

Abstract
Transforming growth factor beta (TGF-β) is the most potent and ubiquitous profibrogenic cytokine and its expression is increased in almost all the fibrotic diseases and in experimental fibrosis models. TGF-β increases ROS production and decreases the concentration of glutathione (GSH), the most abundant intracellular free thiol and an important antioxidant, in various types of cells, which mediates many of TGF-β’s fibrogenic effects. A decreased GSH concentration is also observed in human fibrotic diseases and in experimental fibrosis models. Although the biological significance of GSH depletion in the development of fibrosis remains obscure, GSH and N-acetylcysteine (NAC), a precursor of GSH, have been used in clinics for the treatment of fibrotic diseases. This review summarizes recent findings in the field to address the potential mechanism whereby oxidative stress mediates TGF-β’s fibrogenesis and the potential therapeutic values of antioxidant treatment in fibrotic diseases.
Keywords: TGF-β, oxidative stress, fibrosis, glutathione
INTRODUCTION
Fibrosis, characterized by increased deposition of extracellular matrix (ECM) proteins in basement membrane and interstitial tissue, is a common pathological feature and terminal stage of many diseases involved in almost all the organ systems. Currently, there is no FDA-approved antifibrotic drug or efficacious treatment for fibrotic disease due to incomplete understanding of underlying pathogenesis. Many cytokines, chemokines, and growth factors are involved in the development of fibrosis; however, transforming growth factor beta (TGF-β) is considered to be the most potent and ubiquitous profibrogenic cytokine. TGF-β stimulates the production of reactive oxygen species (ROS) in various types of cells, while ROS activate TGF-β and mediate many of TGF-β’s fibrogenic effects. Glutathione (GSH), a tripeptide, is the most abundant intracellular free thiol and an important antioxidant. GSH concentration decreases in experimental fibrosis models and in human fibrotic diseases. The mechanism and biological significance of GSH depletion in fibrotic diseases, however, remains unclear. Importantly, TGF-β administration decreases GSH in various types of cells in vitro while GSH replenishment suppresses TGF-β’s fibrogenic activity, suggesting an important role of GSH depletion in TGF-β’s fibrogenesis. Nonetheless, although intensive studies have been conducted, the molecular mechanism underlying stimulation of ROS production by TGF-β and whereby ROS mediate TGF-β’s fibrogenic effects remains obscure. This review is intended to summarize recent studies to address the role of oxidative stress and GSH depletion in TGF-β’s fibrogenesis, the underlying molecular mechanism, and the potential therapeutic value of antioxidant treatment in fibrotic diseases.
INCREASED OXIDATIVE STRESS IS A COMMON PATHOLOGICAL FEATURE OF FIBROSIS
There is a substantial body of evidence supporting the hypothesis that increased oxidative stress plays an important role in the development of fibrotic diseases in humans. The lung is a major target for oxidant damage due to its direct exposure to the atmosphere. Accumulating evidence suggests that oxidants generated endogenously or inhaled from the environment play an essential role in the pathogenesis of various pulmonary fibrotic disorders including idiopathic pulmonary fibrosis (IPF), cystic fibrosis (CF), asthma, acute respiratory distress syndrome (ARDS), and chronic obstructive pulmonary disease (COPD) [1–7]. Oxidized lipid and protein products including 8-isoprostane and carbonylated proteins have been identified in the exhaled air, bronchoalveolar lavage fluid (BALF), or lung tissue from patients with these fibrotic diseases; antioxidants, on the other hand, ameliorate the clinical symptoms [1, 3–14]. Silicosis and asbestosis are two important occupational lung fibrotic diseases; ROS and reactive nitrogen species (RNS) have been shown to play a key role in the development of silicosis and asbestosis [15–17]. Workers exposed to silica or asbestos fiber have increased oxidative DNA damage, measured by 8-hydroxy-2′-deoxyguanosine (8-OHdG), and nitrotyrosine adducts [15–17]. Oxidative stress markers have also been detected in silica and/or asbestos-induced lung fibrosis animal models [18–20]. The bleomycin-induced animal model of lung fibrosis has been widely used to study the pathogenesis of fibrosis and the mechanism underlying antifibrotic effects of various compounds. It has been well documented that bleomycin treatment increases ROS production, which mediate bleomycin-induced fibrosis [21–24].
In addition to lung fibrosis, ROS/RNS have also been shown to play critical roles in the development of fibrosis in many other organs. Liver fibrosis/cirrhosis is a final stage of the liver diseases commonly associated with chronic alcohol consumption and hepatitis B or C virus infection. Both alcohol and hepatitis virus core proteins have been shown to increase ROS/RNS production independently and synergistically [25–28]; ROS/RNS play a central role in the development of liver fibrosis/cirrhosis under these pathological conditions [25, 27–36]. Oxidative stress markers have been detected in the serum or biopsy samples from liver cirrhosis patients and in experimental liver fibrosis/cirrhosis animals. In liver biopsies, areas of fibrosis were localized to areas with increased 4-hydroxy-2′-nonenal (4-HNE), a marker of lipid peroxidation [31, 37]. Renal fibrosis occurs under various pathological conditions including diabetes, hypertension, and urinary tract obstruction. Although the primary causes of renal fibrosis are different, oxidative stress has been found to be a common pathological feature of these diseases and contributes importantly to the development of renal fibrosis [38–43]. The levels of serum 8-isoprostane, an oxidation product of lipids, have been shown to correlate with severity of fibrosis in patients with systemic sclerosis [44]. Accumulating evidence also shows that oxidative stress plays a role in several aspects of fibrotic cardiac repair/remodeling after infarction, including apoptosis, inflammatory and fibrogenic responses, by increasing advance glycated end products or inducing the expression of profibrogenic cytokines such as TGF-β [45–49]. Together, the data indicate that oxidative stress is a common pathological feature of fibrosis and plays an important role in the development of fibrosis under various pathological conditions.
ROS AND TGF-β’S FIBROGENESIS
TGF-β and fibrosis
TGF-β, existing in three isoforms (TGF-β1, TGF-β2, and TGF-β3), is a multiple function protein involved in the regulation of cell proliferation, differentiation, apoptosis, adhesion, and migration, and therefore plays a pivotal role in the pathogenesis of many diseases. Various cytokines, chemokines, and growth factors are involved in the development of fibrosis; however, TGF-β is considered to be the most potent and ubiquitous profibrogenic cytokine. TGF-β mRNA and/or protein expression is increased in fibrotic diseases involved in almost all the organ systems, including pulmonary fibrosis, liver cirrhosis, renal fibrosis, systemic sclerosis, and cardiac fibrosis [50–61]. Increased TGF-β expression is also detected in experimental fibrosis models induced by different stimuli [62–68]. Whole body overexpression of TGF-β in TGF-β transgenic mice leads to multiple organ/tissue fibrosis [69–74] while specifically increasing the expression of constitutively active TGF-β in the lung and peritoneum by adenovirus-mediated gene transfer technique leads to lung and peritoneal fibrosis, respectively [75–78]. On the other hand, therapeutic administration of TGF-β binding proteins, treatment of mice with anti-TGF-β antibody or inhibitor to activin receptor-like kinase 5 (ALK5, a TGF-β type 1 receptor), or overexpression of dominant negative TGF-β type II receptor ameliorates fibrosis in different organ systems induced by different stimuli [77, 79–86]. All these lines of evidence indicate that TGF-β is essential for the development of fibrosis.
TGF-β stimulates ROS production
The mechanism underlying the induction of fibrosis by TGF-β has been studied intensively in the past years. Increasing evidence indicates that ROS play a central role in TGF-β’s fibrogenesis although the underlying mechanism remains largely undetermined. TGF-β increases ROS production in numerous types of non-phagocytic cells, such as endothelial cells, epithelial cells, smooth muscle cells, and fibroblasts [87–98]. Although it is still controversial, studies have shown that TGF-β can stimulate ROS production from different cellular compartments in different types of cells. Thannickal et al. reported that TGF-β stimulated ROS production through activation of cell-membrane associated oxidase, which led to an increased release of H2O2 to extracellular space in human lung fibroblasts and bovine pulmonary artery endothelial cells [88–90]. Yoon et al., on the other hand, reported that TGF-β induced a prolonged mitochondrial ROS production through decreasing complex IV activity in Mv1Lu cells, a mink lung epithelial cell line [92]. Herrera et al. also showed that TGF-β increased ROS production in mitochondria in rat hepatocytes [99, 100]. Using different inhibitors, Albright et al. demonstrated that mitochondria and microsomes (cytochrome P450 1A1) were the major source of ROS in TGF-β treated rat hepatocytes [99].
NAPDH oxidase (Nox) enzymes are a family of heme-containing proteins with primary function being transporting electrons from NADPH to oxygen, forming ROS. Seven members have been identified in the Nox family including Nox1, Nox2, Nox3, Nox4, Nox5, Dual oxidase1 (Duox1), and Dual oxidase 2 (Duox2), with different Nox family members being expressed in different cell types and tissues/organs. Emerging evidence suggests that Nox/Duox family members are important ROS producers not only for phagocytic but also for non-phagocytic cells although the biological functions of Nox/Duox in non-phagocyes are still mostly unknown. Importantly, it has been reported that TGF-β activates/induces Nox4 in various types of cells in vitro [91, 93–98, 101–103] and in vivo [103], associated with a stimulation of ROS production, although the effects of TGF-β on the expression/activities of other Nox/Duox family members remain largely unclear at the moment. The prototype NADPH oxidase, Nox2, also called gp91phox, is mainly expressed in phagocytes and located on the plasma membrane; Nox4, however, is expressed in many types of non-phagocytic cells and associated with other cell compartments such as endoplasmic reticulum (ER) [101, 104–106], perinuclear space [107], and nucleus [101, 108, 109]. Nonetheless, the biological function of Nox4 in these cell compartments as well as its role in TGF-β’s signaling process are unclear at the moment and are currently under intensive investigation.
In addition to directly stimulating ROS production through Nox4, mitochondria, and/or microsomes, TGF-β may increase ROS/RNS levels by suppressing the expression of several antioxidant enzymes including glutaredoxin, catalase, superoxide dismutase (SOD), and glutathione peroxidase (GPx) [110–112], and by decreasing the concentration of glutathione, the most abundant intracellular free thiol and an important antioxidant (the effect of TGF-β on GSH metabolism is detailed in the following section). Islam et al. reported that TGF-β inhibited the mRNA expression as well as the activities of catalase and glutathione peroxidase in a hamster pancreatic beta-cell line (HIT), associated with an increase in intracellular ROS level and protein oxidation [111]. Kayanoki et al. showed on the other hand that TGF-β1 suppressed the expression of manganese-superoxide dismutase, copper, zinc-superoxide dismutase, glutathione-S-transferase, and catalase in cultured rat hepatocytes in time- and cell density-dependent manners [110]. Inhibition of the expression of these antioxidant enzymes was followed by an increase in peroxide levels in the cells, suggesting that suppression of antioxidant enzyme expression augments the production of hydrogen peroxide [110].
ROS activate latent TGF-β and induce TGF-β gene expression
TGF-β isoforms are secreted by some cells as a latent complex consisting of TGF-β and its latency association protein (LAP), called the small latent complex. Most cells, however, secrete TGF-β as a large latent complex, in which a latent TGF-β binding protein (LTBP) is associated with the small latent TGF-β complex through a covalent disulfide bond [113, 114]. Currently, 4 LTBP isoforms have been identified and differences in isoforms of the LTBP family proteins account for the differential localization and availability of the latent TGF-β complex [115]. Release of TGF-β from its LAP, a process called latent TGF-β activation, is required for the binding of TGF-β to its receptors. Multiple mechanisms have been proposed for the activation of latent TGF-β, including conformation changes induced by thrombospondin-1 [116, 117], binding to integrins ανβ6 and ανβ8 [118, 119], proteolytic cleavage of LAP by plasmin and matrix metalloproteinase [120–123], and oxidative modification of LAP or activation of MMPs, which then cleave LAP to release active TGF-β [124–128].
ROS have been shown to play important roles in the development of fibrosis under various pathological conditions. One potential mechanism whereby ROS promote fibrosis is activating latent TGF-β [124–126, 128–131]. In a cell-free model, recombinant human latent TGF-β was activated after exposure to ionizing radiation or metal iron plus ascorbate, which lead to ROS production [124]. Pociask et al. also showed that recombinant human latent TGF-β was activated in a cell free system in the presence of asbestos and ascorbic acid and that such an activation was reduced significantly by addition of superoxide dismutase, catalase, or deferoxamine, an iron chelator [126]. ROS produced by activated human CD4+CD25− T cells have also been shown to activate TGF-β and such activation was inhibited by antioxidant MnTBAP [128]. Two potential mechanisms have been proposed for ROS activation of latent TGF-β: direct oxidation of LAP and indirectly through activation of MMPs such as MMP-2 and MMP-9, which in turn cleave LAP to release active TGF-β. Vodovotz et al. reported that treatment of recombinant LAP with nitric oxide (NO) reduced its ability to neutralize active TGF-β1 although NO had no direct effect on the activation or the activity of recombinant latent TGF-β1 [125]. The activation of latent TGF-β by asbestos-ascorbate mediated generation of ROS apparently also resulted from oxidative modification in LAP, leading to a loss of its ability to bind to TGF-β1 [126]. On the other hand, ROS generated by ultraviolent light B (UVB) have been shown to activate TGF-β in keratinocytes through stimulating MMP-2 and MMP-9 activities as a MMP-2 and MMP-9 inhibitor diminished the UVB-induced increase in the amount of the active TGF-β but had no effect on the total amount of TGF-β [129].
