSCs using a proteomic approach. We have revealed a molecular signature of Dicer-deficient NSCs, including decreases in pro-survival and/or anti-apoptosis genes in NSCs lacking the miRNA machinery. Our results demonstrate that the combination of proteomics and signaling-pathway analyses of proteins regulated by Dicer is an effective method to uncover protein-expression regulatory networks and epigenetic control during NSC development.
Results
Cortical Dicer deletion reduces the neural-progenitor pool
To examine Dicer function in mouse cortical NSC development, we selectively disrupted Dicer expression in the cerebral cortex using a Cre-loxP system by breeding floxed Dicer mice (Dicerloxp/loxp) with an Emx1-Cre line in which the Emx1 promoter is active in the cortical dividing zone by embryonic day 10.5 (E10.5) (Gorski et al., 2002) (supplementary material Fig. S1A,B). Dicer conditional-knockout mice (DicerCre/loxp, KO) had smaller cerebral cortices with a significantly thinner cortical wall at postnatal day 5 (P5), and the size of the cortex was greatly reduced at P14, resulting in a gap in the cortical midline (supplementary material Fig. S1C,D).
Because Dicer expression is deleted in the cortical ventricular zone (VZ), we examined the expression pattern of progenitor markers (e.g. the transcription factors Hes5 and Ngn2, and the Wnt signaling molecule β-catenin) (Chenn and Walsh, 2002; Nieto et al., 2001). The expression pattern of neural-progenitor markers seemed normal in E13.5 Dicer KO cortices (supplementary material Fig. S2). However, at E15.5, the expression levels of Hes5, Ngn2 and β-catenin were significantly reduced in the VZ of the Dicer KO mice (supplementary material Fig. S3A). Moreover, cells expressing Pax6 and Ki-67, which are markers for radial glial progenitors and all dividing cells, respectively (Englund et al., 2005; Heins et al., 2002), were greatly reduced in the VZ and the subventricular zone (SVZ) in Dicer KO cortices (supplementary material Fig. S3B-E). Cell death was observed in Dicer-deficient cortices at E13.5, with the highest cell death rate at E15.5, as detected by a TUNEL assay (supplementary material Fig. S3F,G). Expression of phospho-histone H3 (PH3), which labels mitotic cells in the M phase of a cell cycle, was greatly reduced in E15.5 Dicer KO cortices, especially in the SVZ, suggesting a proliferation defect of neural progenitors (supplementary material Fig. S3H,I). The number of intermediate progenitors in the SVZ, colabeled with Tbr2 and BrdU, was also reduced (supplementary material Fig. S3J,K). Our results suggest that Dicer function is essential for controlling the number of cortical neural progenitors.
Identifying self-renewable Dicer-deficient cortical NSCs
To test whether Dicer function affects development of cortical neuroepithelial cells or NSCs, we cultured NSCs, collected from E12.5 and E13.5 cortices, to form neurospheres in a medium containing the mitogens fibroblast growth factor 2 (FGF2) and epidermal growth factor (EGF). We found that very few neurospheres were developed from Dicer-deficient NSCs, which is consistent with our observations of reduced neural progenitors in vivo (Fig. 1A,B). The great reduction of neurospheres suggests that Dicer and miRNAs are essential for expanding early cortical neural progenitors and NSCs.
Fig. 1.

Dicer-deficient NSCs can self-renew. (A,B) A great reduction of neurospheres derived from NSCs of DicerCre/loxp (Ko) mice, as compared with controls (Ctrl), was observed. The primary neurosphere is termed as passage-0 (p-0). n=4 wells; **P<0.001. (C,D) The surviving Dicer-deficient NSCs produced numbers of neurospheres similar to those in controls after passaging, shown as a representative experiment at passage-2 (p-2). n=4 wells. (E) Dicer proteins were absent in Dicer-deficient neurospheres, shown as examples of passage-1 (p-1) and p-2, detected by western blotting assays. Actin served as a loading control. (F) miRNAs, for example miR-181b and miR-125b, were absent in p-1 Dicer-deficient neurospheres, as detected by northern blotting analyses. U6 served as a loading control.
To test whether surviving Dicer-deficient NSCs have the capacity for self-renewal, we dissociated neurospheres into single cells in a clonal density and tested whether they can form new neurospheres. Interestingly, surviving Dicer-deficient NSCs can self-renew for more than five passages. This is indistinguishable from control NSCs, as shown in an example of passage-2 (p-2) neurospheres (the primary neurosphere is termed as passage-0; p-0) (Fig. 1C,D). We found that Dicer protein is undetectable in both p-1 and p-2 surviving Dicer-deficient NSCs, indicating a complete ablation of Dicer protein (Fig. 1E). Additionally, mature miRNAs that are usually expressed in NSCs, such as miR-181b and miR-125b (Rybak et al., 2008), are absent in surviving Dicer-deficient NSCs, suggesting a complete block of miRNA biogenesis (Fig. 1F).
We next assessed whether surviving Dicer-deficient NSCs express stem-cell markers. Expression of the NSC marker nestin was detected in Dicer-deficient neurospheres at p-0 and p-2 (Fig. 2A,C). Similar to controls, under mitogenic conditions more than 98% of Dicer-deficient NSCs were BrdU positive, suggesting that surviving Dicer-deficient NSCs are dividing cells (Fig. 2B,D). Moreover, dissociated p-2 Dicer-deficient neurospheres expressed NSC markers such as nestin and SSEA-1 similarly to control neurospheres (Fig. 2E,F) (Capela and Temple, 2006; Wrobel et al., 2007). We did not detect significant numbers of dead cells in p-1 Dicer-deficient NSCs cultured under mitogenic conditions, compared to control NSCs (Fig. 2G).
Fig. 2.

Dicer-deficient NSCs express stem-cell markers. (A) Primary neurospheres (p-0) derived from Dicer-deficient NSCs (Ko) expressed nestin similarly to control NSCs (Ctrl). (B) More than 98% of Dicer-deficient NSCs (p-0) were BrdU positive, suggesting that they are dividing at the same rate as controls. (C) Nestin was expressed in the passaged neurospheres (p-2) derived from NSCs of KO and control cortices. The insets show neurosphere immunostaining without the primary antibodies for nestin. (D) More than 98% of Dicer-deficient NSCs (p-2) were BrdU positive. (E,F) Dissociated Dicer- deficient NSCs (p-2) expressed NSC markers such as nestin and SSEA-1. (G) Dicer-deficient NSCs (p-1) survived normally as control NSCs in a medium with mitogens. n>3 fields.
Our results indicate that a small population of Dicer-deficient NSCs maintain properties of ‘stemness’ and divide in the same way as the controls. Once this population of Dicer-deficient NSCs can survive in response to mitogens, Dicer function seems to no longer be required for their self-renewal and proliferation.
