

Abstract
In just a few years, glycogen synthase kinase-3 (GSK3) has transformed from an obscure enzyme seldom encountered in the immune literature to one implicated in an improbably large number of roles. GSK3 is a crucial regulator of the balance between pro- and anti-inflammatory cytokine production in both the periphery and the central nervous system, endowing GSK3 inhibitors such as lithium with the capacity to diminish inflammation. T cell proliferation, differentiation, and survival are influenced by GSK3. Many effects stem from GSK3 regulation of critical transcription factors, such as NF-κB, NFAT and STATs. These discoveries led to the rapid application of GSK3 inhibitors to animal models of sepsis, arthritis, colitis, multiple sclerosis, and others that demonstrated their potential for therapeutic interventions.
Introductory overview
The innate and adaptive immune systems are crucial for sustaining life but can also contribute to a host of debilitating diseases. Investigators have wrestled with numerous strategies to maintain or restore a healthy balance in the activities of these systems. During the last few years, the ubiquitous serine/threonine kinase glycogen synthase kinase-3 (GSK3) was identified as a regulator of many components of the immune system, suggesting it may be a plausible therapeutic target in inflammatory and autoimmune diseases. Although unobtrusively named due to its initial identification as an enzyme phosphorylating glycogen synthase, GSK3 has since been found to be a point of convergence of many signaling pathways and to regulate many cellular functions through its capacity to phosphorylate over 50 substrates [1]. The complexity of actions of GSK3 is mirrored by the complex mechanisms that regulate its actions (Box 1). Ironically, GSK3 is inhibited by the cation lithium, the simplest of all drugs used therapeutically in humans [2]. Lithium is the classic therapeutic treatment for bipolar disorder (previously called manic-depression), and exerts a broad range of effects on immune cells (Box 2). The complexities of GSK3 regulation offer multiple strategies to control GSK3, for example by regulating individual kinases that phosphorylate GSK3 or the association of proteins with GSK3 in complexes that are specific for individual signaling pathways, and the availability of an inhibitor approved for human use promises rapid application for new intervention objectives. Here we review current knowledge about the roles of GSK3 in innate and adaptive immunity and summarize preliminary animal testing using GSK3 inhibitors in animal models of a rapidly expanding number of diseases.
Box 1. Regulation of GSK3.
“GSK3” designates two isoforms, GSK3α and GSK3β, that are ubiquitously expressed, highly homologous, and usually have equivalent actions. GSK3 is different from most kinases in that it is constitutively partially active, and the most common regulatory mechanism is inhibition by phosphorylation on serine21-GSK3α and serine9-GSK3β. This inhibitory phosphorylation can be mediated by several kinases, such as Akt/protein kinase B (PKB), protein kinase C (PKC), and protein kinase A (PKA). Thus, many signaling pathways converge on GSK3 to inhibit its activity via serine21/9-phosphorylation. Additionally, the activity of GSK3 is optimal when phosphorylated on the regulatory tyrosine279-GSK3α and tyrosine216-GSK3β. GSK3 is known to phosphorylate more than 50 substrates, so precise regulation is needed to direct or inhibit its phosphorylation of specific substrates. Substrate-selective actions of GSK3 can be regulated by three other mechanisms: (1) by the dynamic association of GSK3 in protein complexes; (2) by the dynamic regulation of the subcellular localization of GSK3 or localized regulation of its inhibitory serine-phosphorylation, such as regulated nuclear transport of GSK3 or regulation of its phosphorylation in mitochondria; and (3) by the phosphorylation state of its substrate. Most substrates of GSK3 must be ‘primed’, i.e., pre-phosphorylated at a residue 4-amino acids C-terminal to the GSK3 phosphorylation site. This necessitates temporal coordination of the activity of the priming kinase along with GSK3 activity for GSK3 to phosphorylate the primed substrate.
Lithium has been used in human patients as a mood stabilizer for the treatment of bipolar disorder for over 50 years [76]. Lithium is a direct inhibitor of GSK3 [2] and also increases the inhibitory serine-phosphorylation of GSK3 [77]. During the last decade, much evidence indicates that inhibition of GSK3 by lithium is important for its therapeutic mood stabilizing action. Thus, lithium is a valuable experimental tool for inhibiting GSK3 in vivo and it provides a feasible therapeutic intervention for conditions requiring GSK3 inhibition, such as inflammation. GSK3 is also inhibited by other drugs currently used therapeutically, such as valproate acid, by new selective inhibitors developed during the last decade, and by a number of hormones (e.g., insulin) and neurotrophins (e.g., brain-derived neurotrophic factor) that may influence inflammation in part by controlling the activity of GSK3 [65,78,79].
Box 1 figure.
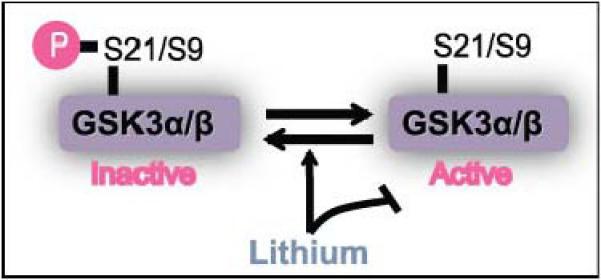
The activity of the GSK3 isoforms α and β are regulated by phosphorylation of serine 21 and 9 respectively. Lithium is the best characterized pharmacological inhibitor of GSK3 activity.
Box 2. Effects of the GSK3 inhibitor lithium on inflammation and immune cells.