TGF-β exists as three isoforms and all of them are secreted as a latent form. Interestingly, Jobling et al. showed that redox-mediated activation was restricted to the LAP/TGF-β1 isoform [130]. They also showed that exposure of TGF-β1 to ROS had no effect on its association with LAP while exposing LAP/TGF-β1 to ROS prohibited their association and that reducing agents restored the ability of LAP to neutralize TGF-β [130]. These data suggest that the oxidative modification leading to activation of latent TGF-β occurs in LAP. Using site-specific mutation, they identified a methionine residue at amino acid position 253 unique to LAP-TGFβ1 as a critical target for ROS-mediated activation of latent TGF-β1 [130]. Therefore, they proposed that latent TGF-β1 contained a redox switch centered at methionine 253, which allowed latent TGF-β1 to act uniquely as an extracellular sensor of oxidative stress in tissues [130].
In addition to activating latent TGF-β, ROS can also stimulate the expression and secretion of TGF-β in many types of cells [49, 131–135]. In macrophages, the lipid peroxidation product 4-HNE upregulated the expression of TGF-β [132]. In human alveolar epithelial cells, ROS generated by xanthine/xanthine oxidase system induced TGF-β expression through a transcription mechanism while RNS increased TGF-β expression by a post-transcription mechanism [133]. Angiotensin II (AngII) induces TGF-β expression and fibrosis in various animal models. Zhao et al. showed that AngII induced Nox2 and TGF-β expression, accompanied by extensive perivascular and interstitial fibrosis in both ventricles of rat heart; the antioxidants apocynin and tempol, on the other hand, blocked AngII-induced Nox2 and TGF-β expression [49]. The iron chelator deforoxamine and the free radical scavenger T-0970 have also been shown to suppress AngII-induced TGF-β expression in rat heart [134]. Single-walled carbon nanotubes induced TGF-β expression, which was significantly attenuated in Nox2 null mice [135]. These data suggest an important role of Nox2 in the induction of TGF-β. A schematic diagram illustrating the potential relationship between TGF-β induction/activation and ROS production is presented in Fig 1.
Fig 1. TGF-β increases ROS production and ROS activate/induce TGF-β.
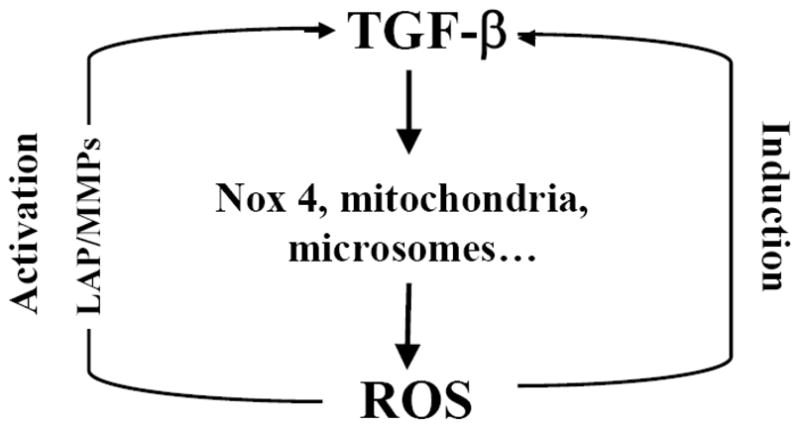
TGF-β increases ROS production by Nox4, mitochondria, and microsomes. On the other hand, ROS induce TGF-β gene expression and activate TGF-β through oxidizing latency associated protein (LAP) or activating MMPs, which in turn release LAP.
Potential mechanisms whereby ROS mediate TGF-β’s fibrogenesis
Stimulation of fibroblast proliferation and activation
Myofibroblasts are the major players in the development of fibrosis. Fibroblast proliferation and activation to become myofibroblasts therefore is a key step in the initiation of fibrosis. One of the major mechanisms by which TGF-β induces fibrosis is stimulating fibroblast proliferation and activation. TGF-β has been shown to stimulate the proliferation and activation of fibroblasts originating from different organ systems [136–141] and ROS/RNS play critical role in this process [98, 103]. Cucoranu et al. reported that TGF-β treatment stimulated the expression of Nox4 and alpha smooth muscle actin (α-SMA), a myofibroblast marker, in primary human cardiac fibroblasts while knockdown of Nox4 by siRNA reduced TGF-β-stimulated production of ROS and α-SMA mRNA expression, suggesting that ROS mediate TGF-β-induced differentiation of cardiac fibroblasts to myofibroblasts [98]. The same study further showed that ROS mediated TGF-β-induced differentiation of myofibroblasts by activating Smad2/3, although the detailed mechanism underlying Smad2/3 activation by ROS was not explored [98]. A study published in Nature Medicine recently further showed that TGF-β1 specifically induced mRNA and protein expression of the Nox4 isoform in human fetal lung mesenchymal cells (hFLMCs), in myofibroblastic foci/mesenchymal cells from IPF patient lungs, and in lung tissue of bleomycin or hapten FITC treated mice [103]. Most importantly, they showed that inhibition of Nox4 expression by siRNA technique or pharmaceutical agent blocked TGF-β1-stimulated ROS production and myofibroblast activation [103]. These studies demonstrated clearly that ROS/RNS mediate TGF-β’s fibrogenesis in part by activating fibroblasts.
In addition to resident fibroblasts, circulating fibrocytes, which derive from bone marrow mesenchymal progenitor cells, also serve as an important source of fibroblasts (myofibroblasts) found in the fibrosis foci [142–145]. TGF-β has been shown to play a critical role in the differentiation of circulating fibrocytes to myofibroblasts [145–147] although it is not clear at the moment whether ROS/RNS are involved in this process.
Induction of EMT
Although a long standing paradigm for the origination of myofibroblasts is that myofibroblasts differentiate from resident fibroblasts upon stimulation, emerging evidence indicates that myofibroblasts may also arise from epithelial cells through epithelial-mesenchymal transition (EMT), a process initially observed in embryogenesis and carcinogenesis and characterized by the loss of epithelial markers and the acquisition of a mesenchymal phenotype [148–152]. EMT has been shown to occur in many fibrotic diseases, including idiopathic pulmonary fibrosis [153–155], and to play an essential role in the development of fibrosis in several animal models induced by different stimuli [78, 143, 156–159]. In addition to epithelial cells, recent studies have shown that endothelial cells may also undergo such mesenchymal transition, termed endothelial-mesenchymal transition (EndMT) and that EndMT contributes importantly to the development of cardiac fibrosis, kidney fibrosis, and corneal fibrosis [160–162]. Many cytokines, chemokines, and growth factors are involved in EMT/EndMT; however, TGF-β is considered to be a key inducer of EMT/EndMT. TGF-β induces EMT and EndMT in various types of cells in vitro [163–167] and in animal models in vivo [78, 158]. Increased TGF-β expression is also associated with EMT phenotypic changes under various human fibrotic disease conditions [153, 154, 157, 167]. Blocking TGF-β signaling with Smad 7 has been shown to suppress EndMT in several fibrosis models including injury-induced corneal fibrosis model and pressure overload or chronic allograft rejection induced cardiac fibrosis model [161, 162]. Together, the evidence suggests that both EMT and EndMT are involved in the development of fibrosis and that one potential mechanism whereby TGF-β stimulates fibrosis is induction of EMT/EndMT.
ROS have been shown to function as signaling molecules mediating the induction of EMT by different stimuli in different cell types [152, 168–170]. Several signaling pathways including Smad [164, 171–174], Ras/Raf/MAPK [170, 174, 175] and PI3K-Akt [176–179] pathways are implicated in the induction of EMT by TGF-β. Some of these pathways including the Ras/Raf/MAPK and PI3K-Akt pathways are redox sensitive. Therefore, it is speculated that ROS/RNS play an important role in TGF-β induction of EMT. This speculation is confirmed by a recent study [170]. Rhyu et al. reported that TGF-β increased ROS production and induced epithelial-mesenchymal transition (increased the expression of α-SMA, a mesenchymal marker, and decreased the expression of E-cadherin, an epithelial marker) in rat renal tubular epithelial cells [170]. Suppression of cellular ROS signaling with antioxidants blocked TGF-β-induced EMT, suggesting that ROS mediate the induction of EMT by TGF-β in renal epithelial cells [170].
Induction of apoptosis
Apoptosis and fibrosis are juxtaposed in some fibrotic diseases. Accumulating evidence suggests that apoptosis may be directly linked to the development of fibrosis [73, 180–182]. Using an externally regulated triple transgenic system, Lee et al. showed that increasing TGF-β1 expression in airway epithelial cells induced a transient wave of epithelial apoptosis that was followed by mononuclear-rich inflammation and tissue fibrosis [73]. They also showed that a caspase inhibitor blocked TGF-β1-induced apoptosis and ameliorated TGF-β1-induced fibrosis [73]. These results suggest that apoptosis is involved in the development of fibrosis in this animal model. Apoptosis is commonly triggered in hepatocytes through activation of death receptors such as Fas death receptor. Canbay showed that 3-day bile duct ligation induced more TUNEL-positive hepatocytes (apoptosis), higher serum ALT values (liver damage), and profound liver fibrotic changes (increased expression of α-SMA, TGF-β, and collagen mRNAs as well as collagen staining) in wild type mice than in Fas-deficient mice, suggesting that Fas-mediated hepatocyte apoptosis is mechanistically linked to liver fibrogenesis [180]. Using a pancaspase inhibitor, IDN-6556, they further concluded that hepatocyte apoptosis initiated cascades culminating in liver injury and fibrosis [181]. TGF-β induces apoptosis in variety of cell types, associated with an increase in ROS production, while antioxidants blocked TGF-β-induced apoptosis [95, 96, 99, 100, 111, 183–186]. These data suggest that ROS mediate TGF-β’s fibrogenic effects in part by inducing apoptosis.
Induction of ECM gene expression and suppression of ECM protein degradation
TGF-β upregulates the expression of extracellular matrix (ECM) proteins, including collagens as well as protease inhibitors such as plasmin activator inhibitor 1 (PAI-1) and tissue inhibitor of metalloproteinases (TIMPs), leading to increased ECM production and decreased ECM degradation. Importantly, ROS have been shown to be involved in the induction of ECM proteins and protease inhibitor expression by TGF-β [187–193]. Both Garcia-Trevijano and Cao showed that TGF-β1 increased H2O2 production and induced procollagen-α1(I) mRNA expression in hepatic stellate cells while treatment of the cells with catalase reduced H2O2 levels and prevented the induction of procollagen by TGF-β, suggesting that ROS mediate TGF-β induction of procollagen expression in these cells [187, 188]. ECM degradation is mediated mainly by the matrix metalloproteinases (MMPs) and the plasminogen activators (PAs)/plasmin systems [194–196]. The activities of MMPs and PAs/plasmin, on the other hand, are inhibited by TIMPs and PAI-1. PAI-1, a physiological inhibitor of tissue type and urokinase type plasminogen activators (tPA and uPA), suppresses ECM degradation by inhibiting tPA/uPA activity and thus the activation of plasminogen and MMPs [197]. TGF-β induces the expression of both TIMPs and PAI-1, which contributes importantly to the development of fibrosis under various pathological conditions. Li et al. reported that TGF-β1 increased ROS production and induced TIMP-3 in primary human and bovine chondrocytes while ROS scavenger and antioxidant NAC blocked TGF-β1-induced TIMP-3 mRNA and protein expression [191]. Studies have also shown that the induction of PAI-1 by TGF-β is mediated by ROS in different types of cells [190, 192, 193]. Our previous studies showed that TGF-β increased the production of ROS as well as the expression of TGF-β1, procollagen α2(I), and PAI-1 mRNAs in murine embryo fibroblast cells (NIH3T3 cells) [192]. Exogenous glutathione selectively inhibited TGF-β-induced PAI-1 expression, accompanied by a restoration of plasmin activity and collagen degradation, with no effect on TGF-β-induced TGF-β1 or procollagen α2(I) mRNA expression [192]. Together, the data suggest that ROS play a central role in TGF-β’s fibrogenesis by mediating TGF-β-induced fibroblast proliferation/activation, EMT, epithelial/endothelial apoptosis, and ECM accumulation (Fig 2).
Fig 2.

ROS mediate many of TGF-β’s fibrogenic effects.
ROS and TGF-β’s signaling
Although numerous studies have shown that ROS mediate the induction of many TGF-β responsive genes the underlying mechanism remains equivocal. TGF-β signaling through the Smad pathway has been well described in the past decades. Following the binding of TGF-β to the type II receptor (TβR-II), the type I receptor (TβR-I) is phosphorylated, which further phosphorylates Smad2 or Smad3. Phosphorylated Smad2 and Smad3 then form a heteromeric complex with Smad4, which translocates to the nucleus and binds to the promoters of TGF-β responsive genes to regulate their expression. In addition to the Smad pathway, several other pathways including mitogen activated protein kinase (MAPK) pathways and PI3K-Akt pathway are also important in TGF-β signaling. MAPK pathways, including mainly JNK, p38, and ERK MAPK pathways, are sensitive to the redox environment. Numerous studies including ours have shown that ROS mediate TGF-β’s fibrogenic effects by modulating the activities of MAPK pathways although the underlying molecular mechanism remains unclear [97, 193, 198–202].