Surviving Dicer-deficient NSCs display abnormal differentiation
To test whether Dicer-deficient NSCs are able to differentiate into neurons and glia, we cultured dissociated NSCs and neurospheres in a medium without mitogens using previously described methods (Shen et al., 2006). Similar to control NSCs, passaged Dicer- deficient NSCs (p-1) gave rise to cells that express the neuronal marker Tuj1, and the glial markers GFAP and O4 (Fig. 3A). However, differentiated Dicer-deficient cells displayed abnormal morphology with shorter neurites and processes (Fig. 3A). Abnormal morphology was also observed in differentiated cells derived from surviving primary Dicer-deficient NSCs (supplementary material Fig. S4). Our results suggest a differentiation defect of Dicer-deficient NSCs.
Fig. 3.

Surviving Dicer-deficient NSCs display abnormal differentiation. (A) Under the differentiation condition without mitogens, passaged (p-1) NSCs collected from Dicer knockout (Ko) cortices gave rise to cells expressing neuronal (Tuj1+) and glial (GFAP+ and O4+) markers. However, their morphology was abnormal, as shown with shorter neurites than controls (Ctrl). (B) The majority of Dicer-deficient NSCs underwent cell death, shown as an example of p-1 NSCs in differentiation condition cultured for 2 days in vitro. n=6 fields; **P<0.0008. (C) Dicer-deficient NSCs did not survive well in a differentiation culture medium without mitogens. Whereas control neurospheres (p-2) survived, most Dicer KO neurospheres died after 48 hours in culture. Many differentiated cells (arrows) migrated away from the control neurosphere but not from the Dicer-deficient neurosphere.
Moreover, under the differentiation culture condition, a majority of Dicer-deficient NSCs underwent cell death, as shown in an example of p-1 dissociated neurospheres cultured for 2 days to induce differentiation (Fig. 3B). Additionally, whereas control neurospheres in the medium without mitogens attached well to the surface of the culture dish, and the differentiated cells migrated away from the neurospheres after 48 hours in culture, Dicer-deficient neurospheres shrank and only very few differentiated cells migrated away from the neurospheres, suggesting that they undergo cell death and fail to differentiate after mitogen withdrawal (Fig. 3C).
Thus, although Dicer deletion does not inhibit differentiation of NSCs, it seems that Dicer deletion and mitogen withdrawal affect the survival and proper differentiation of NSCs.
Surviving Dicer-deficient NSCs display normal karyotype and heterochromatin protein expression but have enlarged nuclei
Because surviving Dicer-deficient NSCs can self-renew but display abnormal differentiation, we first checked whether their karyotype (metaphase spread) is abnormal. As shown, similar to controls, the majority of Dicer-deficient NSCs dissociated from neurospheres (p-1) had 40 chromosomes (Fig. 4A-C).
Fig. 4.

Surviving Dicer-deficient NSCs have normal karyotype and heterochromatin protein expression. (A,B) Karyotype analyses (metaphase spreads) of surviving Dicer-deficient (Ko) and control (Ctrl) NSCs collected from p-1. (C) The percentage of chromosome numbers among counted cells. More than 54% Dicer KO NSCs displayed 40 chromosomes, which is similar to the controls (63%); n=33 cells for either Ctrl or KO NSCs. (D) Immunohistochemistry for heterochromatin proteins in control and Dicer- deficient NSCs (p-1). Both H3K9me3 and HP1β were detected and coexpressed in Dicer-deficient NSCs. The insets in merged images show immunostaining without the primary antibodies for H3K9me3 and HP1β. (E) Almost 100% of control and Dicer-deficient NSCs were H3K9me3- and HP1β-positive. n>250 cells; three Dicer KO and three control animals were used for statistical analyses. (F) H3K9me3- and HP1β-expressing Dicer- deficient NSCs had elongated nuclei. (G) The intensity of H3K9me3 expression levels was similar between the control (blue line) and KO (red line) NSCs, as detected by the flow cytometric analysis. The gray area indicates an isotype-negative control. The representative result shown here is from three independent experiments.
Recent work showed that Dicer is crucial for maintaining heterochromatin structures in ES cells and chicken DT40 cells (Fukagawa et al., 2004; Kanellopoulou et al., 2005). We thus assessed whether the heterochromatin formation is disorganized in Dicer-deficient NSCs. We first examined expression patterns of the heterochromatin protein 1 beta (HP1β) and tri-methylated histone H3 on lysine 9 (H3K9me3), which play an important role in heterochromatin organization (Fischle et al., 2005), in cultured NSCs. Similar to the control NSCs, HP1β and H3K9me3 were coexpressed in almost all analyzed Dicer-deficient NSCs with similar intensity (Fig. 4D,E). However, the nuclei were enlarged and the shape of nucleus was elongated in Dicer-deficient NSCs (Fig. 4F). Our results suggest that Dicer deletion does not significantly alter expression levels of heterochromatin proteins but might affect the heterochromatin structure in surviving NSCs.
Protein expression profiles of surviving Dicer-deficient NSCs
miRNAs, processed by Dicer, silence protein translation of target genes. Because Dicer-deficient NSCs lack miRNA biogenesis, the expression levels of target proteins that are usually regulated by miRNAs might have been altered. We thus generated protein expression profiles and compared protein levels between Dicer-deficient and control NSCs using a proteomic approach. Proteins were extracted from p-2 Dicer-deficient and control NSCs, and identified using tandem mass spectrometry (MS/MS). When applying strict thresholds (see Materials and Methods), 1175 of more than 2900 protein identifications were selected for further analysis (supplementary material Table S1). These 1175 proteins are termed as the ‘reference list’ in the following analyses in this paper (supplementary material Table S2). We separated these proteins into groups of those with >2× differential levels either in Dicer-deficient or in control NSCs (termed as KO-bias or WT-bias list, supplementary material Tables S3, S4). From this categorization, 251 and 311 proteins were identified to be in the KO-bias and WT-bias groups, respectively. The remaining 613 proteins were classified as similar levels between Dicer-deficient and control NSCs (supplementary material Table S5).
Next, we performed functional analyses of these data sets using Ingenuity Pathway Analysis (IPA) and the Database for Annotation, Visualization and Integrated Discovery (DAVID). Enrichment analyses of genes in specific biological functions were performed with DAVID for Gene Ontology (GO) biological processes and GO molecular function using the reference list as a background (supplementary material Fig. S5). It seems that there is an enrichment of genes involved in cell-cycle processes (23, P=0.07) in control NSCs (supplementary material Table S3), whereas there is an enrichment of a more complex repertoire of biological processes in the Dicer-deficient NSCs, such as signal transduction (60, P=0.00002), and protein-mediated (33, P=0.008) and vesicle-mediated (18, P=0.077) transport (supplementary material Fig. S5A,B and Table S4). By contrast, proteins involved in overall metabolic regulation and other so-called ‘house-keeping’ biological processes, such as organelle and cytoskeleton organization and biogenesis, showed similar expression levels between Dicer-deficient and control NSCs (supplementary material Fig. S5C and Table S5).