Inflammation and aberrant immune responses have long been suspected of contributing to certain psychiatric diseases (reviewed in [40,41]). Since psychiatric patients often are treated with lithium (see Box 1), many investigators have examined whether lithium alters immune cells or inflammatory factors in these patient populations. Lithium treatment causes leukocytosis (increased white blood cells derived from hematopoietic stem cells), particularly granulocytosis and neutrophilia, which led to its use to treat multiple causes of neutropenia (neutrophil depletion). This is partially mediated by lithium eliciting granulocyte colony-stimulating factor (G-CSF) production which increases hematopoietic stem cells (reviewed in [80,81]). Lithium also causes lymphopenia, with a reduction in CD4+, but not CD8+ T cells, possibly due to the differentiation of stem cells into granulocytes instead of lymphocytes, however lithium increases IL-2 production and lymphocyte proliferation during in vitro stimulation (reviewed in [80,82]). Wnt signaling or β-catenin accumulation caused by GSK3 inhibitors arrested CD8+ T cell development into effector cells and promoted the generation of self-renewing multipotent CD8+ memory cells, and promoted the survival of T regulatory (Treg) cells [38,39]. In vivo and in vitro studies of lithium on cytokine production have produced mixed results, likely due to the complexity and diversity of the patient populations, the many factors that influence cytokine production, and variations in experimental protocols (reviewed in [81-83]). However, an extensive study reported an inflammatory signature associated with bipolar disorder and broad anti-inflammatory effects of in vivo lithium treatment [84]. Elegant studies have demonstrated that lithium reduces the pro-inflammatory enzyme phospholipase A2 and the turnover of its product arachidonate [85]. Several dermatological effects of lithium, such as acne, rash, exfoliative dermatitis, and psoriasis, may be linked to its regulation of inflammatory and immune responses (reviewed in [86]). In retrospect, inhibition of GSK3 may underlie many of these lithium effects, but GSK3 was not examined because many studies were conducted in vivo or before GSK3 was identified as a target of lithium. Also, some of these effects of lithium may be due to its inhibition of other enzymes, which include phosphoglucomutase, inositol monophosphatase, and bisphosphate 3′-nucleotidase 1 [78,87]. Thus, while lithium clearly inhibits GSK3 and this is likely critical for its therapeutic actions in psychiatric diseases, GSK3 has yet to be identified as the target in many of the effects of lithium on immune cells and inflammatory processes.
The mammalian innate immune system activates defensive mechanisms within minutes of microbial invasion, followed by a slower antigen-specific response mounted by the adaptive immune system. Innate immune cells use pathogen recognition receptors (PRRs), such as Toll-like receptors (TLRs), to recognize pathogen-associated molecular patterns (PAMPs) of microbes. Activation of PRRs upregulates the expression of the major histocompatibility complexes (MHC I and II) and co-stimulatory molecules and stimulates inflammatory cytokine secretion, which help guide the subsequent activation of the adaptive immune response. The activated adaptive immune cells further promote actions on the innate immune system, for instance type I T helper (Th1) cells secrete interferon-γ (IFNγ) to activate macrophages, cytokines secreted by Th2 cells activate eosinophils, and IL-17 secreted by Th17 cells, along with IL-23 secreted by APCs, are suspected of driving autoimmune inflammation in the central nervous system (CNS). Thus, innate and adaptive immune cells are sequentially activated and mutually regulate one another. As summarized below, GSK3 exerts powerful influences on both of these arms of the immune system, and GSK3-dependent actions are associated with the development of a broad range of diseases.
GSK3 regulates innate immunity
The crucial role of GSK3 in inflammation was established by the finding that active GSK3 is necessary for pro-inflammatory cytokine production following stimulation of TLRs [3]. For example, GSK3 deficiency induced pharmacologically with lithium or other GSK3 inhibitors or by molecular manipulations reduced by 67-90% the production of proinflammatory interleukin-6 (IL-6), IL-1β, IL-12p40, IFNγ and tumor necrosis factor (TNF) by TLR-stimulated monocytes [3]. IFNγ amplifies lipopolysaccharide (LPS)-induced inflammatory cytokine production, and this amplification is dependent on active GSK3 through mechanisms involving inhibition of IL-10 production [4] and activation of the transcription factor STAT3 [5]. IFNγ-induced STAT1 activation was reported to be initially independent of GSK3, but persistent STAT1 activation and expression of the proinflammatory mediators TNFα, RANTES, and iNOS were diminished by inhibiting GSK3 in RAW264.7 macrophages [6]. Remarkably, GSK3 can differentially regulate monocyte production of the anti-inflammatory cytokine IL-10 [3,7,8]. Inhibition of GSK3 increases LPS-induced IL-10 production by regulating transcription factors (Figure 1), and increases production of the anti-inflammatory IL-1 receptor antagonist by regulating mitogen activated protein (MAP) kinases [9]. GSK3 inhibition also promotes LPS-stimulated IFNβ production via its ability to regulate c-Jun activity [10]. Cytokine regulation by GSK3 also occurs in cell types other than monocytes, such as natural killer (NK) cells [11], macrophage-derived RAW264.7 cells [5], and dendritic cells (DC) [12,13], where GSK3 regulation of IL-12 expression can control the Th1/Th2 balance [12], as well as in cells of the CNS as discussed below. Much of this regulation by GSK3 is mediated by its control of transcription factors that activate cytokine expression (Figures 1 and 2). Thus, it is now evident that GSK3 inhibitors are powerful tools that are able to shift the balance of cytokines from pro-inflammatory to anti-inflammatory, which may have therapeutic applications for inflammatory conditions.
Figure 1.