MAPK activation results from the activation of upstream MAPK kinase kinase, which leads to phosphorylation of tyrosine and serine/threonine residues on MAPKs, or inactivation of MAPK phosphatases (MKPs), which dephosphorylate tyrosine, serine/threonine, or both. Although it is still not clear whether ROS can directly oxidize MAPK, numerous studies have shown that protein phosphatases including protein tyrosine phosphatases (PTPs) such as PTP1B, serine/threonine phosphatases such as protein phosphatase 2A (PP2A), and some dual specific MAPK phosphatases (DS-MKPs) such as MKP-1 and MKP-3 can be inactivated by ROS through oxidation of critical cysteine residues in their active sites, associated with a sustained activation of MAPK [203–213]. Interestingly, TGF-β has been shown to inhibit the activity of PP2A [214–219], phosphatase and tensin homolog (PTEN) [220, 221], a dual-specificity phosphatase, and protein tyrosine phosphatase [222] in different cell types. Therefore, one potential mechanism whereby ROS mediate TGF-β activation of MAPK pathways and therefore the expression of TGF-β responsive genes could be the inactivation of MKPs (Fig 3).
Fig 3. ROS and TGF-β signaling.

TGF-β increases ROS production, which inactivates MAPK phosphatases (MKPs). Inactivation of MKPs leads to sustained activation of MAPKs, which induces the expression of TGF-β responsive genes by activating (phosphorylating) transcription factors such as AP-1 and SP-1 or by increasing phosphorylation, nuclear translocation, and/or DNA binding of the Smad proteins.
In addition to MAPK pathways, emerging evidence shows that the activity of the Smad pathway can also be affected the redox status of cells [170, 191, 223]. Li et al. reported that the induction of TIMP-3 by TGF-β1 was Smad2-dependent and redox sensitive as NAC or reduced GSH blocked TGF-β1-stimulated Smad2 phosphorylation and TIMP-3 expression [191]. Rhyu also showed that, in TGF-β-treated renal tubular epithelial cells, antioxidants effectively blocked not only the activation of p38 and ERK MAPKs but also the phosphorylation of Smad 2, associated with an inhibition of EMT [170]. The molecular mechanism underlying the activation of the Smad pathway by ROS is unclear. MAPKs have been shown to interact with Smad signaling in different ways. In addition to affecting phosphorylation and nuclear translocation of Smad proteins [170, 191, 224–226], MAPKs may also interfere with the binding of Smad proteins to the promoters through modifying the binding of other transcription factors [193, 227–231]. Our previous studies showed that GSH had no effect on TGF-β-induced Smad 2/3 phosphorylation or Smad2/3/4 nuclear translocation in fibroblasts; however, it blocked TGF-β-induced binding of transcription factors to not only AP-1 and SP-1 but also Smad cis elements in the PAI-1 promoter [193]. Therefore, it is speculated that AP-1 and SP-1 may be involved in the binding of Smad to its cis elements in the PAI-1 promoter and that ROS mediate the induction of PAI-1 by TGF-β through increasing the binding of AP-1 and SP-1, and consequently Smad complex, to the PAI-1 promoter [193].
TGF-β, GSH, and FIBROSIS
Glutathione: functions and homeostasis
Glutathione (GSH), a tripeptide, is the most abundant intracellular free thiol. GSH plays a critical role in regulating a variety of cellular functions including detoxification of xenobiotics, synthesis of DNA and other endogenous compounds, modulation of gene expression, and the regulation of the cell cycle. However, the most important and well-known function of GSH is antioxidant. Glutathione can reduce hydrogen peroxide and lipid peroxide through glutathione peroxidase (GPx) catalyzed reactions, and conjugate and therefore detoxify electrophiles spontaneously or through glutathione S-transferase (GST) catalyzed reactions. Another important mechanism whereby GSH exerts its antioxidant function is to keep protein cysteine residues in their reduced form through reactions catalyzed by glutaredoxin (Grx) and sulfiredoxin (Srx) (Fig 4). Cysteine residues are important for protein structure and function and they are sensitive to oxidation. During oxidant challenge, cysteine residues, especially those in cysteine thiolate (Cys-S−) form, can be oxidized to the sulfenic (RSOH), sulfinic (RSO2H), and sulfonic (RSO3H) acid, depending on the intensity of oxidant insults. Although conversion to sulfonic acid is considered to be irreversible, sulfinic acid can be reduced back to sulfenic acid through Srx catalyzed reaction [232–236]. Sulfenic acid can then react with vicinal protein thiols to form disulfides or with GSH to form protein mixed disulfides (glutathionylation), which can be reduced back to free thiol form by Grx [237–239] or Srx [240, 241] catalyzed reactions using GSH as a reductant (Fig 4). Many transcription factors such as NFkB, AP-1, and NrF-2 as well as signaling molecules such as protein phosphatases including PTPs, PP2A, and MKP-1, and MKP-3 contain redox sensitive cysteine residues in their active sites and undergo reversible oxidative modifications upon stimulation by growth factors or oxidants [204, 208, 210, 212, 217, 242–248]. Such reversible oxidative modifications of protein cysteine residues have been increasingly recognized as an important mechanism whereby reactive oxygen/nitrogen species (ROS/RNS) regulate protein functions and cell signaling [249–252]. As the intracellular GSH concentrations (ranging from 1 to 10 mM) are much higher than most other redox active compounds, the ratio of glutathione disulfide (GSSG) and GSH determines cell redox status [253]. Therefore, by detoxifying H2O2 and lipid peroxides through glutathione peroxidase catalyzed reactions and by modifying the redox status of protein cysteine residues in these signaling molecules through Grx and Srx catalyzed reactions, GSH plays a central role in regulating cell redox signaling [237, 247, 253–257] (Fig 4).
Fig 4. GSH in antioxidant defense and in redox signaling.

GSH reduces hydrogen peroxide (H2O2) and lipid peroxide (ROOH) through glutathione peroxidase (GPx) catalyzed reactions. Oxidized glutathione (GSSG) formed is then reduced back to GSH by glutathione reductase (GR) catalyzed reaction in consumption of NADPH. GSH also plays a critical role in protein redox signaling through glutaredoxin (Grx) and sulfiredoxin (Srx) catalyzed reactions. During oxidant challenge, protein cysteine residues, especially those in thiolate form (Cys-S−), can be oxidized to the sulfenic (RSOH), sulfinic (RSO2H) and sulfonic (RSO3H) acid, depending on the intensity of oxidative insults. Although conversion to sulfonic acid form is considered to be irreversible, sulfinic acid can be reduced back to sulfenic acid through Srx catalyzed reaction. Sulfenic acid then reacts with GSH to form protein mixed disulfides (glutathionylation), which can be reduced back to free-thiol form (deglutathionylation) through Grx or Srx catalyzed reactions using GSH as a reductant.
As GSH is the most highly concentrated antioxidant in the cell and the determinant factor for cellular redox status, maintenance of intracellular GSH homeostasis is vital for normal cell functions [247]. There are several mechanisms by which cells maintain their intracellular GSH homeostasis, mainly by GSH redox cycling, direct uptake, and de novo synthesis. GSH redox cycling, catalyzed by GSSG reductase (GR), prevents the loss of GSH in the form of GSSG that is generated during the reduction of various oxidants with GSH by reducing GSSG back to GSH. A few types of cells can also directly uptake intact GSH from surrounding fluid. Most cells, under both normal and oxidative stress conditions, however, tend to depend on the de novo synthesis to maintain their intracellular GSH levels. De novo GSH synthesis is a two step process catalyzed by glutamate-cysteine ligase (GCL, EC6.3.2.2), also known as γ-glutamylcysteine synthetase (GCS), and GSH synthetase (EC6.3.2.3) [258]. GCL, which catalyzes the first reaction, is the rate-limiting enzyme in de novo GSH synthesis [258]. Accumulating evidence suggests that the regulation of GCL gene expression and activity is a major determinant of intracellular GSH homeostasis under physiological condition and upon oxidant challenge.
Glutathione depletion in fibrotic diseases
GSH concentration has been found to be decreased in human subjects with various fibrotic diseases. In patients with lung fibrotic diseases such as cystic fibrosis [259, 260], COPD [261], ARDS [262–266], IPF [267–273], and sarcoidosis [274], GSH concentration is decreased in the lung lining fluid. Such a decrease in GSH concentration is associated with increased oxidative damage of macromolecules. Exposure to asbestos or silica leads to lung fibrosis. It has been shown that workers exposed to asbestos, silica, or related chemicals have a decreased GSH level in the plasma and in the lung lining fluid [275, 276]. GSH depletion is also a common feature for chronic liver diseases associated with chronic alcohol consumption or hepatitis B or C infection [277–283]. Loguercio et al. reported that liver cirrhosis patients have decreased levels of glutathione and other antioxidants and increased levels of oxidative stress markers [279]. Using a 2-step infusion protocol, Bianchi et al. found that the primary defect responsible for the decrease in glutathione concentration in liver cirrhosis is reduced production [277]. Alcohol consumption leads to decreases in GSH concentrations not only in liver but also in the lung in humans. It was reported that chronic ethanol ingestion in human subjects causes 80–90% depletion of extracellular glutathione and an increase in oxidative stress in the alveoli, which is accompanied by an increased risk of developing ARDS and acute lung injury [284].
GSH depletion has also been observed in various experimental fibrosis models involved in different organ systems and induced by different insults [24, 285–288]. Lung fibrosis is one of the major side effects caused by bleomycin, a chemotherapy drug used for the cancer treatment. It has been reported that bleomycin decreases GSH and induces lung fibrosis in mice and rats [287, 289–293]. Paraquat is a broad spectrum contact herbicide that is known to cause progressive pulmonary fibrosis in human and animals [294, 295]. It has been reported that paraquat-induced lung fibrosis is associated with a decline in GSH content in the lung [286, 295]. A significant depletion of GSH was also observed in the lung and red blood cells in chrysotile-exposed rats during different developmental stages of asbestosis [296]. Decreased GSH level is also a common pathological feature of liver fibrosis/cirrhosis induced by different stimuli including alcohol [297, 298], carbon tetrachloride (CCl4) [299, 300], and dimethylnitrosamine [301, 302]. Such a decrease in GSH level was associated with an elevated ROS/RNA production, lipid peroxidation, and fibrosis [303]. Taken together, the data indicate that decreased GSH concentration is a common pathological feature of fibrosis. Nonetheless, although it has been well documented that GSH concentration decreases in fibrotic diseases, the mechanism and biological significance of GSH depletion in the development of fibrosis remain unclear and warrant further investigation.
TGF-β decreases intracellular GSH
Although the mechanism underlying GSH depletion in fibrotic diseases is unclear TGF-β has been shown to decrease GSH in various types of cells including endothelial cells, hepatocytes, epithelial cells, and fibroblasts in vitro [304–312]. Sanchez et al. reported that TGF-β-induced apoptosis in fetal hepatocytes was preceded by an induction of reactive oxygen species production and a decrease in the intracellular GSH concentration [305]. Cycloheximide, a protein synthesis inhibitor, prevented the depletion of intracellular GSH, increase in ROS production, and apoptosis induced by TGF-β [306]. Our previous studies showed that TGF-β decreased intracellular GSH in murine embryo fibroblasts (NIH3T3 cells), which was accompanied by increased collagen accumulation [312]. Replenishment of intracellular GSH by exogenous GSH, NAC, or GSH ester blocked TGF-β-induced collagen accumulation and PAI-1 expression, and stimulated collagen degradation, suggesting an important role of GSH depletion in TGF-β’s fibrogenesis [192, 193, 312]. Nonetheless, whether increased expression of TGF-β is responsible for the depletion of GSH in fibrotic diseases in humans remains to be determined.
The mechanism whereby TGF-β decreases intracellular GSH has been explored in several studies. GCL is the rate-limiting enzyme in de novo GSH synthesis and is composed of two subunits, the catalytic subunit (GCLC) and the modifier subunit (GCLM). It has been reported that TGF-β suppresses the expression of GCLC mRNA and protein in a human lung epithelial cell line (A549), accompanied by a decrease in GCL activity and GSH level [307]. It has also been reported that TGF-β inhibits GCLC gene transcription, measured by nuclear run-on assay, but had no effect on the stability of GCLC mRNA [307]. Jardine et al. reported that TGF-β suppressed the GCLC promoter activity in alveolar epithelial cells and that the increased binding of c-Jun and Fra-1 dimers to AP-1 site within the antioxidant response element (ARE) in the promoter of GCLC gene might be responsible for the down-regulation of GCLC gene by TGF-β in these cells [309]. Bakin et al. further showed that TGF-β suppressed the activity of the human GCLC proximal promoter bearing ARE; ectopic expression of constitutively active Smad3 (Smad3E) alone inhibited the reporter activity whereas dominant negative Smad3 (Smad3A) blocked TGF-β’s effects [311]. They also demonstrated that TGF-β increased the protein level of activating transcription factor3 (ATF3), a stress-inducible transcription repressor, and that ectopic expression of ATF3 suppressed GCLC promoter activity and endogenous GCLC expression [311]. Their studies suggest that the Smad3-ATF3 pathway mediates TGF-β suppression of GCLC expression. Importantly, Tiitto et al. showed that GCL expression was decreased in the fibroblast foci in IPF lung [313]. Together, the data suggest that decreased GSH biosynthesis capacity resulting from inhibition of GCLC gene expression by TGF-β may underlie the decrease in GSH concentrations observed in human fibrotic diseases. A schematic diagram elucidating the potential mechanism whereby TGF-β depletes GSH and GSH antagonizes TGF-β’s fibrogenesis is presented in Fig 5.
Fig 5. GSH and TGF-β-mediated fibrogenesis.