Moreover, because Dicer-deficient NSCs can self-renew in medium with mitogens, such as EGF and FGF, we analyzed genes involved in the EGF, FGF and VEGF pathways using Panther, Uniprot and IPA (supplementary material Tables S6-S8). We found that, although some of the genes involved in the EGF, FGF and VEGF pathways were upregulated by >twofold in either the Dicer-deficient NSCs (8, 4 and 5 genes, respectively) or control NSCs (8, 5 and 7), the majority of genes (21, 14 and 9) are similarly expressed in Dicer-deficient and control NSCs, suggesting a normal response to mitogens and an ability of self-renewal of Dicer-deficient NSCs (supplementary material Fig. S6).
Cell death of Dicer-deficient NSCs and progenitors
We found that an obvious defect of Dicer-deficient NSCs and progenitors is cell death. To reveal the underlying mechanisms of cell death in Dicer-deficient NSCs, we analyzed all proteins in the reference list and, using Panther, Uniprot and IPA, found 233 proteins to be involved in various well-studied cell-death pathways such as apoptosis, necrosis and necroptosis. These 233 proteins were further analyzed by literature search in regard to their functional effect on either pro-survival or pro-cell-death (supplementary material Table S9). An increase of pro-cell-death proteins and a decrease of pro-survival proteins were detected in Dicer-deficient NSCs (Fig. 5). The ratio of these two classes was shifted from about 0.7 in the case of similarly expressed proteins and about 1.0 for proteins with expression bias in controls to 2.5 in the case of the Dicer-deficient NSCs. Our bioinformatic analyses suggest that Dicer-deficient NSCs are more sensitive to cell death, owing to increased pro-cell-death proteins and the lack of protective pro-survival proteins.
Fig. 5.
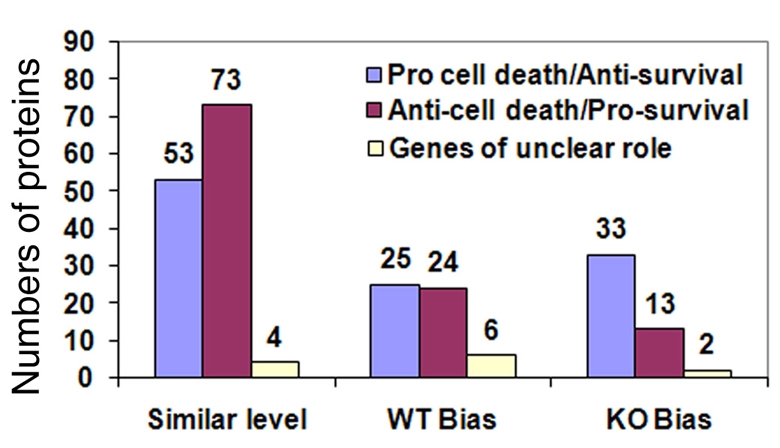
Unbalanced cell-survival and cell-death pathways in Dicer-deficient NSCs. Signaling pathway analyses identified an increase of pro-cell-death proteins and a decrease of pro-survival proteins in Dicer-deficient NSCs.
Consistent with the bioinformatic analysis, increased cell death in neural progenitors was detected by the TUNEL assay and was demonstrated by a higher caspase-3 (Casp3) expression, indicating increased apoptosis in Dicer KO cortices (Fig. 6A; supplementary material Fig. S3). Increased expression of programmed cell death protein 4 (PDCD4) was also observed in Dicer-deficient NSCs, as detected by western blotting assays (Fig. 6B). We also detected a decrease of superoxide dismutase (SOD1) expression in Dicer-deficient NSCs, suggesting a reduced resistance to oxidative stress in NSCs when Dicer is deleted (Fig. 6C).
Fig. 6.

Cell-death pathways affected by Dicer deletion in NSCs and progenitors. (A) An increased amount of apoptotic progenitor cells, detected by activated Casp-3 (arrowheads), was detected in E15.5 cortices of Dicer KO mice compared with controls (Ctrl). (B) Expression of PDCD4 was increased in Dicer-deficient NSCs, as detected by western blotting assays. Actin served as a loading control. Shown as one example experiment, relative levels of PDCD4 expression were normalized by actin expression between Ctrl and KO. (C,D) Decreased expression of SOD1 and increased expression of FMR1 were detected in Dicer-deficient NSCs, as analyzed by western blotting assays. Shown as example experiments; relative levels of SOD1 and FMR1 expression were normalized by actin expression between Ctrl and KO. The representative western blotting results shown here are from neurosphere cultures of at least three Dicer KO and three control animals. (E) A simplified network generated from proteins identified to be differentially expressed in NSCs of Dicer KO and Ctrl. It illustrates FMR1 as a hub protein in miRNA- and Dicer-dependent regulation of cell death and survival. The intensity of the node color indicates the degree of up- (red, representing >twofold higher in Ctrl compared with KO) or down- (green, representing >twofold higher in KO compared with Ctrl) regulation. The complete network is shown in supplementary material Fig. S7.
To further connect the functional relationships between key molecules differentially expressed in Dicer-deficient NSCs, we generated networks consisting of the genes identified in the reference list using IPA (see Materials and Methods). Several hub proteins upregulated in Dicer-deficient NSCs were identified among the networks generated, such as fragile X mental retardation protein 1 (FMR1; also known as FMRP) and Casp3 (Fig. 6E; supplementary material Fig. S7). Similarly, hub proteins upregulated in control NSCs were found, for instance transforming growth factor-beta receptor type II (TGFβR2) and SOD1 (Fig. 6E; supplementary material Fig. S7). Consistent with the proteomic results, using western blotting assays we detected increased expression of FMR1 and PDCD4 and decreased expression of SOD1 in Dicer-deficient NSCs (Fig. 6). Inconsistent with proteomic results, our western blots did not detect reproducible changes of TGFβR2 levels between Dicer-deficient and control NSCs among all samples tested (n=4, data not shown). It remains to be established whether altered expression of several hub proteins affected by Dicer deletion could be sufficient to cause abnormal features of NSCs such as defects in survival and differentiation.
Discussion
To assess the function of the RNAase-III enzyme Dicer in NSC development, we deleted Dicer expression in the dividing zone in the mouse cerebral cortex using genetic approaches, which in turn blocks miRNA biogenesis. Here, we show that the loss of Dicer function severely affects neural-progenitor development and causes smaller cortices. We have discovered a population of self-renewable Dicer-deficient cortical NSCs that is sensitive to mitogen withdrawal. Moreover, we have generated protein expression profiles of Dicer-deficient NSCs using proteomics and identified crucial signaling pathways that are affected by Dicer deletion.
Dicer functions in expanding the cortical neural-progenitor pool
Cortical Dicer deletion reduces the progenitor pool and results in a significant reduction in cortical size. At E15.5, a critical stage of neural-progenitor development, both radial glial cells and intermediate progenitors are greatly reduced in the VZ and SVZ, owing to a higher cell-death rate in Dicer KO cortices than controls (supplementary material Fig. S3). We found that, at E13.5, aside from cell death, neural progenitors display normal numbers in the Dicer KO cortices (supplementary material Fig. S2). On the basis of similar results to ours, a previous report concludes that miRNAs are not essential for early neural-progenitor development (De Pietri Tonelli et al., 2008). However, we believe that the normal Dicer KO cortex at E13.5 does not mean that Dicer and miRNA functions are not essential for early neural-progenitor development.