NF-κB, CREB, and AP-1 transcription factor regulation by GSK3. GSK3 regulates the activities of over 20 transcription factors, including several that are important for immune function. NF-κB is a key inflammatory transcription factor, but the GSK3 regulatory influences on NF-κB are complex due to cell-selective, stimulus-selective, and promoter-selective interactions that can be stimulatory or inhibitory (reviewed in [66]). GSK3 is required for NF-κB-mediated expression of several proinflammatory cytokines [3,16]. Promoter-selective regulation of NF-κB has been demonstrated wherein GSK3 is required for expression of IL-6 and monocyte chemoattractant protein-1 (MCP-1/CCL2), but not of IκBα and macrophage inflammatory protein-2 (MIP-2/CXCL2) [67]. In certain conditions, GSK3 can also inhibit NF-κB [68], and GSK3-mediated phosphorylation of p65-NF-κB promotes the docking of Nurr1 and the CoREST corepressor complex to reduce stimulated NF-κB transcriptional activity to the basal level [69]. Thus, the effects of GSK3 on NF-κB are context-specific, and involve both activation and subsequent feedback inhibition of NF-κB activity. GSK3 dampens the production of anti-inflammatory IL-10 by suppressing transcriptional activity of CREB and AP-1, both of which contribute to IL-10 expression [3,8]. The opposing effects of GSK3 on pro- and anti-inflammatory cytokines are at least partially mediated by competition between NF-κB and CREB for the co-activator CREB-binding protein (CBP), which is well-known to be present in limited amounts and for which transcription factors compete. Red arrows signify actions regulated by GSK3.
Figure 2.

GSK3 regulation of STATs, Smads, β-catenin, and NFAT. Among the signal transducer and activator of transcription (STAT) family of transcription factors that are critical mediators of responses to cytokines and growth factors, GSK3 selectively promotes STAT3 and STAT5 activation [5,46,49]. Thus, regulation of transcription factors by GSK3 modulates both the expression of inflammatory molecules and cellular responses stimulated by cytokines. GSK3 also regulates Smad transcription factors that transduce signals generated by TGF-β and other stimulants of cell surface receptors to regulate gene expression [70-72]. The transcriptional co-activator β-catenin is phosphorylated by GSK3 in the Wnt signaling pathway, which targets β-catenin for proteosomal degradation. Inhibitors of GSK3 or activation of Wnt signaling induce stabilization of β-catenin that leads to its nuclear transport and accumulation to promote Tcf/LEF-mediated transcription. β-catenin also modulates activities of other transcription factors. For example, β-catenin negatively regulates bacteria-induced inflammation by inhibiting NF-κB [73], providing an indirect mechanism for GSK3 to promote NF-κB activity by down-regulating β-catenin. Increased β-catenin activity often contributes to the proliferation and survival of immune cells. β-catenin levels are low in unstimulated B cells, but B cell receptor activation triggers accumulation of β-catenin after inducing inhibition of GSK3 [74]. The T cell receptor (TCR) activates the NFATc (nuclear factor of activated T cells) family of transcription factors that promote antigenic and tolerogenic genetic programs. TCR signals induce nuclear localization of NFAT and GSK3 inactivates NFATc by phosphorylation-dependent stimulation of NFATc nuclear export [75]. Therefore, inhibition of GSK3 keeps NFATc active, mimicking the CD28 costimulatory signal for T cell proliferation [26,29,31]. Red arrows signify actions regulated by GSK3.
Although GSK3 predominantly abets inflammatory signaling, exceptions have been reported. For example, activation of the IL-17 receptor inhibits GSK3, and GSK3 inhibitors promote IL-17-induced IL-6 production [14], LPS-induced TNFα expression in cardiomyocytes [15], and TNF-induced IL-6 expression in endothelial cells [16]. These paradoxical pro-inflammatory actions of GSK3 inhibitors are likely attributable to complex interactions of multiple transcription factors that contribute to inflammatory signals that are differentially regulated by GSK3 dependent on the context of cell type and stimulus.
Other inflammatory molecules are also regulated by GSK3. Lipid mediators, such as prostaglandin E2 (PGE2), are generated from arachidonic acid by cyclooxygenases (COX) and PGE synthases. PGE2 exerts anti-inflammatory actions on macrophages and T cells, profoundly influencing T cell differentiation toward Th2 by inhibiting the production of IL-12p70, TNFα, IL-6, and the chemokines CCL3, and CCL4, and promoting IL-10 production, these actions are all associated with PGE2-induced inhibition of GSK3 [17]. PGD2-induced inhibition of GSK3 also may modulate Th2-mediated cytokine production and chemotaxis [18]. These and other findings indicate that inhibition of GSK3 induced by prostaglandins is critical for several cellular effects. The production of nitric oxide (NO), synthesized from L-arginine by NO synthase, also is dependent on GSK3 [19], and NO influences the production of cytokines by macrophages [4,20]. Other regulatory actions of GSK3 on the innate immune system have begun to be identified, although they have not been as extensively investigated as cytokine production.
The adaptive immune response depends on successful antigen presentation by MHC and MHC-like molecules, and recent findings raise the possibility that GSK3 is involved in antigen presentation by antigen-presenting cells. Stable expression of MHC I or II on the cell surface involves antigen-presenting cells, such as dendritic cells, displaying peptides in association with MHC I molecules for presentation to CD8+ T cells, while MHC II molecules present antigenic peptides derived from exogenous proteins to CD4+ T cells. GSK3 is essential for the expression of the NK cell ligands MHC I-related chain A (MICA) and B (MICB) expression on cancer cells [21]. Further, the major histocompatibility class II transactivator (CIITA), a master regulator of MHC II expression, is a substrate of GSK3 [22]. GSK3 also profoundly influences DC differentiation and maturation by suppressing spontaneous maturation and enhancing cytokine production after CD40L- or E. coli-dependent activation [13]. GSK3 phosphorylates the adhesion-associated protein paxillin after LPS stimulation of RAW264.7 macrophages [23], which suggests that GSK3 may regulate migration of innate immune cells, as it does many other cell types [as reviewed in [24]], an action potentially critical for focusing inflammatory molecule production at a site of infection.
Taken together, GSK3 regulates many functions of the innate immune system that will be further clarified in the near future, and could be exploited to develop therapies for diseases involving inflammation.