TGF-β decreases intracellular GSH by suppressing the expression of glutamate cysteine ligase (GCL), the rate-limiting enzyme in de novo GSH synthesis. On the other hand, GSH suppresses TGF-β-induced fibrogenic effects by reducing intracellular ROS levels and by preventing oxidative modifications/inactivation of MAPK phosphatases and thus MAPK activation. GSH may also block TGF-β-induced Smad pathway activity indirectly by inhibiting MAPK pathways.
THERAPEUTIC VALUE OF THIOLS IN FIBROTIC DISEASES
Antifibrotic effects of NAC and GSH in animal models
As GSH concentration decreases in many fibrotic diseases and in experimental fibrosis models, it is reasonable to assume that treatments that increase the concentration of GSH and other antioxidants in the target tissue would prevent the progression of fibrotic diseases. Numerous studies have been conducted to elucidate the antifibrotic potential of thiol reagents in experimental fibrosis models. NAC, which easily passes the cell membrane and is hydrolyzed intracellularly to donate sulfhydryl groups for GSH synthesis [314], is commercially available with few known side effects. Therefore, NAC has been extensively studied as a potential treatment for fibrotic diseases. It has been reported that NAC attenuates liver cirrhosis in different animal models [315–318]. In a Nonalcoholic Steatohepatitis (NASH) model, NAC was found to be able to prevent many aspects of NASH and decrease Picrosirius red staining of collagen, a marker of fibrosis, although it did not block development of steatosis [315]. The bile-duct ligation (BDL) model has been widely used to induce liver cirrhosis to study the pathology of liver cirrhosis and the mechanism of antifibrotic effects of chemicals. Yang et al. showed that NAC administration decreased BDL-induced expression of procollagen III mRNA and protein, associated with a reduction of portal venous pressure and intrahepatic resistance, two liver cirrhosis indicators [316]. NAC has also been shown to reduce CCl4 [318] and dimethylnitrosamine [317] induced liver cirrhosis in animal models.
The antifibrotic effect of NAC has also been well documented in lung fibrosis models. Oral administration of NAC at doses between 2.45 and 3 mmol/kg/day reduced bleomycin-induced lung fibrosis in mice [319] and in rats [22, 320, 321] associated with an increase in GSH concentration. Aerosol administration of NAC also attenuated bleomycin-induced lung fibrosis in mice [322]. Nonetheless, although many studies have demonstrated the protective effects of NAC/GSH in various fibrosis models the underlying mechanism remains unclear. In almost all of these studies, NAC treatment was initiated before bleomycin treatment and a significant inhibition of the inflammatory response was observed. Therefore, it is not clear whether GSH/NAC attenuated fibrosis by suppressing early-stage inflammatory response or whether they also have direct anti-fibrotic effect.
NAC and GSH clinical trials
Despite severe outcomes (e.g. the patients usually die within 3–5 years after diagnosis of IPF), there is no efficacious treatment for these devastating fibrotic diseases such as IPF and liver cirrhosis due to a poor understanding of the complex pathological process. It was speculated in the past that fibrosis results from an unremitting inflammatory response to an exogenous insult, which leads to fibroblast activation/proliferation and eventually culminates in progressive fibrosis. Therefore, anti-inflammatory agents alone or in combination with cytotoxic drugs have been used in clinics as a standard therapeutic regimen for the treatment of fibrotic diseases. However, there is little evidence indicating that these agents alter the natural history of the disease or improve survival of the patients [323–326]. Although other therapeutic strategies are in development or in early clinical trials, including interferon gamma (INFγ) and anti-TGF-β or anti-connective tissue growth factor (CTGF) antibody, the efficacies of these treatments remain unclear [327–330].
Based on the findings that GSH concentration is decreased in the lung lining fluid of fibrotic disease patients and oxidative stress plays a critical role in the development of fibrosis, GSH and NAC have been used clinically for the treatment of various lung fibrotic diseases including IPF and CF. Studies have shown that administration of GSH or NAC increased GSH concentration in the lung lining fluid in IPF and CF patients. GSH/NAC has also been shown to slow deterioration of the lung functions of CF or IPF patients in some clinic trials. However, the efficacy of GSH and NAC in the treatment of lung fibrotic diseases remains to be further evaluated. Behr et al. reported that there was an increase in the total and reduced glutathione in native BAL fluid and in the epithelial lining fluid in fibrosing alveolitis patients after treatment with 600 mg NAC three times daily for 12 weeks plus immunosuppressive therapy. The increase in GSH concentration was accompanied by a decrease of methionine sulfoxide content, an indicator of oxidative stress at the alveolar surface, and a significant improvement of pulmonary function [270]. In another pilot study [331], 30 patients with IPF were randomly assigned to either NAC or placebo group and received NAC (176 mg, twice a day) or bromhexine hydrochloride (4 mg daily) for 12 months. Efficacy was assessed by the changes in pulmonary function, the 6-min walking test, high-resolution CT, health-related quality of life, and serum KL-6 (Krebs von den Lungen-6) value before and after the treatment. The results showed that there were significant differences between the NAC and control groups in terms of mean changes in lowest Saturation of Oxygen (SaO2) during the 6-min walking test, serum KL-6, and the ground-glass score on high-resolution CT. However, differences between two groups in pulmonary function, 6-min walking distance, or quality of life were not significant [331]. In a double-blind randomized placebo-controlled multicenter trial, IPF patients were divided into two groups with one group receiving the standard therapy of prednisone plus azathioprine while another group receiving NAC (600 mg 3 times daily) plus standard therapy. The changes between baseline and after 12-month therapy in vital capacity (VC) and single-breath carbon monoxide diffusing capacity (DLco) were monitored. The results showed that NAC treatment slowed the deterioration of VC and DLco (9% and 24% improvement for VC and DLco, respectively). However, the differences in the mortalities between two groups during the study were not statistically significant (9% for NAC plus standard therapy group vs. 11% for standard therapy only group) [332].
Cystic fibrosis (CF) is caused by an autosomal recessive mutation of CF transmembrane conductance regulator (CFTR) protein, which has been shown to function as a GSH transporter [333–335]. It has been well documented that GSH concentration decreases in the lung lining fluid of CF patients and that oxidative stress contributes importantly to the airway pathogenesis of CF. Therefore, GSH and NAC have been on clinical trial for the treatment of CF patients for many years. A report from a double-blind placebo controlled clinical trial showed that the lung function was improved in CF patients who received NAC for six months as compared with placebo controls [336]. In a 14-day glutathione inhalation trial (thrice-daily of 300–400 mg GSH), Griese et al. reported that GSH concentration in lavage of CF patients was increased 3–4 folds 1 hour post inhalation and was still double 12 hours post inhalation. FEV1 was increased compared with pretreatment values although a transient drop was observed at beginning [337]. The same group of investigators further showed that although inhalation of GSH has no effect on oxidative stress markers, it decreased BALF levels of PGE2 and increased CD4+ and CD8+ lymphocytes, which correlated inversely and positively with lung function, respectively [338]. In a randomized, double-blind, placebo-controlled pilot study, Bishop et al. found that after inhalation of buffered reduced glutathione at dose of 66 mg/kg/day for 8 weeks, the peak flow was dramatically increased in GSH group as compared with placebo group [339]. Self-reported average improvement in GSH group was also bigger than placebo group. Of 13 primary and secondary outcomes examined, 11 outcomes favored GSH treatment, suggesting a potential therapeutic value of GSH in the treatment of CF [339].
Overall, while NAC and GSH show a clear inhibitory effect on the development of fibrosis in animal models, their effects in clinics are not as dramatic as in animal studies, although promising. It should be emphasized that, in almost all animal studies, GSH/NAC supplement started before or simultaneously with treatment of bleomycin or other fibrogenic stimuli, which usually induce an early stage inflammatory response. Although inflammation may not be essential in the progression of IPF, animal studies have shown that early inflammatory response is critical for the development of fibrosis. As GSH and NAC are potent anti-inflammation agents, GSH and NAC may exert their antifibrotic effects in animal models by suppressing the early inflammatory response and potentially the later fibrotic response. In IPF, on the other hand, the early inflammatory stage has passed and lung fibrosis has developed when patients seek medical help. Under those circumstances, the efficacy of GSH/NAC may not be as dramatic as that observed in pretreated animal models. One important question that needs to be addressed therefore is whether antioxidant treatment is still effective when given in the later stages of fibrosis in animal models. Otherwise, identification of early stages of IPF patients is critical for efficacious treatment with antioxidants. It should also be emphasized that in addition to its role in antioxidant defense, GSH has many other functions, including detoxification of various toxicants, especially electrophiles, by forming conjugates with these compounds. NAC may provide a similar protective effect after conversion to GSH. Therefore, the therapeutic effects observed with NAC in clinical trials may result from the detoxification of other drugs, such as immunosuppresive drugs administered at the same time [325, 332]. An ongoing clinical trial [PANTHER (Prednisone, Azathioprine and N-acetylcysteine: A Study THat Evaluates Responses in IPF) to be conduced by IPFnet] will compare the therapeutic effects of NAC versus placebo versus NAC plus immunosuppressive therapy (prednisone and azathioprine) in the early stage of IPF patients. The results from this clinical trial should provide direct evidence whether NAC has direct benefit for IPF.
CONCLUSION
ROS/RNS play an important role in the development of fibrosis and in TGF-β-mediated fibrogenesis. The molecular mechanism by which ROS/RNS mediate TGF-β-induced fibrosis, however, remains undetermined. GSH is the most abundant intracellular free thiol and an important antioxidant. GSH concentration decreases in the fibrotic diseases and in experimental fibrosis models. Although TGF-β decreases GSH in various types of cells in vitro, whether increased TGF-β expression is responsible for the decrease in GSH concentration in fibrotic diseases in humans is unknown. Furthermore, the biological significance of GSH depletion in the development of fibrosis is also unclear. GSH and NAC have been used in clinics for the treatment of various fibrotic diseases. Although promising, the therapeutic value and mechanism underlying their therapeutic effects remain to be further evaluated. As TGF-β plays a vital role in the development of fibrosis under almost all the pathological conditions and ROS mediate many of TGF-β’s fibrogenic effects, to uncover the molecular mechanism underlying redox regulation of TGF-β’s fibrogenesis will be critical for the development of efficacious treatment for these devastating diseases.