The following factors might contribute to differing interpretations of Dicer functions in expanding cortical neural progenitors from us and from De Pietri Tonelli et al. (De Pietri Tonelli et al., 2008): (1) miRNAs processed prior to Dicer deletion. At E10.5, when Emx1-Cre is active, some miRNAs might have already been processed in NSCs before the Dicer deletion. Our studies and others have shown that the processed miRNAs might continue to function for some time before being completely degraded and might contribute to normal progenitor development in E13.5 Dicer KO cortices (Harfe et al., 2005; Kawase-Koga et al., 2009). (2) Different Emx1-Cre lines used. The earliest activity of our Emx1-Cre is detected by E10.5 (Gorski et al., 2002). De Pietri Tonelli et al. (De Pietri Tonelli et al., 2008) used Emx1-Cre knock-in mouse line that is active by E9.5 (Iwasato et al., 2000). Different Cre lines might have distinct activities in excising floxed Dicer. Moreover, Dicer proteins might not be ablated completely when Emx1-Cre is active. We still detected a very low level of Dicer protein in E13.5 Dicer KO cortices (Kawase-Koga et al., 2009). This low level of Dicer can continue to process miRNAs, which might maintain normal neural-progenitor development until E13.5. (3) NSC culture conditions. Our primary NSC cultures collected from E12.5 and E13.5 Dicer KO cortices show a greatly reduced ability to form neurospheres (Fig. 1). miRNAs processed prior to Dicer deletion might be degraded in NSCs and cause a reduced neurosphere formation. Additionally, the cell culture conditions might magnify the effects of cell death caused by Dicer ablation and lead to decreased expansion of NSCs. Together, our results indicate that Dicer function is essential for expanding both the early neural-progenitor pool and NSCs.
Molecular signature of Dicer-deficient cortical NSCs
Previous work has shown that Dicer plays an important role in stem-cell development by regulating proliferation and differentiation (Hatfield et al., 2005; Jin and Xie, 2007; Kanellopoulou et al., 2005; Murchison et al., 2005; Park et al., 2007). Specific miRNAs that are essential for pluripotency and differentiation of ES cells have been demonstrated (Foshay and Gallicano, 2009; Houbaviy et al., 2003; Judson et al., 2009; Krichevsky et al., 2006; Viswanathan et al., 2008; Xu et al., 2009). Given that Dicer is essential for the production of mature miRNAs, which in turn regulates protein translation, we analyzed the changes of protein levels in normal and Dicer-deficient NSCs using a mass-spectrometry-based proteomic approach. Similar to altering a single miRNA in cell lines (Baek et al., 2008; Selbach et al., 2008), deleting Dicer has released the repression of hundreds of proteins, perhaps usually silenced by miRNAs via direct or indirect effects, as indicated by increased levels of some proteins in the Dicer-deficient NSCs (supplementary material Table S1). Our results suggest that miRNAs indeed regulate the synthesis of many proteins in NSCs. At the same time, we also observed examples of reduced protein levels in Dicer-deficient NSCs, which could be a secondary effect of repression through proteins with increased expression. Our results also identify several important biological processes that are not affected by Dicer deletion, for instance the overall metabolic regulation and ‘house-keeping’ biological processes such as organelle and cytoskeleton organization and biogenesis. In summary, our studies have revealed a molecular signature of proteins affected by proper Dicer expression and miRNA functions in NSCs (supplementary material Fig. S5).
In our studies, we have identified a population of Dicer-deficient NSCs, which is distinct from previously reported germline and tissue-specific stem cells lacking Dicer. These NSCs, once they have survived beyond p-0 in a medium with mitogens, have the properties of self-renewal but display abnormal differentiation when mitogens are withdrawn. Our signaling-pathway analyses have not identified significant differences of numbers of proteins involved in mitogens such as FGF, EGF or VEGF pathways in Dicer-deficient versus control NSCs, implying the normal proliferation of surviving Dicer-deficient NSCs. Additionally, it is possible that normal responses to mitogens such as FGF in Dicer-deficient NSCs are the balanced result, caused by the loss of miRNAs, of altered expression of many genes that are usually up- or downregulated by mitogens.
It seems that the majority of Dicer-deficient NSCs either dies or differentiates in the primary culture (p-0), but a small population of Dicer-deficient NSCs, perhaps responding equally to mitogens as do control NSCs, can bypass the effect of Dicer deletion and expand normally (Fig. 1A-D). However, without mitogens, surviving Dicer-deficient NSCs undergo cell death and differentiate into cells that express neuronal and glial markers but show abnormal morphology, with shorter neurites and processes. Given such abnormal morphology, proteins that usually maintain cell skeleton structures are probably affected by Dicer deletion. For instance, we have found decreased expression of the neurofilament component NEFM in Dicer-deficient NSCs (data not shown).
Recent work shows that Dicer is required for maintaining the heterochromatin assembly, probably by the short interfering RNA (siRNA) pathway (Fukagawa et al., 2004; Kanellopoulou et al., 2005). The karyotype and heterochromatin protein expression levels seem normal in surviving Dicer-deficient NSCs. In the p-0 neurosphere culture, we cannot rule out that the heterochromatin assembly might be abnormal in this population of NSCs because most NSCs either fail to survive and proliferate, or differentiate early. Moreover, the abnormal size and shape of nuclei in passaged Dicer-deficient NSCs further suggest some abnormalities of heterochromatin structures (Fig. 4). The severe survival and differentiation defects in Dicer KO cortices and NSCs are probably the conjugated effects of both lack of miRNAs and abnormal heterochromatin maintenance.
Dicer functions in preventing cell death
Our studies and others have shown that cell death occurs in NSCs, progenitors and postmitotic neurons when Dicer is ablated in the mouse brain using genetic tools (Cuellar et al., 2008; Davis et al., 2008; De Pietri Tonelli et al., 2008; Kawase-Koga et al., 2009; Schaefer et al., 2007). These results indicate that cell death is a common feature in Dicer-ablated cells. Our proteomic and signaling-pathway analyses of differentially expressed proteins have revealed an increase of pro-cell-death and, perhaps more importantly, a decrease of pro-survival proteins in Dicer-deficient NSCs; these changes might affect the overall balance of cell death and survival (Fig. 4). We have shown that almost all surviving Dicer-deficient NSCs are nestin and BrdU positive, suggesting that, although the culture might not contain 100% pure NSCs, a great majority of them are dividing cells and not mixed with differentiated cells. Thus, altered cell-death pathways detected by proteomics and bioinformatics assays should represent the biological changes in surviving proliferative Dicer-deficient NSCs.