GSK3 regulates adaptive immunity
GSK3 modulates both of the key aspects of adaptive immunity, i.e., specificity and clonal expansion, by direct actions on proliferation and survival, and indirectly by modifying the repertoire of cytokines produced and influencing differentiation and anergy.
The antigen-specific response of the adaptive immune system is delayed relative to the innate response. T cell activation is induced by ligation of the antigen-receptor complex (T cell receptor (TCR)/CD3). Stimulation of co-receptors, such as CD28, provides maximal and sustained activation and proliferation of T cells, influences the response to T cell-dependent antigens, prevents anergy, and supports cell survival. Stimulation of CD28 inhibits GSK3 by increased inhibitory serine-phosphorylation mediated by the phosphatidylinositol 3-kinase (PI3K) pathway but independently of guanine nucleotide exchange factor Vav-1 [25]. By inhibiting GSK3, CD28 relieves the inhibition on NFAT, therefore providing a mechanism for cells to monitor receptor occupancy to maintain T cell responses [25-28]. Drug-induced inhibition of GSK3 in vitro mimics the CD28 costimulatory signal, at least partially substituting for T cell costimulation [26,29]. Maintenance of GSK3 inhibition is critical for CD4+ and CD8+ T cell survival after activation [30]. However, memory CD4+ T cells are less dependent than naïve CD4+ T cells on inhibition of GSK3 for proliferative responses [27]. Expression of constitutively active GSK3β decreased proliferation of CD8+ T cells and suppressed TCR-induced IL-2 production [31], whereas inhibition of GSK3 increased IL-2 production in both CD4+ and CD8+ T cells [27,31]. Similar to the innate immune system, GSK3 inhibition reduced the production of several proinflammatory cytokines by splenocytes stimulated by myelin oligodendrocyte glycoprotein peptide (MOG33-55) after isolation from experimental autoimmune encephalitis (EAE)-induced mice [32] and increased the production of anti-inflammatory IL-10 by memory CD4+ T cells [27] and by B cells [33]. T cell migration across endothelial cell barriers separating blood from tissue is critical for accessing targets, and failure to clear pathogens can result from blocked migration, e.g., due to the lack of necessary recognition molecules. T cell motility is promoted by the chemokine CXCL12 which inhibits GSK3-mediated phosphorylation of CRMP2 [34], and by GSK3 inhibition by Wnt signaling, which induces expression of metalloproteinases that remodel the extracellular matrix [35]. Thus, in the last few years, investigators have found several critical T cell processes that are regulated by GSK3, and identified actions of GSK3 in the adaptive immune system that will undoubtedly expand as this topic receives more attention.
Although it is clear that active GSK3 impairs T cell proliferation, differentiation, survival, and several other functions, the targets phosphorylated by GSK3 to cause these effects remain to be clearly established. However, several transcription factors have been shown to be critical for the regulation of T cell function by GSK3. As described in Figures 1 and 2, the transcription factors NF-κB, CREB, AP-1, STATs, NFAT, Smads, and β-catenin are each regulated by GSK3 to influence immune responses. For example, Wnt-induced dissociation of GSK3 from the β-catenin destruction complex leads to β-catenin activation that promotes survival, proliferation, and differentiation of T lymphocyte progenitors [36,37]. β-catenin also enhances Treg and CD8+ memory T cell survival, providing a potential mechanism by which inhibitors of GSK3 could down-modulate T cell-mediated immune responses [38,39]. GSK3 also impedes T cell proliferation by stabilizing the cyclin-dependent kinase inhibitor p27kip1 [26,27]. Furthermore, mitogenic stimulation of T lymphocytes causes a rapid activation of protein synthesis, in part due to increased expression of many translational components such as the initiation factor eIF2B. eIF2B is phosphorylated by GSK3, which inhibits nucleotide exchange, and this inhibition is released by TCR activation-induced inhibition of GSK3 [40]. Further mechanistic studies are likely to identify additional substrates of GSK3, including transcription factors and other proteins that mediate its functional effects on adaptive immune responses.
GSK3 regulates neuroinflammation
Neuroinflammation refers to inflammatory events in the CNS that may include infiltration of cells (e.g., macrophages, lymphocytes), as well as increased inflammatory molecules that are generated centrally or peripherally and subsequently in the CNS. Neuroinflammation is evident in trauma and many neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis and likely contributes to psychiatric diseases, such as major depression, and during pregnancy or early postnatal periods it appears to contribute to developmental disorders, such as autism and schizophrenia [41,42]. Inflammation exacerbates neuronal damage induced by ischemia, which is lessened in TLR4 knockout mice that exhibit inhibited GSK3 in the hippocampus [43], and GSK3 inhibitors provide significant neuronal protection from ischemia [44]. Inflammation is an important pathological component of diabetes that might be controlled by GSK3 inhibitors [24] and treatment with insulin, which causes inhibition of GSK3, or a selective GSK3 inhibitor reduced ischemia/reperfusion-induced inflammation and infarct volume in diabetic rats [45]. In the CNS, inflammatory molecules can be produced by both microglia (often called the “macrophages of the CNS”) and astrocytes. Similar to peripheral cells, GSK3 in astrocytes and microglia promotes the production of several proinflammatory cytokines (IL-6, TNF), chemokines (IL-8, RANTES, CXCL10), and NO [19,46-48], whereas production of the anti-inflammatory cytokine IL-10 is inhibited by GSK3 [48]. GSK3 promotes astrocyte activation, which is characterized by upregulation of GFAP (glial fibrillary acidic protein) expression, and migration [46,49], and microglia migration and activation [19]. Inhibition of GSK3 is critical for promoting survival of microglia during oxygen-glucose deprivation [50], and induced by erythropoietin [51], which is thought to be critical for effective tissue repair and reorganization necessary for avoiding permanent CNS damage. Furthermore, GSK3 inhibitors provide protection from inflammation-induced neuronal toxicity [19]. These findings indicate that GSK3 inhibitors that can enter the CNS, such as lithium, may provide protection from the deleterious effects of neuroinflammation occurring after trauma and infection, as well as the neuroinflammation that is associated with neurodegenerative and psychiatric diseases.