Acknowledgments
The work was supported by grants from National Institute of Environmental Health Sciences (NIH ES011831 and NHLBI 5R01HL088141-02) and a grant from the American Lung Association (CI-1190-N) to RM Liu.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, van Herwaarden CL, Bast A. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1996;154:813–816. doi: 10.1164/ajrccm.154.3.8810624. [DOI] [PubMed] [Google Scholar]
- 2.Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med. 1996;154:1055–1060. doi: 10.1164/ajrccm.154.4.8887607. [DOI] [PubMed] [Google Scholar]
- 3.Pratico D, Basili S, Vieri M, Cordova C, Violi F, Fitzgerald GA. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2alpha-III, an index of oxidant stress. Am J Respir Crit Care Med. 1998;158:1709–1714. doi: 10.1164/ajrccm.158.6.9709066. [DOI] [PubMed] [Google Scholar]
- 4.Barreiro E, de la Puente B, Minguella J, Corominas JM, Serrano S, Hussain SNA, Gea J. Oxidative Stress and Respiratory Muscle Dysfunction in Severe Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2005;171:1116–1124. doi: 10.1164/rccm.200407-887OC. [DOI] [PubMed] [Google Scholar]
- 5.Juul K, Tybjaerg-Hansen A, Marklund S, Lange P, Nordestgaard BG. Genetically Increased Antioxidative Protection and Decreased Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2006;173:858–864. doi: 10.1164/rccm.200509-1387OC. [DOI] [PubMed] [Google Scholar]
- 6.Kuleci S, Hanta I, Kocabas A, Canacankatan N. The effect of different treatment modalities on oxidative stress in COPD. Advances in Therapy. 2008;25:710–717. doi: 10.1007/s12325-008-0064-4. [DOI] [PubMed] [Google Scholar]
- 7.Louhelainen N, Myllarniemi M, Rahman I, Kinnula VL. Airway biomarkers of the oxidant burden in asthma and chronic obstructive pulmonary disease: current and future perspectives. Int J Chron Obstruct Pulmon Dis. 2008;3:585–603. doi: 10.2147/copd.s3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maier K, Leuschel L, Costabel U. Increased levels of oxidized methionine residues in bronchoalveolar lavage fluid proteins from patients with idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1991;143:271–274. doi: 10.1164/ajrccm/143.2.271. [DOI] [PubMed] [Google Scholar]
- 9.Paola Rottoli BM, Cianti Riccardo, Bargagli Elena, Vagaggini Cecilia, Nikiforakis Nikolaos, Pallini Vitaliano, Bini Luca. Carbonylated proteins in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and idiopathic pulmonary fibrosis. PROTEOMICS. 2005;5:2612–2618. doi: 10.1002/pmic.200401206. [DOI] [PubMed] [Google Scholar]
- 10.Psathakis K, Mermigkis D, Papatheodorou G, Loukides S, Panagou P, Polychronopoulos V, Siafakas NM, Bouros D. Exhaled markers of oxidative stress in idiopathic pulmonary fibrosis. Eur J Clin Invest. 2006;36:362–367. doi: 10.1111/j.1365-2362.2006.01636.x. [DOI] [PubMed] [Google Scholar]
- 11.Starosta V, Rietschel E, Paul K, Baumann U, Griese M. Oxidative Changes of Bronchoalveolar Proteins in Cystic Fibrosis. Chest. 2006;129:431–437. doi: 10.1378/chest.129.2.431. [DOI] [PubMed] [Google Scholar]
- 12.Rosias PPR, Hartog GJMD, Robroeks CMHHT, Bast A, Donckerwolcke RAMG, Heynens JWCM, Suykerbuyk J, Hendriks HJE, Jöbsis Q, Dompeling E. Free radicals in exhaled breath condensate in cystic fibrosis and healthy subjects. Free Radical Research. 2006;40:901–909. doi: 10.1080/10715760500522648. [DOI] [PubMed] [Google Scholar]
- 13.REID DW, MISSO N, AGGARWAL S, THOMPSON PJ, WALTERS EH. Oxidative stress and lipid-derived inflammatory mediators during acute exacerbations of cystic fibrosis. Respirology. 2007;12:63–69. doi: 10.1111/j.1440-1843.2006.00962.x. [DOI] [PubMed] [Google Scholar]
- 14.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YHC, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JSM, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pilger A, Germadnik D, Schaffer A, Theiler A, Pils P, Sluka F, Winker N, Rudiger HW. 8-Hydroxydeoxyguanosine in leukocyte DNA and urine of quartz-exposed workers and patients with silicosis. Int Arch Occup Environ Health. 2000;73:305–310. doi: 10.1007/s004200000117. [DOI] [PubMed] [Google Scholar]
- 16.Kim J. Urinary 8-hydroxy-2′-deoxyguanosine as a biomarker of oxidative DNA damage in workers exposed to fine particulates. Environmental Health Perspectives. 2004;112:666. doi: 10.1289/ehp.6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pelclova D, Fenclova Z, Kacer P, Kuzma M, Navratil T, Lebedova J. Increased 8-isoprostane, a marker of oxidative stress in exhaled breath condensate in subjects with asbestos exposure. Ind Health. 2008;46:484–489. doi: 10.2486/indhealth.46.484. [DOI] [PubMed] [Google Scholar]
- 18.Srivastava KD, Rom WN, Jagirdar J, Yie TA, Gordon T, Tchou-Wong KM. Crucial role of interleukin-1beta and nitric oxide synthase in silica-induced inflammation and apoptosis in mice. Am J Respir Crit Care Med. 2002;165:527–533. doi: 10.1164/ajrccm.165.4.2106009. [DOI] [PubMed] [Google Scholar]
- 19.Porter DW, Millecchia LL, Willard P, Robinson VA, Ramsey D, McLaurin J, Khan A, Brumbaugh K, Beighley CM, Teass A, Castranova V. Nitric oxide and reactive oxygen species production causes progressive damage in rats after cessation of silica inhalation. Toxicol Sci. 2006;90:188–197. doi: 10.1093/toxsci/kfj075. [DOI] [PubMed] [Google Scholar]
- 20.Sato T, Takeno M, Honma K, Yamauchi H, Saito Y, Sasaki T, Morikubo H, Nagashima Y, Takagi S, Yamanaka K, Kaneko T, Ishigatsubo Y. Heme Oxygenase-1, a Potential Biomarker of Chronic Silicosis, Attenuates Silica-induced Lung Injury. Am J Respir Crit Care Med. 2006;174:906–914. doi: 10.1164/rccm.200508-1237OC. [DOI] [PubMed] [Google Scholar]
- 21.Manoury B, Nenan S, Leclerc O, Guenon I, Boichot E, Planquois JM, Bertrand CP, Lagente V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir Res. 2005;6:11. doi: 10.1186/1465-9921-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yildirim Z, Kotuk M, Iraz M, Kuku I, Ulu R, Armutcu F, Ozen S. Attenuation of bleomycin-induced lung fibrosis by oral sulfhydryl containing antioxidants in rats: erdosteine and N-acetylcysteine. Pulm Pharmacol Ther. 2005;18:367–373. doi: 10.1016/j.pupt.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Liu R, Ahmed KM, Nantajit D, Rosenthal FS, Hai CX, Li JJ. Therapeutic effects of alpha-lipoic acid on bleomycin-induced pulmonary fibrosis in rats. Int J Mol Med. 2007;19:865–873. [PubMed] [Google Scholar]
- 24.Iyer SS, Ramirez AM, Ritzenthaler JD, Torres-Gonzalez E, Roser-Page S, Mora AL, Brigham KL, Jones DP, Roman J, Rojas M. Oxidation of extracellular cysteine/cystine redox state in bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2009;296:L37–45. doi: 10.1152/ajplung.90401.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perlemuter G, Letteron P, Carnot F, Zavala F, Pessayre D, Nalpas B, Brechot C. Alcohol and hepatitis C virus core protein additively increase lipid peroxidation and synergistically trigger hepatic cytokine expression in a transgenic mouse model. J Hepatol. 2003;39:1020–1027. doi: 10.1016/s0168-8278(03)00414-8. [DOI] [PubMed] [Google Scholar]
- 26.Purohit V, Brenner DA. Mechanisms of alcohol-induced hepatic fibrosis: a summary of the Ron Thurman Symposium. Hepatology. 2006;43:872–878. doi: 10.1002/hep.21107. [DOI] [PubMed] [Google Scholar]
- 27.Samuele De Minicis DAB. Oxidative stress in alcoholic liver disease: Role of NADPH oxidase complex. Journal of Gastroenterology and Hepatology. 2008;23:S98–S103. doi: 10.1111/j.1440-1746.2007.05277.x. [DOI] [PubMed] [Google Scholar]
- 28.Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S, Majano PL, Benedicto I, Rosado JA, Salido GM, Gonzalez-Gallego J. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. Journal of Hepatology. 2009;50:872–882. doi: 10.1016/j.jhep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 29.Yadav D, Hertan HI, Schweitzer P, Norkus EP, Pitchumoni CS. Serum and liver micronutrient antioxidants and serum oxidative stress in patients with chronic hepatitis C. Am J Gastroenterol. 2002;97:2634–2639. doi: 10.1111/j.1572-0241.2002.06041.x. [DOI] [PubMed] [Google Scholar]
- 30.Bedossa P, Paradis V. Approaches for treatment of liver fibrosis in chronic hepatitis C. Clin Liver Dis. 2003;7:195–210. doi: 10.1016/s1089-3261(02)00076-4. [DOI] [PubMed] [Google Scholar]
- 31.Seki S, Kitada T, Sakaguchi H. Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatology Research. 2005;33:132–134. doi: 10.1016/j.hepres.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 32.Fujita N, Horiike S, Sugimoto R, Tanaka H, Iwasa M, Kobayashi Y, Hasegawa K, Ma N, Kawanishi S, Adachi Y, Kaito M. Hepatic oxidative DNA damage correlates with iron overload in chronic hepatitis C patients. Free Radical Biology and Medicine. 2007;42:353–362. doi: 10.1016/j.freeradbiomed.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44:1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urtasun R, de la Rosa LC, Nieto N. Oxidative and Nitrosative Stress and Fibrogenic Response. Clinics in Liver Disease Hepatic Fibrosis: Pathogenesis, Diagnosis, and Emerging Therapies. 2008;12:769–790. doi: 10.1016/j.cld.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das KS, Balakrishnan V, Mukherjee S, Vasudevan DM. Evaluation of blood oxidative stress-related parameters in alcoholic liver disease and non-alcoholic fatty liver disease. Scand J Clin Lab Invest. 2008;68:323–334. doi: 10.1080/00365510701673383. [DOI] [PubMed] [Google Scholar]
- 36.Pawlak K, Zolbach K, Borawski J, Mysliwiec M, Kovalchuk O, Chyczewski L, Pawlak D. Chronic viral hepatitis C, oxidative stress and the coagulation/fibrinolysis system in haemodialysis patients. Thrombosis Research. 2008;123:166–170. doi: 10.1016/j.thromres.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 37.Graeme A Macdonald KRB, Ward Patrick J, Walker Neal I, Houglum Karl, George D Keith, Smith Jeffery L, Powell Lawrie W, Crawford Darrell Hg, Ramm Grant A. Lipid peroxidation in hepatic steatosis in humans is associated with hepatic fibrosis and occurs predominately in acinar zone 3. Journal of Gastroenterology and Hepatology. 2001;16:599–606. doi: 10.1046/j.1440-1746.2001.02445.x. [DOI] [PubMed] [Google Scholar]
- 38.Ha H, Lee HB. Reactive Oxygen Species and Matrix Remodeling in Diabetic Kidney. J Am Soc Nephrol. 2003;14:S246–249. doi: 10.1097/01.asn.0000077411.98742.54. [DOI] [PubMed] [Google Scholar]
- 39.Ha H, Lee HB. Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrology (Carlton) 2005;10(Suppl):S7–10. doi: 10.1111/j.1440-1797.2005.00448.x. [DOI] [PubMed] [Google Scholar]
- 40.Agarwal R. Proinflammatory effects of oxidative stress in chronic kidney disease: role of additional angiotensin II blockade. Am J Physiol Renal Physiol. 2003;284:F863–869. doi: 10.1152/ajprenal.00385.2002. [DOI] [PubMed] [Google Scholar]
- 41.Nangaku M. Chronic Hypoxia and Tubulointerstitial Injury: A Final Common Pathway to End-Stage Renal Failure. J Am Soc Nephrol. 2006;17:17–25. doi: 10.1681/ASN.2005070757. [DOI] [PubMed] [Google Scholar]
- 42.Zhao W, Chen SS, Chen Y, Ahokas RA, Sun Y. Kidney fibrosis in hypertensive rats: role of oxidative stress. Am J Nephrol. 2008;28:548–554. doi: 10.1159/000115289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Djamali A, Vidyasagar A, Adulla M, Hullett D, Reese S. Nox-2 is a modulator of fibrogenesis in kidney allografts. Am J Transplant. 2009;9:74–82. doi: 10.1111/j.1600-6143.2008.02463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogawa F, Shimizu K, Muroi E, Hara T, Hasegawa M, Takehara K, Sato S. Serum levels of 8-isoprostane, a marker of oxidative stress, are elevated in patients with systemic sclerosis. Rheumatology. 2006;45:815–818. doi: 10.1093/rheumatology/kel012. [DOI] [PubMed] [Google Scholar]
- 45.Murdoch CE, Zhang M, Cave AC, Shah AM. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res. 2006;71:208–215. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 46.Sirker A, Zhang M, Murdoch C, Shah AM. Involvement of NADPH oxidases in cardiac remodelling and heart failure. Am J Nephrol. 2007;27:649–660. doi: 10.1159/000109148. [DOI] [PubMed] [Google Scholar]
- 47.Sun Y. Oxidative stress and cardiac repair/remodeling following infarction. Am J Med Sci. 2007;334:197–205. doi: 10.1097/MAJ.0b013e318157388f. [DOI] [PubMed] [Google Scholar]
- 48.Aragno M, Mastrocola R, Alloatti G, Vercellinatto I, Bardini P, Geuna S, Catalano MG, Danni O, Boccuzzi G. Oxidative stress triggers cardiac fibrosis in the heart of diabetic rats. Endocrinology. 2008;149:380–388. doi: 10.1210/en.2007-0877. [DOI] [PubMed] [Google Scholar]
- 49.Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol Cell Biochem. 2008;317:43–50. doi: 10.1007/s11010-008-9803-8. [DOI] [PubMed] [Google Scholar]
- 50.Broekelmann TJ, Limper AH, Colby TV, McDonald JA. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci U S A. 1991;88:6642–6646. doi: 10.1073/pnas.88.15.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khalil N, O’Connor RN, Flanders KC, Unruh H. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am J Respir Cell Mol Biol. 1996;14:131–138. doi: 10.1165/ajrcmb.14.2.8630262. [DOI] [PubMed] [Google Scholar]