We found an increase of FMR1 expression in Dicer-deficient NSCs (Fig. 6). Increased FMR1 expression might alter various downstream pathways. Notably, our study shows that altered FMR1 expression affects the expression of SOD1, and might in turn activate the apoptotic activity mediated by Casp3 (Fig. 6; supplementary material Fig. S7). FMR1 is an RNA-binding protein and has been shown to be associated with the miRNA processing pathway (Jin et al., 2004). Recent studies have shown that FMR1 regulates neuronal-specific miR-124 expression and is involved in maintaining germline stem cells in Drosophila (Xu et al., 2008; Yang et al., 2007; Yang et al., 2009). Moreover, FMR1 is a putative target for several miRNAs, such as miR-194 and miR-326 (John et al., 2004). Our study and others indicate that precise expression levels of FMR1 are important in normal stem-cell development in both mice and Drosophila.
Moreover, our results suggest a general role for Dicer in protecting cells from cell death. Even though our proteomic approach in this study is not able to reveal specific miRNA and target-protein pairs that might be important in survival of NSCs, the protein expression profiles created here, particularly proteins upregulated in Dicer-deficient NSCs, will serve as a reference for further identifying direct targets of miRNAs.
Revealing functions and pathways of miRNA-regulated proteins, as identified in this study, enhances our understanding of processes that are crucial for survival, self-renewal and differentiation of NSCs. miRNAs are also important for human neurological disease conditions (Chang and Mendell, 2007; Stark et al., 2008). Taking the advantage of the fast development of miRNA delivery techniques, miRNAs have become novel and promising targets for gene therapies (Couzin, 2008). Understanding the role of specific miRNAs in NSC development in future work will develop practical tools for manipulating proliferation, survival and differentiation of NSCs for the treatment of neurodegenerative diseases using stem-cell-based methods.
Materials and Methods
Generation of Dicer conditional-knockout mice
The floxed Dicer transgenic mice (Dicerflox/flox) (Murchison et al., 2005) were bred with the Emx1-Cre mice (The Jackson Laboratory) to generate Emx1-Cre:Dicerflox/flox animals (called DicerCre/flox, KO). The activity of Emx1-Cre was tested by breeding Emx1-Cre mice with the Rosa26-GFP reporter line (The Jackson Laboratory).
For staging of embryos, midday of the day of vaginal-plug formation is considered as E0.5, the first 24 hours after birth are defined as P0. Animal use was overseen by the Animal Facility at the Weill Cornell Medical College.
Genotyping of Dicer conditional-knockout mice
Mouse tail-tip biopsies were used for genotyping by PCR reactions using the following primer pairs: for Cre, 5′-TAAAGATATCTCACGTACTGACGGTG-3′ and 5′-TCTCTGACCAGAGTCATCCTTAGC-3′ (product size: 350 bp); for Dicer, 5′-ATTGTTACCAGCGCTTAGAATTCC-3′ and 5′-GTACGTCTACAATTGTCTATG-3′ (product sizes: 767 bp from Dicerflox allele and 560 bp from the wild-type Dicer gene).
Nissl staining
Sections (10 μm) were processed through incubation in the following solutions in order: ethanol/chloroform (1:1, overnight), 100% ethanol (30 seconds), 95% ethanol (30 seconds), distilled water (30 seconds, twice), cresyl violet (3-5 minutes), distilled water (2 minutes, three times), 50% ethanol (2 minutes), 95% ethanol (5-30 minutes), 100% ethanol (5 minutes, twice), xylene (3 minutes, twice), and then mounted with a coverslip.
Tissue preparation and immunohistochemistry
Mouse brains were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) over night, incubated in 25-30% sucrose in PBS, embedded in OCT and stored at −80°C until use. Brains were sectioned (10 μm) using a cryostat. For antigen recovery, sections were incubated in heated (95-100°C) antigen recovery solution (1 mM EDTA, 5 mM Tris, pH 8.0) for 15-20 minutes, and cooled down for 20-30 minutes. Before applying antibodies, sections were blocked in 10% normal goat serum (NGS) in PBS with 0.1% Tween-20 (PBT) for 1 hour. Sections were incubated with primary antibodies at 4°C overnight and visualized using goat anti-rabbit IgG—Alexa-Fluor-488 and/or goat anti-mouse IgG—Alexa-Fluor-594 (1:350, Molecular Probes) for 1 hour at room temperature. Images were captured using a Leica digital camera under a fluorescent microscope (Leica DMI6000B).
Primary antibodies against the following antigens were used: phospho-histone H3 (PH3) (1:1000, Upstate), active Casp3 (1:80, Chemicon), bromodeoxyuridine (BrdU) (1:100, Molecular Probes), Ki-67 (1:500, Abcam), Pax6 (1:30, DSHB), Tbr1 (1:2500) and Tbr2 (1:2000, kindly provided by Robert Hevner, University of Washington, Seattle, WA), SSEA-1 (1:100; Cell Signaling Technology), β-tubulin III (TuJ1) (1:600, Chemicon), Map2 (1:750, Chemicon), GFAP (1:600, DAKO), O4 (1:2, DSHB) and nestin (1:8, DSHB).
TUNEL assay
To identify apoptotic cells in the cortex, we performed a TUNEL assay using the Apop Tag Fluorescein in situ Apoptosis detection kit (Chemicon) on 10-μm frozen sections. This assay was performed according to the manufacturer's instructions.
Cell counting in the cortical wall
Coronal sections were collected in the medial cortical region (at levels between the anterior commissure and the anterior hippocampus). At least four sections from each brain and three brains from different litter were chosen for antibody labeling and TUNEL assay. Positive cells were counted in each bin in a field (total six bins from the ventricular surface to the pial surface) using the method described previously (Takahashi et al., 1993). Positive cells and percentage of positive cells are presented as in a field in the cortical wall in the cortex.
Neurosphere culture
The preparation of neurosphere culture was performed as described previously with minor modifications (Reynolds et al., 1992). The embryos were removed at E12.5 and E13.5. The cerebral cortex from each embryo was dissected and transferred to artificial cerebrospinal fluid (aCCSF: 124 mM NaCl, 5 mM KCl, 2 mM CaCl2, 26 mM NaHCO3, 1.3 mM MgCl2, 10 mM glucose) and mechanically triturated into single cells. These cells were plated at a cell density of 5 cells/μl on uncoated 24-well dishes and cultured in DMEM/F12 (Gibco) with N2 supplement (Gibco), B27 supplement (Gibco), 10 ng/ml FGF2 (Invitrogen) and 20 ng/ml EGF (Invitrogen). The number of primary neurospheres per well was counted after 7 days of culture.
For the self-renewal analysis of NSCs, primary neurospheres were dissociated and passaged at a cell density of 1 cell/μl using the same culture conditions as in the primary culture. For the differentiation analysis of NSCs, dissociated cells from neurospheres were cultured in DMEM/F12, supplemented with B27 supplement, N2 supplement and 1% fetal bovine serum (FBS) (Gibco) on poly-L-lysine (Sigma)- and laminin (Invitrogen)-coated glass coverslips in 24-well dishes for 2-5 days (Shen et al., 2004). For the BrdU-incorporation analysis, dissociated cells from neurospheres were cultured on poly-L-lysine- and laminin-coated glass coverslips in 24-well dishes with mitogens FGF2 and EGF for 2 days. 10 μg/ml BrdU was added to the culture for 24 hours and then washed out. Cells were stained for anti-BrdU antibodies (1:100).