Rodent models of disease: amelioration of symptoms by GSK3 inhibitors
Endotoxic shock, commonly known as sepsis and its sequela septic shock, is a challenging inflammatory condition because it involves a massive production of cytokines and other inflammatory mediators that cause high mortality. Strikingly, in vivo administration of GSK3 inhibitors provided protection from endotoxic shock sufficiently enough to allow survival of 70% of mice from an otherwise 100% lethal LPS dose [3]. GSK3 inhibitor-induced survival was accompanied by reduced pro-inflammatory and increased anti-inflammatory cytokine production. These findings showed for the first time the powerful ability of GSK3 inhibitors to shift the balance of an in vivo pro-inflammatory response to an anti-inflammatory one, and revealed the therapeutic potential for these drugs in inflammatory conditions [3].
Application of GSK3 inhibitors to control inflammation rapidly expanded to other conditions, often resulting in large, “clinically” significant outcomes. Collagen-induced arthritis was ameliorated by in vivo administration of GSK3 inhibitors, with ~80% reduction of joint swelling [8] and diminished histologically-graded damage [52]. GSK3 inhibitor administration counteracted TLR2-dependent peritonitis in mice, reducing peritoneal cells by ~60% [8]. In other rodent models, GSK3 inhibitors reduced by ~75% the systemic inflammation, renal dysfunction, and hepatotoxicity in endotoxemia [53], attenuated asthma [54], diminished sepsis-associated muscle protein degradation [55], reduced inflammation in experimentally-induced colitis [56], and reduced by 67-84% inflammation-induced organ injury caused by hemorrhage and resuscitation [57]. GSK3 inhibitors reduced inflammation associated with the injury of several organs, including airway [58], heart [59], and kidney [60]. The GSK3 inhibitor lithium also promoted the survival of lupus-prone mice [61] and of mice challenged with a lethal dose of the bacteria Francisella tularensis [62]. Thus, in vivo administration of GSK3 inhibitors provides a new strategy to reduce inflammation and has proven effective in a wide range of peripheral inflammatory conditions in rodent models.
Administration of GSK3 inhibitors also has been shown to control neuroinflammation in the CNS. Treatment with GSK3 inhibitors reduced the development of inflammation and tissue injury associated with spinal cord trauma, significantly blocking the development of hind limb motor impairments [63]. Administration of the GSK3 inhibitor lithium was remarkably effective in treating EAE, which models in mice some of the clinical, immunological, and neuropathological features of multiple sclerosis [32]. Multiple sclerosis is the most common inflammatory demyelinating disease of the CNS and is widely thought to be an autoimmune disease involving proinflammatory Th1 and Th17 cells that infiltrate the CNS [64]. EAE is induced in susceptible mice by eliciting immune responses to myelin antigens. The initial response encompasses activation of the immune system, resulting in the production of many inflammatory molecules, such as the cytokines IL-6 and IL-17. Both infiltrating immune cells and CNS-resident glia produce multiple cytokines and other inflammatory molecules to amplify neuroinflammation and cause demyelination and axonal damage. Pretreatment with the GSK3 inhibitor lithium in vivo suppressed clinical symptoms of EAE, demyelination, microglia activation, and leukocyte infiltration in the spinal cord of mice [32]. In relapsing/remitting EAE, lithium administered after the first relapse episode maintained long-term protection, and after lithium withdrawal the disease rapidly relapsed. In vitro, lithium suppressed MOG35-55-induced IFNγ, IL-6 and IL-17 production by splenocytes isolated from MOG35-55-immunized mice [32]. These findings demonstrate that lithium suppresses EAE and may be useful for therapeutic intervention for neuroinflammatory conditions, such as multiple sclerosis.
Conclusions
During the last few years, GSK3 leapt from being an obscure metabolic kinase to being recognized as profoundly regulating many components of the innate and adaptive immune systems, and to be considered as a valid therapeutic target in a rapidly growing number of diseases. Inhibitors of GSK3 greatly influence the cytokine and chemokine repertoire induced by inflammatory stimuli, generally promoting anti-inflammatory outcomes. Being constitutively active, GSK3 possesses an optimal characteristic for contributing to rapid innate responses to PAMPs since no intermediary steps are required for its activation. This logical utilization of GSK3 in the innate immune system provides a therapeutic opportunity to dampen such responses by diminishing the activity of GSK3 via pharmacological or molecular approaches during conditions of persistent or excessive inflammation, including conditions involving neuroinflammation. It is already evident that GSK3 inhibitors can have therapeutic effects in a number of animal models of inflammatory conditions. It is also evident that GSK3 modulates aspects of the adaptive immune system, particularly at the level of antigen-presenting cells and T cells. The broad array of immune actions affected by GSK3 is partly, but not entirely, attributable to the remarkable number of crucial transcription factors regulated by GSK3. Since lithium is already clinically approved and effective, its use in human inflammatory and autoimmune diseases could be taken up rapidly. Also, many new selective inhibitors of GSK3 have been developed in the last few years that might prove useful for controlling inflammatory diseases, although only one GSK-3 inhibitor called NP12, has advanced to trials in human patients [65]. It appears that the field is only at the initial cusp of understanding the roles of GSK3 in immune responses and the potential application of GSK3-modifying interventions for therapies. Many questions remain unanswered, such as identifying potential differential sensitivities of immune responses to GSK3 inhibition and the impact on therapeutic applications, the extent of GSK3 inhibition that will be necessary to be effective clinically, and if this inhibition is tolerated, that should be addressed in the near future.
Acknowledgements
Supported by a Young Investigator Award from NARSAD (to E.B.) and the National Institutes of Health grants MH38752 (to R.S.J.) and DE09081 (to S.M.). We apologize to those investigators whose work could not be cited because of space limitations.