- 52.Jagirdar J, Lee TC, Reibman J, Gold LI, Aston C, Begin R, Rom WN. Immunohistochemical localization of transforming growth factor beta isoforms in asbestos-related diseases. Environ Health Perspect. 1997;105(Suppl 5):1197–1203. doi: 10.1289/ehp.97105s51197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, Hance AJ, Tazi A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J. 2003;22:69–76. doi: 10.1183/09031936.03.00014703. [DOI] [PubMed] [Google Scholar]
- 54.Rimal B, Greenberg AK, Rom WN. Basic pathogenetic mechanisms in silicosis: current understanding. Curr Opin Pulm Med. 2005;11:169–173. doi: 10.1097/01.mcp.0000152998.11335.24. [DOI] [PubMed] [Google Scholar]
- 55.Brook NR, Waller SAWJR, Bicknell GR, Nicholson ML. Fibrosis-associated gene expression in renal transplant glomeruli after acute renal allograft rejection. British Journal of Surgery. 2003;90:1009–1014. doi: 10.1002/bjs.4133. [DOI] [PubMed] [Google Scholar]
- 56.Ali MA, Koura BA, el-Mashad N, Zaghloul MH. The Bcl-2 and TGF-beta1 levels in patients with chronic hepatitis C, liver cirrhosis and hepatocellular carcinoma. Egypt J Immunol. 2004;11:83–90. [PubMed] [Google Scholar]
- 57.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tzortzaki EG, Antoniou KM, Zervou MI, Lambiri I, Koutsopoulos A, Tzanakis N, Plataki M, Maltezakis G, Bouros D, Siafakas NM. Effects of antifibrotic agents on TGF-[beta]1, CTGF and IFN-[gamma] expression in patients with idiopathic pulmonary fibrosis. Respiratory Medicine. 2007;101:1821–1829. doi: 10.1016/j.rmed.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 59.Brosius F. New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Reviews in Endocrine & Metabolic Disorders. 2008;9:245–254. doi: 10.1007/s11154-008-9100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varga J. Systemic sclerosis: an update.(Disease/Disorder overview) Bulletin of the NYU Hospital for Joint Diseases. 2008;66:198. [PubMed] [Google Scholar]
- 61.Fahy RJ, Lichtenberger F, McKeegan CB, Nuovo GJ, Marsh CB, Wewers MD. The acute respiratory distress syndrome: a role for transforming growth factor-beta 1. Am J Respir Cell Mol Biol. 2003;28:499–503. doi: 10.1165/rcmb.2002-0092OC. [DOI] [PubMed] [Google Scholar]
- 62.Williams AO, Flanders KC, Saffiotti U. Immunohistochemical localization of transforming growth factor-beta 1 in rats with experimental silicosis, alveolar type II hyperplasia, and lung cancer. Am J Pathol. 1993;142:1831–1840. [PMC free article] [PubMed] [Google Scholar]
- 63.Coker RK, Laurent GJ, Shahzeidi S, Lympany PA, du Bois RM, Jeffery PK, McAnulty RJ. Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am J Pathol. 1997;150:981–991. [PMC free article] [PubMed] [Google Scholar]
- 64.Rube CE, Uthe D, Schmid KW, Richter KD, Wessel J, Schuck A, Willich N, Rube C. Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol Biol Phys. 2000;47:1033–1042. doi: 10.1016/s0360-3016(00)00482-x. [DOI] [PubMed] [Google Scholar]
- 65.Kumar RK, Herbert C, Foster PS. Expression of growth factors by airway epithelial cells in a model of chronic asthma: regulation and relationship to subepithelial fibrosis. Clin Exp Allergy. 2004;34:567–575. doi: 10.1111/j.1365-2222.2004.1917.x. [DOI] [PubMed] [Google Scholar]
- 66.Ellmers LJ, Scott NJA, Medicherla S, Pilbrow AP, Bridgman PG, Yandle TG, Richards AM, Protter AA, Cameron VA. Transforming Growth Factor-{beta} Blockade Down-Regulates the Renin-Angiotensin System and Modifies Cardiac Remodeling after Myocardial Infarction. Endocrinology. 2008;149:5828–5834. doi: 10.1210/en.2008-0165. [DOI] [PubMed] [Google Scholar]
- 67.Churg A, Zhou S, Preobrazhenska O, Tai H, Wang R, Wright JL. Expression of Profibrotic Mediators in Small Airways versus Parenchyma after Cigarette Smoke Exposure. Am J Respir Cell Mol Biol. 2009;40:268–276. doi: 10.1165/rcmb.2007-0367OC. [DOI] [PubMed] [Google Scholar]
- 68.Ng YY, Chen YM, Tsai TJ, Lan XR, Yang WC, Lan HY. Pentoxifylline inhibits transforming growth factor-beta signaling and renal fibrosis in experimental crescentic glomerulonephritis in rats. Am J Nephrol. 2009;29:43–53. doi: 10.1159/000150600. [DOI] [PubMed] [Google Scholar]
- 69.Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clouthier DE, Comerford SA, Hammer RE. Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Invest. 1997;100:2697–2713. doi: 10.1172/JCI119815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mozes MM, Bottinger EP, Jacot TA, Kopp JB. Renal expression of fibrotic matrix proteins and of transforming growth factor-beta (TGF-beta) isoforms in TGF-beta transgenic mice. J Am Soc Nephrol. 1999;10:271–280. doi: 10.1681/ASN.V102271. [DOI] [PubMed] [Google Scholar]
- 72.Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L741–749. doi: 10.1152/ajplung.00208.2003. [DOI] [PubMed] [Google Scholar]
- 73.Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J, Homer RJ, Elias JA. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vicencio AG, Lee CG, Cho SJ, Eickelberg O, Chuu Y, Haddad GG, Elias JA. Conditional overexpression of bioactive transforming growth factor-beta1 in neonatal mouse lung: a new model for bronchopulmonary dysplasia? Am J Respir Cell Mol Biol. 2004;31:650–656. doi: 10.1165/rcmb.2004-0092OC. [DOI] [PubMed] [Google Scholar]
- 75.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kolb M, Bonniaud P, Galt T, Sime PJ, Kelly MM, Margetts PJ, Gauldie J. Differences in the fibrogenic response after transfer of active transforming growth factor- beta1 gene to lungs of “fibrosis-prone” and “fibrosis-resistant” mouse strains. Am J Respir Cell Mol Biol. 2002;27:141–150. doi: 10.1165/ajrcmb.27.2.4674. [DOI] [PubMed] [Google Scholar]
- 77.Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, Murphy A, Chakravarty S, Dugar S, Higgins L, Protter AA, Gauldie J. Progressive Transforming Growth Factor {beta}1-induced Lung Fibrosis Is Blocked by an Orally Active ALK5 Kinase Inhibitor. Am J Respir Crit Care Med. 2005;171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- 78.Margetts PJ, Bonniaud P, Liu L, Hoff CM, Holmes CJ, West-Mays JA, Kelly MM. Transient overexpression of TGF-{beta}1 induces epithelial mesenchymal transition in the rodent peritoneum. J Am Soc Nephrol. 2005;16:425–436. doi: 10.1681/ASN.2004060436. [DOI] [PubMed] [Google Scholar]
- 79.Giri SN, Hyde DM, Hollinger MA. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax. 1993;48:959–966. doi: 10.1136/thx.48.10.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Higashiyama H, Yoshimoto D, Kaise T, Matsubara S, Fujiwara M, Kikkawa H, Asano S, Kinoshita M. Inhibition of activin receptor-like kinase 5 attenuates bleomycin-induced pulmonary fibrosis. Exp Mol Pathol. 2007;83:39–46. doi: 10.1016/j.yexmp.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 81.Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346:281–284. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 82.Wang Q, Wang Y, Hyde DM, Gotwals PJ, Koteliansky VE, Ryan ST, Giri SN. Reduction of bleomycin induced lung fibrosis by transforming growth factor beta soluble receptor in hamsters. Thorax. 1999;54:805–812. doi: 10.1136/thx.54.9.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kolb M, Margetts PJ, Galt T, Sime PJ, Xing Z, Schmidt M, Gauldie J. Transient transgene expression of decorin in the lung reduces the fibrotic response to bleomycin. Am J Respir Crit Care Med. 2001;163:770–777. doi: 10.1164/ajrccm.163.3.2006084. [DOI] [PubMed] [Google Scholar]
- 84.Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1327–1334. doi: 10.1152/ajplung.2001.280.6.L1327. [DOI] [PubMed] [Google Scholar]
- 85.Yoo BM, Yeo M, Oh TY, Choi JH, Kim WW, Kim JH, Cho SW, Kim SJ, Hahm KB. Amelioration of pancreatic fibrosis in mice with defective TGF-beta signaling. Pancreas. 2005;30:e71–79. doi: 10.1097/01.mpa.0000157388.54016.0a. [DOI] [PubMed] [Google Scholar]
- 86.Ambalavanan N, Nicola T, Hagood J, Bulger A, Serra R, Murphy-Ullrich J, Oparil S, Chen YF. Transforming growth factor-beta signaling mediates hypoxia-induced pulmonary arterial remodeling and inhibition of alveolar development in newborn mouse lung. Am J Physiol Lung Cell Mol Physiol. 2008;295:L86–95. doi: 10.1152/ajplung.00534.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shibanuma M, Kuroki T, Nose K. Release of H2O2 and phosphorylation of 30 kilodalton proteins as early responses of cell cycle-dependent inhibition of DNA synthesis by transforming growth factor beta 1. Cell Growth Differ. 1991;2:583–591. [PubMed] [Google Scholar]
- 88.Thannickal VJ, Day RM, Klinz SG, Bastien MC, Larios JM, Fanburg BL. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-beta1. Faseb J. 2000;14:1741–1748. doi: 10.1096/fj.99-0878com. [DOI] [PubMed] [Google Scholar]
- 89.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 90.Thannickal VJ, Hassoun PM, White AC, Fanburg BL. Enhanced rate of H2O2 release from bovine pulmonary artery endothelial cells induced by TGF-beta 1. Am J Physiol. 1993;265:L622–626. doi: 10.1152/ajplung.1993.265.6.L622. [DOI] [PubMed] [Google Scholar]
- 91.Hu T, RamachandraRao SP, Siva S, Valancius C, Zhu Y, Mahadev K, Toh I, Goldstein BJ, Woolkalis M, Sharma K. Reactive oxygen species production via NADPH oxidase mediates TGF-{beta}-induced cytoskeletal alterations in endothelial cells. Am J Physiol Renal Physiol. 2005;289:F816–825. doi: 10.1152/ajprenal.00024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yoon YS, Lee JH, Hwang SC, Choi KS, Yoon G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene. 2005;24:1895–1903. doi: 10.1038/sj.onc.1208262. [DOI] [PubMed] [Google Scholar]
- 93.Goettsch C, Goettsch W, Muller G, Seebach J, Schnittler HJ, Morawietz H. Nox4 overexpression activates reactive oxygen species and p38 MAPK in human endothelial cells. Biochem Biophys Res Commun. 2009 doi: 10.1016/j.bbrc.2009.01.107. [DOI] [PubMed] [Google Scholar]
- 94.Murillo MM, Carmona-Cuenca I, Del Castillo G, Ortiz C, Roncero C, Sanchez A, Fernandez M, Fabregat I. Activation of NADPH oxidase by transforming growth factor-beta in hepatocytes mediates up-regulation of epidermal growth factor receptor ligands through a nuclear factor-kappaB-dependent mechanism. Biochem J. 2007;405:251–259. doi: 10.1042/BJ20061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carmona-Cuenca I, Roncero C, Sancho P, Caja L, Fausto N, Fernandez M, Fabregat I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J Hepatol. 2008;49:965–976. doi: 10.1016/j.jhep.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 96.Sancho P, Bertran E, Caja L, Carmona-Cuenca I, Murillo MM, Fabregat I. The inhibition of the epidermal growth factor (EGF) pathway enhances TGF-beta-induced apoptosis in rat hepatoma cells through inducing oxidative stress coincident with a change in the expression pattern of the NADPH oxidases (NOX) isoforms. Biochim Biophys Acta. 2009;1793:253–263. doi: 10.1016/j.bbamcr.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 97.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 98.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 99.Albright CD, Salganik RI, Craciunescu CN, Mar MH, Zeisel SH. Mitochondrial and microsomal derived reactive oxygen species mediate apoptosis induced by transforming growth factor-beta1 in immortalized rat hepatocytes. J Cell Biochem. 2003;89:254–261. doi: 10.1002/jcb.10498. [DOI] [PubMed] [Google Scholar]
- 100.Herrera B, Murillo MM, Alvarez-Barrientos A, Beltran J, Fernandez M, Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-[beta] in fetal rat hepatocytes. Free Radical Biology and Medicine. 2004;36:16–26. doi: 10.1016/j.freeradbiomed.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 101.Sturrock A, Huecksteadt TP, Norman K, Sanders K, Murphy TM, Chitano P, Wilson K, Hoidal JR, Kennedy TP. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1543–1555. doi: 10.1152/ajplung.00430.2006. [DOI] [PubMed] [Google Scholar]
- 102.Ismail S, Sturrock A, Wu P, Cahill B, Norman K, Huecksteadt T, Sanders K, Kennedy T, Hoidal J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am J Physiol Lung Cell Mol Physiol. 2009;296:L489–499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Buul JDV, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and Localization of NOX2 and NOX4 in Primary Human Endothelial Cells. Antioxidants & Redox Signaling. 2005;7:308. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- 105.Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF., Jr Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Helmcke I, Heumuller S, Tikkanen R, Schroder K, Brandes RP. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2383. [DOI] [PubMed] [Google Scholar]
- 107.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz A-C, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MAR, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HHHW, Weissmann N. Hypoxia-Dependent Regulation of Nonphagocytic NADPH Oxidase Subunit NOX4 in the Pulmonary Vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 108.Natarajan V, Pendyala S, Gorshkova IA, Usatyuk P, He D, Pennathur A, Lambeth JD, Thannickal VJ. Role of Nox4 and Nox2 in Hyperoxia-Induced Reactive Oxygen Species Generation and Migration of Human Lung Endothelial Cells. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Junya Kuroda KN, Yamasaki Tomoko, Nakamura Kei-ichiro, Takeya Ryu, Kuribayashi Futoshi, Imajoh-Ohmi Shinobu, Igarashi Kazuhiko, Shibata Yosaburo, Sueishi Katsuo, Sumimoto Hideki. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes to Cells. 2005;10:1139–1151. doi: 10.1111/j.1365-2443.2005.00907.x. [DOI] [PubMed] [Google Scholar]