Western blotting analysis
15-25 μg protein extracts from cortical neurosphere cultures were fractionated by SDS-PAGE, immunoblotted and probed with a rabbit anti-Dicer antibody (ab1416, 1:1000; kindly provided by David Livingston and Chryssa Kanellopoulou, Dana-Farber Cancer Institute, Boston, MA) (Kanellopoulou et al., 2005), a rabbit anti-FMRP antibody (1:500; Sigma), a rabbit anti-SOD1 antibody (1:1000; Abcam), a rabbit anti-PDCD4 antibody (1:1000; Cell Signaling Technology) and a rabbit anti-actin antibody (1:200, Sigma), and subsequently with peroxidase-conjugated secondary antibodies (1:20,000; Sigma). The signals were detected with Supersignal West Pico Chemiluminescent Substrate (Pierce).
Heterochromatin protein expression
Dissociated cells from neurospheres were cultured on poly-L-lysine- and laminin-coated 24-well dishes with 1% FBS and mitogens FGF2 and EGF for 6 days. Cells were stained with antibodies for anti-H3K9me3 (1:500, Upstate) and anti-HP1β (1:2500, Chemicon). H3K9me3- and HP1β-positive cells were counted.
Flow cytometric analysis
To compare cell-body size between control and DicerCre/flox cells, cells isolated from cultured neurospheres were suspended as single cells, and washed with PBS and blocked in 10% NGS in PBS with mouse BD FC Block (1:100, BD Pharmingen) for 10 minutes at 4°C. Cells were then fixed in 4% PFA for 10 minutes at 4°C.
H3K9me3 intracellular staining was performed using fixation and permeabilization buffer (eBioscience) before incubation with anti-H3K9me3 antibodies (1:200) and anti-rabbit IgG—Alexa-Fluor-488 (1:400) mixture.
The fixed cells were acquired (30,000 events) on an LSR II (BD Pharmingen) using the FACS DiVa software (BD Pharmingen). Data were analyzed using the FlowJo analysis software (Tree Star).
Karyotype analysis (metaphase chromosome spreads)
Metaphase chromosomes were prepared as previously described with minor modifications (Mills et al., 2004). Mitotic cells from neurosphere cultures were treated with colcemid (0.03 μg/ml, Invitrogen) for 30 minutes and mechanically dissociated into single cells. Cells were suspended in 75 mM KCl at room temperature for 20 minutes then fixed in fixation solution (3:1 methanol/acetic acid). Cells were then dropped onto the slides, passed through steam and air dried. Metaphase cell spreads were covered with mounting solution containing Hoechst and imaged using a Leica digital camera under a fluorescent microscope (Leica DMI6000B). For quantification of chromosome numbers, metaphase cell spreads were randomly chosen for counting.
Northern blotting analysis
Total RNA was isolated from the cortical neurosphere cultures using the Trizol reagent (Invitrogen) according to manufacturer's instructions. 5 μg total RNA was loaded onto 13% denatured polyacrylamide gels and separated at 65 mA for 1.5 hours at room temperature, and transferred into nitrocellulose membrane overnight using a semi-dry transfer system. After cross-linking for 4 hours at 80°C, membranes were hybridized using locked nucleic acid (LNA) antisense probes specific for miR-181b, miR-125b and U6 (Exiqon). The probes were 3′-end labeled with digoxigenin (DIG)-ddUTP with terminal transferase using the DIG-3′-end labeling kit (Roche). After hybridization at 50°C overnight, membranes were washed 10 minutes in 2× SSC with 0.1% SDS, six times. The miRNA and U6 signals were detected using the CDP-star chemiluminescent substrate (Roche).
Statistic analysis
At least three DicerCre/flox (KO) and three control animals were used for all statistical analyses. Data were shown as mean ± s.e.m. Statistical comparison was made by analysis of variance (unpaired t-test or ANOVAs).
Proteomic assays
Neurospheres from p-2 were washed with cold PBS twice. They were transferred into homogenization buffer [final concentration: 15 mM Tris-HCl (pH 7.7), 0.5 mM PMSF, 0.25 M sucrose, 15 mM NaCl, 1.5 mM MgCl2, 2.5 mM EDTA (pH 8.0), 1 mM EGTA (pH 8.0), 25 mM NaF, 2 mM Nappi, Protease inhibitor Cocktail Complete (Roche)] for homogenization. 50 μg of total protein were transferred into proteomics buffer (final concentration: 20 mM Tris-HCl (pH 8.0), 6 M urea, 2 M thiourea, 4% CHAPS, 1 mM EDTA (pH 8.0), 1 mM PMSF, Protease inhibitor Cocktail Complete, 0.2 mM Na3VO4, 1 mM NaF). The same volume as the homogenization buffer was used to mix completely. Samples of 50 μg of total proteins were fractionated by SDS-PAGE. The gels were stained with Coomassie Blue (PIERCE) for 24 hours. Gel fragments were digested with trypsin. The resulting peptides were analyzed using MS/MS.
Mass spectrometry data sorting
A total of 2962 different proteins were identified by proteomic mass-spectrometry as described above. A ‘protein score’ threshold of >25 was applied, retaining 1175 reliable protein identifications (hereafter referred to as the reference list). For quantification of these protein hits, we used ‘protein matches’ reflecting the count of interpretable MS/MS spectra per protein and this number of peptides identified for each protein is roughly proportional to the quantity of the protein in the sample. Using these protein matches, we separated the proteins into three sub-lists based on a >twofold higher level of protein matches identified in either the wild-type (WT) or Dicer KO samples, resulting in the categorization of 311 and 251 proteins in the WT- and KO-bias list, respectively. The remaining 613 proteins were classified as having similar levels (hereafter referred to as similar-level list).
Protein functional analyses
Functional analyses were performed through the use of IPA (Ingenuity Systems; www.ingenuity.com) and DAVID (http://david.abcc.ncifcrf.gov/home.jsp).
The reference list containing gene identifiers and corresponding expression values was uploaded into the IPA application. Each gene identifier was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base. A cut-off of twofold change of the protein matches value was set to identify genes whose expression was significantly differentially regulated. Biological functions and related pathways of genes that met the twofold change of protein matches value (up- or downregulated, e.g. KO bias and WT bias) were compared against the remainder of the reference list corresponding to the genes identified at similar levels. IPA allows filtering to only consider functions and interactions in protein networks and/or pathways that are known for the defined species and tissue or cell-line range. The stringent filter set for mammals and relaxed filter for nervous system and CNS cell lines were used for the core analyses. Fischer's exact test was used to calculate a P-value determining the probability that each biological function and/or pathway assigned to that data set would be due to chance alone. In order to describe functions specific for the genes remaining at a similar level, we repeated the same analysis with inverted sets using the WT-bias and KO-bias lists as background. Comparison analyses were then done between the core analyses of WT bias, KO bias and similar level. Details on IPA network generation are described in the subsequent section.