References
- 1.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin M, et al. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin CF, et al. IFN-γ synergizes with LPS to induce nitric oxide biosynthesis through glycogen synthase kinase-3-inhibited IL-10. J Cell Biochem. 2008;105:746–755. doi: 10.1002/jcb.21868. [DOI] [PubMed] [Google Scholar]
- 5.Beurel E, Jope RS. Glycogen synthase kinase-3 promotes the synergistic action of interferon-γ on lipopolysaccharide-induced IL-6 production in RAW264.7 cells. Cell Signal. 2009;21:978–985. doi: 10.1016/j.cellsig.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai CC, et al. Glycogen synthase kinase-3β facilitates IFN-γ-induced STAT1 activation by regulating Src homology-2 domain-containing phosphatase 2. J Immunol. 2009;183:856–864. doi: 10.4049/jimmunol.0804033. [DOI] [PubMed] [Google Scholar]
- 7.Chan MM, et al. A role for glycogen synthase kinase-3 in antagonizing mycobacterial immune evasion by negatively regulating IL-10 induction. J Leukoc Biol. 2009;86:283–291. doi: 10.1189/jlb.0708442. [DOI] [PubMed] [Google Scholar]
- 8.Hu X, et al. IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 9.Rehani K, et al. Toll-like receptor-mediated production of IL-1Ra is negatively regulated by GSK3 via the MAPK ERK1/2. J Immunol. 2009;182:547–553. doi: 10.4049/jimmunol.182.1.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, et al. IFN-β production by TLR4-stimulated innate immune cells is negatively regulated by GSK3-β. J Immunol. 2008;181:6797–6802. doi: 10.4049/jimmunol.181.10.6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aoukaty A, Tan R. Role for glycogen synthase kinase-3 in NK cell cytotoxicity and X-linked lymphoproliferative disease. J Immunol. 2005;174:4551–4558. doi: 10.4049/jimmunol.174.8.4551. [DOI] [PubMed] [Google Scholar]
- 12.Ohtani M, et al. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood. 2008;112:635–643. doi: 10.1182/blood-2008-02-137430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodionova E, et al. GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood. 2007;109:1584–1592. doi: 10.1182/blood-2006-06-028951. [DOI] [PubMed] [Google Scholar]
- 14.Shen F, et al. IL-17 receptor signaling inhibits C/EBPβ by sequential phosphorylation of the regulatory 2 domain. Sci Signal. 2009;2:ra8. doi: 10.1126/scisignal.2000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen E, et al. Glycogen synthase kinase-3β suppresses tumor necrosis factor-α expression in cardiomyocytes during lipopolysaccharide stimulation. J Cell Biochem. 2008;104:329–338. doi: 10.1002/jcb.21629. [DOI] [PubMed] [Google Scholar]
- 16.Vines A, et al. Novel anti-inflammatory role for glycogen synthase kinase-3β in the inhibition of tumor necrosis factor-α- and interleukin-1β-induced inflammatory gene expression. J Biol Chem. 2006;281:16985–16990. doi: 10.1074/jbc.M602446200. [DOI] [PubMed] [Google Scholar]
- 17.Jing H, et al. A novel signaling pathway mediates the inhibition of CCL3/4 expression by prostaglandin E2. J Biol Chem. 2004;279:55176–55186. doi: 10.1074/jbc.M409816200. [DOI] [PubMed] [Google Scholar]
- 18.Xue L, et al. Inhibition of PI3K and calcineurin suppresses chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2)-dependent responses of Th2 lymphocytes to prostaglandin D(2) Biochem Pharmacol. 2007;73:843–853. doi: 10.1016/j.bcp.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 19.Yuskaitis CJ, Jope RS. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell Signal. 2009;21:264–273. doi: 10.1016/j.cellsig.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuzzocrea S, et al. Effects of glycogen synthase kinase-3β inhibition on the development of cerulein-induced acute pancreatitis in mice. Crit Care Med. 2007;35:2811–2821. doi: 10.1097/01.ccm.0000295303.62996.9f. [DOI] [PubMed] [Google Scholar]
- 21.Skov S, et al. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005;65:11136–11145. doi: 10.1158/0008-5472.CAN-05-0599. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, et al. CIITA mediates interferon-γ repression of collagen transcription through phosphorylation-dependent interactions with co-repressor molecules. J Biol Chem. 2008;283:1243–1256. doi: 10.1074/jbc.M707180200. [DOI] [PubMed] [Google Scholar]
- 23.Cai X, et al. Glycogen synthase kinase 3- and extracellular signal-regulated kinase-dependent phosphorylation of paxillin regulates cytoskeletal rearrangement. Mol Cell Biol. 2006;26:2857–2868. doi: 10.1128/MCB.26.7.2857-2868.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jope RS, et al. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood JE, et al. TcR and TcR-CD28 engagement of protein kinase B (PKB/AKT) and glycogen synthase kinase-3 (GSK-3) operates independently of guanine nucleotide exchange factor VAV-1. J Biol Chem. 2006;281:32385–32394. doi: 10.1074/jbc.M604878200. [DOI] [PubMed] [Google Scholar]
- 26.Appleman LJ, et al. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J Immunol. 2002;168:2729–2736. doi: 10.4049/jimmunol.168.6.2729. [DOI] [PubMed] [Google Scholar]
- 27.Garcia CA, et al. Antigenic experience dictates functional role of glycogen synthase kinase-3 in human CD4+ T cell responses. J Immunol. 2008;181:8363–8371. doi: 10.4049/jimmunol.181.12.8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neilson J, et al. Monitoring the duration of antigen-receptor occupancy by calcineurin/glycogen-synthase-kinase-3 control of NF-AT nuclear shuttling. Curr Opin Immunol. 2001;13:346–350. doi: 10.1016/s0952-7915(00)00225-9. [DOI] [PubMed] [Google Scholar]