- 110.Kayanoki Y, Fujii J, Suzuki K, Kawata S, Matsuzawa Y, Taniguchi N. Suppression of antioxidative enzyme expression by transforming growth factor-beta 1 in rat hepatocytes. J Biol Chem. 1994;269:15488–15492. [PubMed] [Google Scholar]
- 111.Islam KN, Kayanoki Y, Kaneto H, Suzuki K, Asahi M, Fujii J, Taniguchi N. TGF-beta1 triggers oxidative modifications and enhances apoptosis in HIT cells through accumulation of reactive oxygen species by suppression of catalase and glutathione peroxidase. Free Radic Biol Med. 1997;22:1007–1017. doi: 10.1016/s0891-5849(96)00493-5. [DOI] [PubMed] [Google Scholar]
- 112.Peltoniemi M, Kaarteenaho-Wiik R, Saily M, Sormunen R, Paakko P, Holmgren A, Soini Y, Kinnula VL. Expression of glutaredoxin is highly cell specific in human lung and is decreased by transforming growth factor-beta in vitro and in interstitial lung diseases in vivo. Hum Pathol. 2004;35:1000–1007. doi: 10.1016/j.humpath.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 113.Saharinen J, Taipale J, Monni O, Keski-Oja J. Identification and Characterization of a New Latent Transforming Growth Factor-beta -binding Protein, LTBP-4. J Biol Chem. 1998;273:18459–18469. doi: 10.1074/jbc.273.29.18459. [DOI] [PubMed] [Google Scholar]
- 114.Todorovic V, Jurukovski V, Chen Y, Fontana L, Dabovic B, Rifkin DB. Latent TGF-[beta] binding proteins. The International Journal of Biochemistry & Cell Biology. 2005;37:38–41. doi: 10.1016/j.biocel.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 115.Koli K, Hyytiäinen M, Ryynänen MJ, Keski-Oja J. Sequential deposition of latent TGF-[beta] binding proteins (LTBPs) during formation of the extracellular matrix in human lung fibroblasts. Experimental Cell Research. 2005;310:370–382. doi: 10.1016/j.yexcr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 116.Schultz-Cherry S, Murphy-Ullrich JE. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol. 1993;122:923–932. doi: 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 118.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 119.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol. 1988;106:1659–1665. doi: 10.1083/jcb.106.5.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270:4689–4696. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- 122.Taipale J, Koli K, Keski-Oja J. Release of transforming growth factor-beta 1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. J Biol Chem. 1992;267:25378–25384. [PubMed] [Google Scholar]
- 123.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 124.Barcellos-Hoff M, Dix T. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 125.Vodovotz Y, Chesler L, Chong H, Kim SJ, Simpson JT, DeGraff W, Cox GW, Roberts AB, Wink DA, Barcellos-Hoff MH. Regulation of transforming growth factor beta1 by nitric oxide. Cancer Res. 1999;59:2142–2149. [PubMed] [Google Scholar]
- 126.Pociask DA, Sime PJ, Brody AR. Asbestos-derived reactive oxygen species activate TGF-[beta]1. 2004;84:1013–1023. doi: 10.1038/labinvest.3700109. [DOI] [PubMed] [Google Scholar]
- 127.Wang L, Clutter S, Benincosa J, Fortney J, Gibson LF. Activation of TGF-{beta}1/p38/Smad3 signaling in stromal cells requires ROS-mediated MMP-2 activity during bone marrow damage. Stem Cells. 2005 doi: 10.1634/stemcells.2004-0354. 2004-0354. [DOI] [PubMed] [Google Scholar]
- 128.Amarnath S, Dong L, Li J, Wu Y, Chen W. Endogenous TGF-beta activation by reactive oxygen species is key to Foxp3 induction in TCR-stimulated and HIV-1-infected human CD4+CD25− T cells. Retrovirology. 2007;4:57. doi: 10.1186/1742-4690-4-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang H, Kochevar IE. Involvement of UVB-induced reactive oxygen species in TGF-beta biosynthesis and activation in keratinocytes. Free Radic Biol Med. 2005;38:890–897. doi: 10.1016/j.freeradbiomed.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 130.Jobling MF, Mott JD, Finnegan MT, Jurukovski V, Erickson AC, Walian PJ, Taylor SE, Ledbetter S, Lawrence CM, Rifkin DB, Barcellos-Hoff MH. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat Res. 2006;166:839–848. doi: 10.1667/RR0695.1. [DOI] [PubMed] [Google Scholar]
- 131.Sullivan DE, Ferris M, Pociask D, Brody AR. The latent form of TGFbeta(1) is induced by TNFalpha through an ERK specific pathway and is activated by asbestos-derived reactive oxygen species in vitro and in vivo. J Immunotoxicol. 2008;5:145–149. doi: 10.1080/15476910802085822. [DOI] [PubMed] [Google Scholar]
- 132.Leonarduzzi G, Scavazza A, Biasi F, Chiarpotto E, Camandola S, Vogl S, Dargel R, Poli G. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor b1 expression in the macrophage lineage: a link between oxidative injury and fibrosclerosis. FASEB Journal. 1997;11:851–857. doi: 10.1096/fasebj.11.11.9285483. [DOI] [PubMed] [Google Scholar]
- 133.Bellocq A, Azoulay E, Marullo S, Flahault A, Fouqueray B, Philippe C, Cadranel J, Baud L. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am J Respir Cell Mol Biol. 1999;21:128–136. doi: 10.1165/ajrcmb.21.1.3379. [DOI] [PubMed] [Google Scholar]
- 134.Saito K, Ishizaka N, Aizawa T, Sata M, Iso-o N, Noiri E, Mori I, Ohno M, Nagai R. Iron chelation and a free radical scavenger suppress angiotensin II-induced upregulation of TGF-beta1 in the heart. Am J Physiol Heart Circ Physiol. 2005;288:H1836–1843. doi: 10.1152/ajpheart.00679.2004. [DOI] [PubMed] [Google Scholar]
- 135.Shvedova AA, Kisin ER, Murray AR, Kommineni C, Castranova V, Fadeel B, Kagan VE. Increased accumulation of neutrophils and decreased fibrosis in the lung of NADPH oxidase-deficient C57BL/6 mice exposed to carbon nanotubes. Toxicol Appl Pharmacol. 2008;231:235–240. doi: 10.1016/j.taap.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 136.Clark RA, McCoy GA, Folkvord JM, McPherson JM. TGF-beta 1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: a fibronectin matrix-dependent event. J Cell Physiol. 1997;170:69–80. doi: 10.1002/(SICI)1097-4652(199701)170:1<69::AID-JCP8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 137.Kay EP, Lee MS, Seong GJ, Lee YG. TGF-beta s stimulate cell proliferation via an autocrine production of FGF-2 in corneal stromal fibroblasts. Curr Eye Res. 1998;17:286–293. doi: 10.1076/ceyr.17.3.286.5212. [DOI] [PubMed] [Google Scholar]
- 138.Thannickal VJ, Aldweib KD, Rajan T, Fanburg BL. Upregulated expression of fibroblast growth factor (FGF) receptors by transforming growth factor-beta1 (TGF-beta1) mediates enhanced mitogenic responses to FGFs in cultured human lung fibroblasts. Biochem Biophys Res Commun. 1998;251:437–441. doi: 10.1006/bbrc.1998.9443. [DOI] [PubMed] [Google Scholar]
- 139.Strutz F, Zeisberg M, Renziehausen A, Raschke B, Becker V, van Kooten C, Muller G. TGF-beta 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2) Kidney Int. 2001;59:579–592. doi: 10.1046/j.1523-1755.2001.059002579.x. [DOI] [PubMed] [Google Scholar]
- 140.L Gallelli, Pelaia DFG, Renda T, Terracciano R, Malara N, Vatrella A, Sanduzzi A, D’Agostino B, Rossi F, Vancheri C, Maselli R, Marsico SA, Savino R. Interleukin-6 receptor superantagonist Sant7 inhibits TGF-β-induced proliferation of human lung fibroblasts. Cell Proliferation. 2008;41:393–407. doi: 10.1111/j.1365-2184.2008.00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cho JW, Cha YC, Lee KS. AP-1 transcription factor decoy reduces the TGF-beta1-induced cell growth in scleroderma fibroblasts through inhibition of cyclin E. Oncol Rep. 2008;19:737–741. [PubMed] [Google Scholar]
- 142.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of Circulating Fibrocytes as Precursors of Bronchial Myofibroblasts in Asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 143.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Strieter RM, Keeley EC, Hughes MA, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J Leukoc Biol. 2009:jlb.0309132. doi: 10.1189/jlb.0309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xu J, Gonzalez ET, Iyer SS, Mac V, Mora AL, Sutliff RL, Reed A, Brigham KL, Kelly P, Rojas M. Use of Senescence-Accelerated Mouse Model in Bleomycin-Induced Lung Injury Suggests That Bone Marrow-Derived Cells Can Alter the Outcome of Lung Injury in Aged Mice. J Gerontol A Biol Sci Med Sci. 2009;64A:731–739. doi: 10.1093/gerona/glp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral Blood Fibrocytes: Differentiation Pathway and Migration to Wound Sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 147.Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep. 2006;8:145–150. doi: 10.1007/s11926-006-0055-x. [DOI] [PubMed] [Google Scholar]
- 148.Masszi A, Fan L, Rosivall L, McCulloch CA, Rotstein OD, Mucsi I, Kapus A. Integrity of cell-cell contacts is a critical regulator of TGF-beta 1-induced epithelial-to-myofibroblast transition: role for beta-catenin. Am J Pathol. 2004;165:1955–1967. doi: 10.1016/s0002-9440(10)63247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. Embo J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005;179:11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]
- 151.Han G, Lu SL, Li AG, He W, Corless CL, Kulesz-Martin M, Wang XJ. Distinct mechanisms of TGF-beta1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest. 2005;115:1714–1723. doi: 10.1172/JCI24399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ward C, Forrest IA, Murphy DM, Johnson GE, Robertson H, Cawston TE, Fisher AJ, Dark JH, Lordan JL, Kirby JA, Corris PA. Phenotype of airway epithelial cells suggests epithelial to mesenchymal cell transition in clinically stable lung transplant recipients. Thorax. 2005;60:865–871. doi: 10.1136/thx.2005.043026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Slattery C, Campbell E, McMorrow T, Ryan MP. Cyclosporine A-induced renal fibrosis: a role for epithelial-mesenchymal transition. Am J Pathol. 2005;167:395–407. doi: 10.1016/S0002-9440(10)62984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Saika S, Kono-Saika S, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Flanders KC, Yoo J, Anzano M, Liu CY, Kao WW, Roberts AB. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am J Pathol. 2004;164:651–663. doi: 10.1016/S0002-9440(10)63153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wu Z, Yang L, Cai L, Zhang M, Cheng X, Yang X, Xu J. Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha-smooth muscle actin-Cre transgenic mouse. Respir Res. 2007;8:1. doi: 10.1186/1465-9921-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 162.Sumioka T, Ikeda K, Okada Y, Yamanaka O, Kitano A, Saika S. Inhibitory effect of blocking TGF-beta/Smad signal on injury-induced fibrosis of corneal endothelium. Mol Vis. 2008;14:2272–2281. [PMC free article] [PubMed] [Google Scholar]
- 163.Yao HW, Xie QM, Chen JQ, Deng YM, Tang HF. TGF-beta1 induces alveolar epithelial to mesenchymal transition in vitro. Life Sci. 2004;76:29–37. doi: 10.1016/j.lfs.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 164.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vyas-Read S, Shaul PW, Yuhanna IS, Willis BC. Nitric oxide attenuates epithelial-mesenchymal transition in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L212–221. doi: 10.1152/ajplung.00475.2006. [DOI] [PubMed] [Google Scholar]
- 166.Jain R, Shaul PW, Borok Z, Willis BC. Endothelin-1 induces alveolar epithelial-mesenchymal transition through endothelin type A receptor-mediated production of TGF-beta1. Am J Respir Cell Mol Biol. 2007;37:38–47. doi: 10.1165/rcmb.2006-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kitao A, Sato Y, Sawada-Kitamura S, Harada K, Sasaki M, Morikawa H, Shiomi S, Honda M, Matsui O, Nakanuma Y. Endothelial to Mesenchymal Transition via Transforming Growth Factor-{beta}1/Smad Activation Is Associated with Portal Venous Stenosis in Idiopathic Portal Hypertension. Am J Pathol. 2009;175:616–626. doi: 10.2353/ajpath.2009.081061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang A, Jia Z, Guo X, Yang T. Aldosterone induces epithelial-mesenchymal transition via ROS of mitochondrial origin. Am J Physiol Renal Physiol. 2007;293:F723–731. doi: 10.1152/ajprenal.00480.2006. [DOI] [PubMed] [Google Scholar]
- 169.Zeng R, Yao Y, Han M, Zhao X, Liu XC, Wei J, Luo Y, Zhang J, Zhou J, Wang S, Ma D, Xu G. Biliverdin reductase mediates hypoxia-induced EMT via PI3-kinase and Akt. J Am Soc Nephrol. 2008;19:380–387. doi: 10.1681/ASN.2006111194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667–675. doi: 10.1681/ASN.2004050425. [DOI] [PubMed] [Google Scholar]
- 171.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, Dockrell ME. The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J. 2006;393:601–607. doi: 10.1042/BJ20051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yang J, Dai C, Liu Y. A novel mechanism by which hepatocyte growth factor blocks tubular epithelial to mesenchymal transition. J Am Soc Nephrol. 2005;16:68–78. doi: 10.1681/ASN.2003090795. [DOI] [PubMed] [Google Scholar]