For enrichment analyses using DAVID, the whole reference list was uploaded and used as the background list. Individual lists of WT bias, KO bias and similar level were similarly uploaded and analyzed against the background list using the default settings with EASE P-value <0.1. Genes were then classified by their biological processes and molecular function categories according to the GO classification system as well as their canonical pathway according to the KEGG pathway database. The categories for unknown biological processes or molecular function were omitted in the results.
Ingenuity network generation
The network was generated through the use of IPA. The gene lists were uploaded and separated as described in the previous section. The respective gene sets were then overlaid onto a global molecular network developed from information contained in the Ingenuity Pathways Knowledge Base. Networks of these genes were then algorithmically generated based on their connectivity. Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as an edge (line; Fig. 6E). All edges are supported by at least one reference from the literature, from a textbook or from canonical information stored in the Ingenuity Pathways Knowledge Base. Human, mouse and rat orthologs of a gene are stored as separate objects in the Ingenuity Pathways Knowledge Base, but are represented as a single node in the network. The intensity of the node color indicates the degree of up- (red, representing >twofold higher in WT) or down- (green, representing >twofold higher in KO) regulation (Fig. 6E). Nodes are displayed using various shapes that represent the functional class of the gene product. The FMRP hub network was generated by iterative addition of genes out of the reference list connected to FMR1 with traceable direct or indirect interactions using the IPA ‘grow network’ functionality.
Focused analyses
Gene databases focusing either on cell death (including apoptosis, necrosis and necroptosis), EGF, FGF, VEGF, oxidative-stress response or chromatin-related genes were obtained from Panther (http://www.pantherdb.org/), Uniprot (http://www.uniprot.org/) and IPA (Ingenuity Systems, www.ingenuity.com) using keyword searches. Databases from all three sources were compared and combined with redundancy removed. A database for the concept of necroptosis was obtained from the published supplementary data of Hitomi et al. (Hitomi et al., 2008).
Using these collated databases, a perl script was written to compare them with the reference list. Genes identified to be part of the respective focused database were subsequently separated into the KO bias, WT bias or similar level based on the protein matches values.
Supplementary Material
Acknowledgments
We thank Robin Davisson for critical reading of this manuscript. We thank Gregory Hannon for floxed Dicer mice, Robert Hevner for anti-Tbr1 and Tbr2 antibodies, and David Livingston and Chryssa Kanellopoulou for anti-Dicer antibodies. We are grateful for scientific insights from Yufuko Akamatsu and Hitoshi Aihara, and technique support from Kosco Benerjee. This work was supported in part by the A*STAR (Agency for Science Technology And Research, Singapore) (F.E., S.M.-S.), the Whitehall Foundation (T.S.), the Ellison Medical Foundation (T.S.), the Alice Bohmfalk Charitable Trust (T.S.) and a grant R01MH083680 from the NIH/NIMH (T.S.). Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/4/586/DC1
References
- Anthony T. E., Klein C., Fishell G., Heintz N. (2004). Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron 41, 881-890 [DOI] [PubMed] [Google Scholar]
- Baek D., Villen J., Shin C., Camargo F. D., Gygi S. P., Bartel D. P. (2008). The impact of microRNAs on protein output. Nature 455, 64-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capela A., Temple S. (2006). LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev. Biol. 291, 300-313 [DOI] [PubMed] [Google Scholar]
- Carmell M. A., Hannon G. J. (2004). RNase III enzymes and the initiation of gene silencing. Nat. Struct. Mol. Biol. 11, 214-218 [DOI] [PubMed] [Google Scholar]
- Chang T. C., Mendell J. T. (2007). microRNAs in vertebrate physiology and human disease. Annu. Rev. Genomics Hum. Genet. 8, 215-239 [DOI] [PubMed] [Google Scholar]
- Cheng L. C., Tavazoie M., Doetsch F. (2005). Stem cells: from epigenetics to microRNAs. Neuron 46, 363-367 [DOI] [PubMed] [Google Scholar]
- Chenn A., McConnell S. K. (1995). Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 82, 631-641 [DOI] [PubMed] [Google Scholar]
- Chenn A., Walsh C. A. (2002). Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365-369 [DOI] [PubMed] [Google Scholar]
- Couzin J. (2008). MicroRNAs make big impression in disease after disease. Science 319, 1782-1784 [DOI] [PubMed] [Google Scholar]
- Cuellar T. L., Davis T. H., Nelson P. T., Loeb G. B., Harfe B. D., Ullian E., McManus M. T. (2008). Dicer loss in striatal neurons produces behavioral and neuroanatomical phenotypes in the absence of neurodegeneration. Proc. Natl. Acad. Sci. USA 105, 5614-5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis T. H., Cuellar T. L., Koch S. M., Barker A. J., Harfe B. D., McManus M. T., Ullian E. M. (2008). Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J. Neurosci. 28, 4322-4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pietri Tonelli D., Pulvers J. N., Haffner C., Murchison E. P., Hannon G. J., Huttner W. B. (2008). miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 135, 3911-3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C., Fink A., Lau C., Pham D., Daza R. A., Bulfone A., Kowalczyk T., Hevner R. F. (2005). Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci. 25, 247-251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W., Tseng B. S., Dormann H. L., Ueberheide B. M., Garcia B. A., Shabanowitz J., Hunt D. F., Funabiki H., Allis C. D. (2005). Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116-1122 [DOI] [PubMed] [Google Scholar]
- Foshay K. M., Gallicano G. I. (2009). miR-17 family miRNAs are expressed during early mammalian development and regulate stem cell differentiation. Dev. Biol. 326, 431-443 [DOI] [PubMed] [Google Scholar]
- Fukagawa T., Nogami M., Yoshikawa M., Ikeno M., Okazaki T., Takami Y., Nakayama T., Oshimura M. (2004). Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat. Cell Biol. 6, 784-791 [DOI] [PubMed] [Google Scholar]
- Gorski J. A., Talley T., Qiu M., Puelles L., Rubenstein J. L., Jones K. R. (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 22, 6309-6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F. (2005). Cellular and molecular control of neurogenesis in the mammalian telencephalon. Curr. Opin. Cell Biol. 17, 639-647 [DOI] [PubMed] [Google Scholar]
- Harfe B. D., McManus M. T., Mansfield J. H., Hornstein E., Tabin C. J. (2005). The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc. Natl. Acad. Sci. USA 102, 10898-10903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield S. D., Shcherbata H. R., Fischer K. A., Nakahara K., Carthew R. W., Ruohola-Baker H. (2005). Stem cell division is regulated by the microRNA pathway. Nature 435, 974-978 [DOI] [PubMed] [Google Scholar]
- Heins N., Malatesta P., Cecconi F., Nakafuku M., Tucker K. L., Hack M. A., Chapouton P., Barde Y. A., Gotz M. (2002). Glial cells generate neurons: the role of the transcription factor Pax6. Nat. Neurosci. 5, 308-315 [DOI] [PubMed] [Google Scholar]