- 29.Diehn M, et al. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc Natl Acad Sci U S A. 2002;99:11796–11801. doi: 10.1073/pnas.092284399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sengupta S, et al. Unrestrained glycogen synthase kinase-3β activity leads to activated T cell death and can be inhibited by natural adjuvant. J Immunol. 2007;178:6083–6091. doi: 10.4049/jimmunol.178.10.6083. [DOI] [PubMed] [Google Scholar]
- 31.Ohteki T, et al. Negative regulation of T cell proliferation and interleukin 2 production by the serine threonine kinase GSK-3. J Exp Med. 2000;192:99–104. doi: 10.1084/jem.192.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Sarno P, et al. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2008;181:338–345. doi: 10.4049/jimmunol.181.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lambert SL, Martinez OM. Latent membrane protein 1 of EBV activates phosphatidylinositol 3-kinase to induce production of IL-10. J Immunol. 2007;179:8225–8234. doi: 10.4049/jimmunol.179.12.8225. [DOI] [PubMed] [Google Scholar]
- 34.Varrin-Doyer M, et al. Phosphorylation of collapsin response mediator protein 2 on Tyr-479 regulates CXCL12-induced T lymphocyte migration. J Biol Chem. 2009;284:13265–13276. doi: 10.1074/jbc.M807664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu B, et al. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity. 2007;26:227–239. doi: 10.1016/j.immuni.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gounari F, et al. Somatic activation of β-catenin bypasses pre-TCR signaling and TCR selection in thymocyte development. Nat Immunol. 2001;2:863–869. doi: 10.1038/ni0901-863. [DOI] [PubMed] [Google Scholar]
- 37.Ioannidis V, et al. The β-catenin--TCF-1 pathway ensures CD4(+)CD8(+) thymocyte survival. Nat Immunol. 2001;2:691–697. doi: 10.1038/90623. [DOI] [PubMed] [Google Scholar]
- 38.Ding Y, et al. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med. 2008;14:162–169. doi: 10.1038/nm1707. [DOI] [PubMed] [Google Scholar]
- 39.Gattinoni L, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welsh GI, et al. T-cell activation leads to rapid stimulation of translation initiation factor eIF2B and inactivation of glycogen synthase kinase-3. J Biol Chem. 1996;271:11410–11413. doi: 10.1074/jbc.271.19.11410. [DOI] [PubMed] [Google Scholar]
- 41.Miller AH, et al. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 43.Hua F, et al. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chuang DM. The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann N Y Acad Sci. 2005;1053:195–204. doi: 10.1196/annals.1344.018. [DOI] [PubMed] [Google Scholar]
- 45.Collino M, et al. Insulin reduces cerebral ischemia/reperfusion injury in the hippocampus of diabetic rats: a role for glycogen synthase kinase-3β. Diabetes. 2009;58:235–242. doi: 10.2337/db08-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beurel E, Jope RS. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J Neuroinflammation. 2009;6:9–20. doi: 10.1186/1742-2094-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hashioka S, et al. Antidepressants inhibit interferon-γ-induced microglial production of IL-6 and nitric oxide. Exp Neurol. 2007;206:33–42. doi: 10.1016/j.expneurol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 48.Huang WC, et al. Glycogen synthase kinase-3 negatively regulates anti-inflammatory interleukin-10 for lipopolysaccharide-induced iNOS/NO biosynthesis and RANTES production in microglial cells. Immunology. 2009;128:e275–e286. doi: 10.1111/j.1365-2567.2008.02959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beurel E, Jope RS. Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol Chem. 2008;283:21934–21944. doi: 10.1074/jbc.M802481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chong ZZ, et al. The pro-survival pathways of mTOR and protein kinase B target glycogen synthase kinase-3β and nuclear factor-κB to foster endogenous microglial cell protection. Int J Mol Med. 2007;19:263–272. [PMC free article] [PubMed] [Google Scholar]
- 51.Li F, et al. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3β, β-catenin, and nuclear factor-κB. Curr Neurovasc Res. 2006;3:187–201. doi: 10.2174/156720206778018758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cuzzocrea S, et al. Glycogen synthase kinase-3β inhibition attenuates the degree of arthritis caused by type II collagen in the mouse. Clin Immunol. 2006;120:57–67. doi: 10.1016/j.clim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 53.Dugo L, et al. GSK-3β inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit Care Med. 2005;33:1903–1912. doi: 10.1097/01.ccm.0000178350.21839.44. [DOI] [PubMed] [Google Scholar]
- 54.Bao Z, et al. Glycogen synthase kinase-3β inhibition attenuates asthma in mice. Am J Respir Crit Care Med. 2007;176:431–438. doi: 10.1164/rccm.200609-1292OC. [DOI] [PubMed] [Google Scholar]
- 55.Evenson AR, et al. GSK-3β inhibitors reduce protein degradation in muscles from septic rats and in dexamethasone-treated myotubes. Int J Biochem Cell Biol. 2005;37:2226–2238. doi: 10.1016/j.biocel.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 56.Whittle BJ, et al. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3β. Br J Pharmacol. 2006;147:575–582. doi: 10.1038/sj.bjp.0706509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dugo L, et al. Glycogen synthase kinase-3β inhibitors protect against the organ injury and dysfunction caused by hemorrhage and resuscitation. Shock. 2006;25:485–491. doi: 10.1097/01.shk.0000209545.29671.31. [DOI] [PubMed] [Google Scholar]