- Hevner R. F., Hodge R. D., Daza R. A., Englund C. (2006). Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci. Res. 55, 223-233 [DOI] [PubMed] [Google Scholar]
- Hitomi J., Christofferson D. E., Ng A., Yao J., Degterev A., Xavier R. J., Yuan J. (2008). Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311-1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houbaviy H. B., Murray M. F., Sharp P. A. (2003). Embryonic stem cell-specific MicroRNAs. Dev. Cell 5, 351-358 [DOI] [PubMed] [Google Scholar]
- Iwasato T., Datwani A., Wolf A. M., Nishiyama H., Taguchi Y., Tonegawa S., Knopfel T., Erzurumlu R. S., Itohara S. (2000). Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406, 726-731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P., Zarnescu D. C., Ceman S., Nakamoto M., Mowrey J., Jongens T. A., Nelson D. L., Moses K., Warren S. T. (2004). Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat. Neurosci. 7, 113-117 [DOI] [PubMed] [Google Scholar]
- Jin Z., Xie T. (2007). Dcr-1 maintains Drosophila ovarian stem cells. Curr. Biol. 17, 539-544 [DOI] [PubMed] [Google Scholar]
- John B., Enright A. J., Aravin A., Tuschl T., Sander C., Marks D. S. (2004). Human MicroRNA targets. PLoS Biol. 2, e363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson R. L., Babiarz J. E., Venere M., Blelloch R. (2009). Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat. Biotechnol. 27, 459-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C., Muljo S. A., Kung A. L., Ganesan S., Drapkin R., Jenuwein T., Livingston D. M., Rajewsky K. (2005). Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 19, 489-501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase-Koga Y., Otaegi G., Sun T. (2009). Different timings of dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev. Dyn. 238, 2800-2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokovay E., Shen Q., Temple S. (2008). The incredible elastic brain: how neural stem cells expand our minds. Neuron 60, 420-429 [DOI] [PubMed] [Google Scholar]
- Kosik K. S. (2006). The neuronal microRNA system. Nat. Rev. Neurosci. 7, 911-920 [DOI] [PubMed] [Google Scholar]
- Krichevsky A. M., Sonntag K. C., Isacson O., Kosik K. S. (2006). Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells 24, 857-864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A. R. (2005). Constructing circuits: neurogenesis and migration in the developing neocortex. Epilepsia 46Suppl. 7, 15-21 [DOI] [PubMed] [Google Scholar]
- Lee R. C., Feinbaum R. L., Ambros V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843-854 [DOI] [PubMed] [Google Scholar]
- Merkle F. T., Alvarez-Buylla A. (2006). Neural stem cells in mammalian development. Curr. Opin. Cell Biol. 18, 704-709 [DOI] [PubMed] [Google Scholar]
- Mills K. D., Ferguson D. O., Essers J., Eckersdorff M., Kanaar R., Alt F. W. (2004). Rad54 and DNA Ligase IV cooperate to maintain mammalian chromatid stability. Genes Dev. 18, 1283-1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux B. J., Arlotta P., Menezes J. R., Macklis J. D. (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427-437 [DOI] [PubMed] [Google Scholar]
- Murchison E. P., Partridge J. F., Tam O. H., Cheloufi S., Hannon G. J. (2005). Characterization of Dicer-deficient murine embryonic stem cells. Proc. Natl. Acad. Sci. USA 102, 12135-12140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M., Schuurmans C., Britz O., Guillemot F. (2001). Neural bHLH genes control the neuronal versus glial fate decision in cortical progenitors. Neuron 29, 401-413 [DOI] [PubMed] [Google Scholar]
- Noctor S. C., Martinez-Cerdeno V., Ivic L., Kriegstein A. R. (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 7, 136-144 [DOI] [PubMed] [Google Scholar]
- Park J. K., Liu X., Strauss T. J., McKearin D. M., Liu Q. (2007). The miRNA pathway intrinsically controls self-renewal of Drosophila germline stem cells. Curr. Biol. 17, 533-538 [DOI] [PubMed] [Google Scholar]
- Perrier A. L., Studer L. (2003). Making and repairing the mammalian brain-in vitro production of dopaminergic neurons. Semin. Cell Dev. Biol. 14, 181-189 [DOI] [PubMed] [Google Scholar]
- Reynolds B. A., Tetzlaff W., Weiss S. (1992). A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 12, 4565-4574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak A., Fuchs H., Smirnova L., Brandt C., Pohl E. E., Nitsch R., Wulczyn F. G. (2008). A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat. Cell Biol. 10, 987-993 [DOI] [PubMed] [Google Scholar]
- Schaefer A., O'Carroll D., Tan C. L., Hillman D., Sugimori M., Llinas R., Greengard P. (2007). Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 204, 1553-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M., Schwanhausser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58-63 [DOI] [PubMed] [Google Scholar]
- Shen Q., Goderie S. K., Jin L., Karanth N., Sun Y., Abramova N., Vincent P., Pumiglia K., Temple S. (2004). Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 304, 1338-1340 [DOI] [PubMed] [Google Scholar]
- Shen Q., Wang Y., Dimos J. T., Fasano C. A., Phoenix T. N., Lemischka I. R., Ivanova N. B., Stifani S., Morrisey E. E., Temple S. (2006). The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat. Neurosci. 9, 743-751 [DOI] [PubMed] [Google Scholar]
- Stark K. L., Xu B., Bagchi A., Lai W. S., Liu H., Hsu R., Wan X., Pavlidis P., Mills A. A., Karayiorgou M., et al. (2008). Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751-760 [DOI] [PubMed] [Google Scholar]
- Takahashi T., Nowakowski R. S., Caviness V. S., Jr (1993). Cell cycle parameters and patterns of nuclear movement in the neocortical proliferative zone of the fetal mouse. J. Neurosci. 13, 820-833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan S. R., Daley G. Q., Gregory R. I. (2008). Selective blockade of microRNA processing by Lin28. Science 320, 97-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B., Ha I., Ruvkun G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855-862 [DOI] [PubMed] [Google Scholar]
- Wrobel C. N., Mutch C. A., Swaminathan S., Taketo M. M., Chenn A. (2007). Persistent expression of stabilized beta-catenin delays maturation of radial glial cells into intermediate progenitors. Dev. Biol. 309, 285-297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N., Papagiannakopoulos T., Pan G., Thomson J. A., Kosik K. S. (2009). MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 137, 647-658 [DOI] [PubMed] [Google Scholar]
- Xu X. L., Li Y., Wang F., Gao F. B. (2008). The steady-state level of the nervous-system-specific microRNA-124a is regulated by dFMR1 in Drosophila. J. Neurosci. 28, 11883-11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Duan R., Chen D., Wang J., Jin P. (2007). Fragile X mental retardation protein modulates the fate of germline stem cells in Drosophila. Hum. Mol. Genet. 16, 1814-1820 [DOI] [PubMed] [Google Scholar]
- Yang Y., Xu S., Xia L., Wang J., Wen S., Jin P., Chen D. (2009). The bantam microRNA is associated with drosophila fragile X mental retardation protein and regulates the fate of germline stem cells. PLoS Genet. 5, e1000444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Deng W., Gage F. H. (2008). Mechanisms and functional implications of adult neurogenesis. Cell 132, 645-660 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}