- 58.Zhu M, et al. Glycogen synthase kinase 3β and β-catenin are involved in the injury and repair of bronchial epithelial cells induced by scratching. Exp Mol Pathol. 2007;83:30–38. doi: 10.1016/j.yexmp.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 59.Gao HK, et al. Glycogen synthase kinase 3 inhibition protects the heart from acute ischemia-reperfusion injury via inhibition of inflammation and apoptosis. J Cardiovasc Pharmacol. 2008;52:286–292. doi: 10.1097/FJC.0b013e318186a84d. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, et al. Inhibiting glycogen synthase kinase-3 reduces endotoxaemic acute renal failure by down-regulating inflammation and renal cell apoptosis. Br J Pharmacol. 2009;157:1004–1013. doi: 10.1111/j.1476-5381.2009.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lenz SP, et al. Lithium chloride enhances survival of NZB/W lupus mice: influence of melatonin and timing of treatment. Int J Immunopharmacol. 1995;17:581–592. doi: 10.1016/0192-0561(95)00032-w. [DOI] [PubMed] [Google Scholar]
- 62.Zhang P, et al. Glycogen synthase kinase-3β (GSK3β) inhibition suppresses the inflammatory response to Francisella infection and protects against tularemia in mice. Mol Immunol. 2009;46:677–687. doi: 10.1016/j.molimm.2008.08.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cuzzocrea S, et al. Glycogen synthase kinase-3β inhibition reduces secondary damage in experimental spinal cord trauma. J Pharmacol Exp Ther. 2006;318:79–89. doi: 10.1124/jpet.106.102863. [DOI] [PubMed] [Google Scholar]
- 64.Weiner HL. A shift from adaptive to innate immunity: a potential mechanism of disease progression in multiple sclerosis. J Neurol. 2008;255(Suppl 1):3–11. doi: 10.1007/s00415-008-1002-8. [DOI] [PubMed] [Google Scholar]
- 65.Martinez A. Preclinical efficacy on GSK-3 inhibitors: towards a future generation of powerful drugs. Med Res Rev. 2008;28:773–796. doi: 10.1002/med.20119. [DOI] [PubMed] [Google Scholar]
- 66.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steinbrecher KA, et al. Glycogen synthase kinase 3β functions to specify gene-specific, NF-κB-dependent transcription. Mol Cell Biol. 2005;25:8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buss H, et al. Phosphorylation of serine 468 by GSK-3β negatively regulates basal p65 NF-kappaB activity. J Biol Chem. 2004;279:49571–49574. doi: 10.1074/jbc.C400442200. [DOI] [PubMed] [Google Scholar]
- 69.Saijo K, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clifford RL, et al. Novel regulation of vascular endothelial growth factor-A (VEGF-A) by transforming growth factorβ1: requirement for Smads, β-CATENIN, AND GSK3β. J Biol Chem. 2008;283:35337–35353. doi: 10.1074/jbc.M803342200. [DOI] [PubMed] [Google Scholar]
- 71.Guo X, et al. Axin and GSK3β control Smad3 protein stability and modulate TGFβ signaling. Genes Dev. 2008;22:106–120. doi: 10.1101/gad.1590908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang MH, et al. Lithium inhibits Smad3/4 transactivation via increased CREB activity induced by enhanced PKA and AKT signaling. Mol Cell Neurosci. 2008;37:440–453. doi: 10.1016/j.mcn.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duan Y, et al. β-Catenin activity negatively regulates bacteria-induced inflammation. Lab Invest. 2007;87:613–624. doi: 10.1038/labinvest.3700545. [DOI] [PubMed] [Google Scholar]
- 74.Christian SL, et al. The B cell antigen receptor regulates the transcriptional activator β-catenin via protein kinase C-mediated inhibition of glycogen synthase kinase-3. J Immunol. 2002;169:758–769. doi: 10.4049/jimmunol.169.2.758. [DOI] [PubMed] [Google Scholar]
- 75.Pan M, et al. Enhanced NFATc1 nuclear occupancy causes T cell activation independent of CD28 costimulation. J Immunol. 2007;178:4315–4321. doi: 10.4049/jimmunol.178.7.4315. [DOI] [PubMed] [Google Scholar]
- 76.Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Mol Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 77.Jope RS. Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003;24:441–443. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- 78.Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- 79.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Focosi D, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85:20–28. doi: 10.1189/jlb.0608388. [DOI] [PubMed] [Google Scholar]
- 81.Rybakowski JK, et al. Potentiation of antidepressants with lithium or carbamazepine in treatment-resistant depression. Neuropsychobiology. 1999;40:134–139. doi: 10.1159/000026610. [DOI] [PubMed] [Google Scholar]
- 82.Rapaport MH, Manji HK. The effects of lithium on ex vivo cytokine production. Biol Psychiatry. 2001;50:217–224. doi: 10.1016/s0006-3223(01)01144-1. [DOI] [PubMed] [Google Scholar]
- 83.Haack M, et al. Plasma levels of cytokines and soluble cytokine receptors in psychiatric patients upon hospital admission: effects of confounding factors and diagnosis. J Psychiatr Res. 1999;33:407–418. doi: 10.1016/s0022-3956(99)00021-7. [DOI] [PubMed] [Google Scholar]
- 84.Padmos RC, et al. A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry. 2008;65:395–407. doi: 10.1001/archpsyc.65.4.395. [DOI] [PubMed] [Google Scholar]
- 85.Rao JS, et al. Mode of action of mood stabilizers: is the arachidonic acid cascade a common target? Mol Psychiatry. 2008;13:585–596. doi: 10.1038/mp.2008.31. [DOI] [PubMed] [Google Scholar]
- 86.Jafferany M. Lithium and skin: dermatologic manifestations of lithium therapy. Int J Dermatol. 2008;47:1101–1111. doi: 10.1111/j.1365-4632.2008.03873.x. [DOI] [PubMed] [Google Scholar]
- 87.Jope RS. Glycogen synthase kinase 3 (GSK-3) and its inhibitors. John Wiley and Sons, Inc.; 2006. Lithium, the seminal GSK3 inhibitor. [Google Scholar]