- 174.Ivanova L, Butt MJ, Matsell DG. Mesenchymal transition in kidney collecting duct epithelial cells. Am J Physiol Renal Physiol. 2008;294:F1238–1248. doi: 10.1152/ajprenal.00326.2007. [DOI] [PubMed] [Google Scholar]
- 175.Grande M, Franzen A, Karlsson JO, Ericson LE, Heldin NE, Nilsson M. Transforming growth factor-beta and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J Cell Sci. 2002;115:4227–4236. doi: 10.1242/jcs.00091. [DOI] [PubMed] [Google Scholar]
- 176.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta- mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 177.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002;115:3193–3206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- 178.Cho HJ, Baek KE, Saika S, Jeong MJ, Yoo J. Snail is required for transforming growth factor-beta-induced epithelial-mesenchymal transition by activating PI3 kinase/Akt signal pathway. Biochem Biophys Res Commun. 2007;353:337–343. doi: 10.1016/j.bbrc.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 179.Yao K, Ye PP, Tan J, Tang XJ, Shen Tu XC. Involvement of PI3K/Akt pathway in TGF-beta2-mediated epithelial mesenchymal transition in human lens epithelial cells. Ophthalmic Res. 2008;40:69–76. doi: 10.1159/000113884. [DOI] [PubMed] [Google Scholar]
- 180.Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: A link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 181.Canbay A, Feldstein A, Baskin-Bey E, Bronk SF, Gores GJ. The Caspase Inhibitor IDN-6556 Attenuates Hepatic Injury and Fibrosis in the Bile Duct Ligated Mouse. J Pharmacol Exp Ther. 2004;308:1191–1196. doi: 10.1124/jpet.103.060129. [DOI] [PubMed] [Google Scholar]
- 182.Kasper M, Seidel D, Knels L, Morishima N, Neisser A, Bramke S, Koslowski R. Early signs of lung fibrosis after in vitro treatment of rat lung slices with CdCl2 and TGF-β1. Histochemistry and Cell Biology. 2004;121:131–140. doi: 10.1007/s00418-003-0612-6. [DOI] [PubMed] [Google Scholar]
- 183.Herrera B, Alvarez AM, Sanchez A, Fernandez M, Roncero C, Benito M, Fabregat I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. Faseb J. 2001;15:741–751. doi: 10.1096/fj.00-0267com. [DOI] [PubMed] [Google Scholar]
- 184.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. Faseb J. 2005;19:854–856. doi: 10.1096/fj.04-2882fje. [DOI] [PubMed] [Google Scholar]
- 185.Yao K, Tan J, Gu W-z, Ye P-P, Wang K-j. Reactive oxygen species mediates the apoptosis induced by transforming growth factor [beta]2 in human lens epithelial cells. Biochemical and Biophysical Research Communications. 2007;354:278–283. doi: 10.1016/j.bbrc.2006.12.198. [DOI] [PubMed] [Google Scholar]
- 186.Nadja M Meindl-Beinker SD. Transforming growth factor-β and hepatocyte transdifferentiation in liver fibrogenesis. Journal of Gastroenterology and Hepatology. 2008;23:S122–S127. doi: 10.1111/j.1440-1746.2007.05297.x. [DOI] [PubMed] [Google Scholar]
- 187.Garcia-Trevijano ER, Iraburu MJ, Fontana L, Dominguez-Rosales JA, Auster A, Covarrubias-Pinedo A, Rojkind M. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:960–970. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- 188.Cao Q, Mak KM, Lieber CS. DLPC decreases TGF-beta 1-induced collagen mRNA by inhibiting p38 MAPK in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1051–1061. doi: 10.1152/ajpgi.00128.2002. [DOI] [PubMed] [Google Scholar]
- 189.Gooch JL, Gorin Y, Zhang BX, Abboud HE. Involvement of calcineurin in transforming growth factor-beta-mediated regulation of extracellular matrix accumulation. J Biol Chem. 2004;279:15561–15570. doi: 10.1074/jbc.M308759200. [DOI] [PubMed] [Google Scholar]
- 190.Jiang Z, Seo JY, Ha H, Lee EA, Kim YS, Han DC, Uh ST, Park CS, Lee HB. Reactive oxygen species mediate TGF-beta1-induced plasminogen activator inhibitor-1 upregulation in mesangial cells. Biochem Biophys Res Commun. 2003;309:961–966. doi: 10.1016/j.bbrc.2003.08.102. [DOI] [PubMed] [Google Scholar]
- 191.Li WQ, Qureshi HY, Liacini A, Dehnade F, Zafarullah M. Transforming growth factor [beta]1 induction of Tissue Inhibitor of MetalloProteinases 3 in articular chondrocytes is mediated by reactive oxygen species. Free Radical Biology and Medicine. 2004;37:196–207. doi: 10.1016/j.freeradbiomed.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 192.Vayalil PK, Olman M, Murphy-Ullrich JE, Postlethwait EM, Liu RM. Glutathione restores collagen degradation in TGF-beta-treated fibroblasts by blocking plasminogen activator inhibitor-1 expression and activating plasminogen. Am J Physiol Lung Cell Mol Physiol. 2005;289:L937–945. doi: 10.1152/ajplung.00150.2005. [DOI] [PubMed] [Google Scholar]
- 193.Vayalil PK, Iles KE, Choi J, Yi A-K, Postlethwait EM, Liu R-M. Glutathione suppresses TGF-{beta}-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am J Physiol Lung Cell Mol Physiol. 2007 doi: 10.1152/ajplung.00128.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Laurent GJ. Dynamic state of collagen: pathways of collagen degradation in vivo and their possible role in regulation of collagen mass. Am J Physiol. 1987;252:C1–9. doi: 10.1152/ajpcell.1987.252.1.C1. [DOI] [PubMed] [Google Scholar]
- 195.Arthur MJ. Degradation of matrix proteins in liver fibrosis. Pathol Res Pract. 1994;190:825–833. doi: 10.1016/S0344-0338(11)80985-4. [DOI] [PubMed] [Google Scholar]
- 196.Davidson JM. Biochemistry and turnover of lung interstitium. Eur Respir J. 1990;3:1048–1063. [PubMed] [Google Scholar]
- 197.Liu RM. Oxidative stress, plasminogen activator inhibitor 1, and lung fibrosis. Antioxid Redox Signal. 2008;10:303–319. doi: 10.1089/ars.2007.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kutz SM, Hordines J, McKeown-Longo PJ, Higgins PJ. TGF-beta1-induced PAI-1 gene expression requires MEK activity and cell-to-substrate adhesion. J Cell Sci. 2001;114:3905–3914. doi: 10.1242/jcs.114.21.3905. [DOI] [PubMed] [Google Scholar]
- 199.Wang L, Ma R, Flavell RA, Choi ME. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38alpha and p38delta MAPK isoforms by TGF-beta 1 in murine mesangial cells. J Biol Chem. 2002;277:47257–47262. doi: 10.1074/jbc.M208573200. [DOI] [PubMed] [Google Scholar]
- 200.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 201.Woodward RN, Finn AV, Dichek DA. Identification of intracellular pathways through which TGF-[beta]1 upregulates PAI-1 expression in endothelial cells. Atherosclerosis. 2006;186:92–100. doi: 10.1016/j.atherosclerosis.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 202.Guo B, Inoki K, Isono M, Mori H, Kanasaki K, Sugimoto T, Akiba S, Sato T, Yang B, Kikkawa R, Kashiwagi A, Haneda M, Koya D. MAPK//AP-1-dependent regulation of PAI-1 gene expression by TGF-[beta] in rat mesangial cells. Kidney International. 2005;68:972–984. doi: 10.1111/j.1523-1755.2005.00491.x. [DOI] [PubMed] [Google Scholar]
- 203.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 204.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 205.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 206.Denu JM, Tanner KG. Redox regulation of protein tyrosine phosphatases by hydrogen peroxide: detecting sulfenic acid intermediates and examining reversible inactivation. Methods Enzymol. 2002;348:297–305. doi: 10.1016/s0076-6879(02)48648-x. [DOI] [PubMed] [Google Scholar]
- 207.Dixon DP, Fordham-Skelton AP, Edwards R. Redox regulation of a soybean tyrosine-specific protein phosphatase. Biochemistry. 2005;44:7696–7703. doi: 10.1021/bi047324a. [DOI] [PubMed] [Google Scholar]
- 208.Rao RK, Clayton LW. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem Biophys Res Commun. 2002;293:610–616. doi: 10.1016/S0006-291X(02)00268-1. [DOI] [PubMed] [Google Scholar]
- 209.Foley TD, Armstrong JJ, Kupchak BR. Identification and H2O2 sensitivity of the major constitutive MAPK phosphatase from rat brain. Biochem Biophys Res Commun. 2004;315:568–574. doi: 10.1016/j.bbrc.2004.01.096. [DOI] [PubMed] [Google Scholar]
- 210.Foley TD, Kintner ME. Brain PP2A is modified by thiol-disulfide exchange and intermolecular disulfide formation. Biochem Biophys Res Commun. 2005;330:1224–1229. doi: 10.1016/j.bbrc.2005.03.108. [DOI] [PubMed] [Google Scholar]
- 211.Levinthal DJ, Defranco DB. Reversible oxidation of ERK-directed protein phosphatases drives oxidative toxicity in neurons. J Biol Chem. 2005;280:5875–5883. doi: 10.1074/jbc.M410771200. [DOI] [PubMed] [Google Scholar]
- 212.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 213.Foley T, Petro L, Stredny C, Coppa T. Oxidative Inhibition of Protein Phosphatase 2A Activity: Role of Catalytic Subunit Disulfides. Neurochemical Research. 2007 doi: 10.1007/s11064-007-9394-x. [DOI] [PubMed] [Google Scholar]
- 214.Fontenay M, Bryckaert M, Tobelem G. Transforming growth factor-beta 1 inhibitory effect of platelet-derived growth factor-induced signal transduction on human bone marrow fibroblasts: possible involvement of protein phosphatases. J Cell Physiol. 1992;152:507–519. doi: 10.1002/jcp.1041520310. [DOI] [PubMed] [Google Scholar]
- 215.Mire-Sluis AR, Page L, Wadhwa M, Thorpe R. Transforming growth factor-beta 1 blocks interleukin 4 induced cell proliferation by inhibiting a protein tyrosine phosphatase essential for signal transduction. Cytokine. 1994;6:389–398. doi: 10.1016/1043-4666(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 216.Petritsch C, Beug H, Balmain A, Oft M. TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev. 2000;14:3093–3101. doi: 10.1101/gad.854200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Young MR. Tumor-derived prostaglandin E2 and transforming growth factor-beta stimulate endothelial cell motility through inhibition of protein phosphatase-2A and involvement of PTEN and phosphatidylinositide 3-kinase. Angiogenesis. 2004;7:123–131. doi: 10.1007/s10456-004-1027-2. [DOI] [PubMed] [Google Scholar]
- 218.Lires-Deán M, Caramés B, Cillero-Pastor B, Galdo F, López-Armada MJ, Blanco FJ. Anti-apoptotic effect of transforming growth factor-[beta]1 on human articular chondrocytes: role of protein phosphatase 2A. Osteoarthritis and Cartilage. 2008;16:1370–1378. doi: 10.1016/j.joca.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 219.Kim SI, Kwak JH, Wang L, Choi ME. Protein Phosphatase 2A Is a Negative Regulator of Transforming Growth Factor-{beta}1-induced TAK1 Activation in Mesangial Cells. J Biol Chem. 2008;283:10753–10763. doi: 10.1074/jbc.M801263200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ebert MPA, Fei G, Schandl L, Mawrin C, Dietzmann K, Herrera P, Friess H, Gress TM, Malfertheiner P. Reduced PTEN expression in the pancreas overexpressing transforming growth factor-beta 1. Br J Cancer. 2002;86:257–262. doi: 10.1038/sj.bjc.6600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chow JYC, Dong H, Quach KT, Van Nguyen PN, Chen K, Carethers JM. TGF-{beta} mediates PTEN suppression and cell motility through calcium-dependent PKC-{alpha} activation in pancreatic cancer cells. Am J Physiol Gastrointest Liver Physiol. 2008;294:G899–905. doi: 10.1152/ajpgi.00411.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 223.Rocic P, Lucchesi PA. NAD(P)H Oxidases and TGF-{beta}-Induced Cardiac Fibroblast Differentiation: Nox-4 Gets Smad. Circ Res. 2005;97:850–852. doi: 10.1161/01.RES.0000190403.87462.bf. [DOI] [PubMed] [Google Scholar]
- 224.Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M, Fujisawa J, Okazaki K. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23:7416–7429. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- 226.Kamaraju AK, Roberts AB. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J Biol Chem. 2005;280:1024–1036. doi: 10.1074/jbc.M403960200. [DOI] [PubMed] [Google Scholar]
- 227.Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742–5750. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- 228.Yingling JM, Datto MB, Wong C, Frederick JP, Liberati NT, Wang XF. Tumor suppressor Smad4 is a transforming growth factor beta-inducible DNA binding protein. Mol Cell Biol. 1997;17:7019–7028. doi: 10.1128/mcb.17.12.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sano Y, Harada J, Tashiro S, Gotoh-Mandeville R, Maekawa T, Ishii S. ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-beta signaling. J Biol Chem. 1999;274:8949–8957. doi: 10.1074/jbc.274.13.8949. [DOI] [PubMed] [Google Scholar]
- 230.Hua X, Liu X, Ansari DO, Lodish HF. Synergistic cooperation of TFE3 and smad proteins in TGF-beta-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. 1998;12:3084–3095. doi: 10.1101/gad.12.19.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liu P, Zhang C, Feng JB, Zhao YX, Wang XP, Yang JM, Zhang MX, Wang XL, Zhang Y. Cross Talk Among Smad, MAPK, and Integrin Signaling Pathways Enhances Adventitial Fibroblast Functions Activated by Transforming Growth Factor-{beta}1 and Inhibited by Gax. Arterioscler Thromb Vasc Biol. 2008;28:725–731. doi: 10.1161/ATVBAHA.107.159889. [DOI] [PubMed] [Google Scholar]
- 232.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 233.Basu MK, Koonin EV. Evolution of eukaryotic cysteine sulfinic acid reductase, sulfiredoxin (Srx), from bacterial chromosome partitioning protein ParB. Cell Cycle. 2005;4:947–952. doi: 10.4161/cc.4.7.1786. [DOI] [PubMed] [Google Scholar]
- 234.Rhee SG, Jeong W, Chang TS, Woo HA. Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance. Kidney Int Suppl. 2007:S3–8. doi: 10.1038/sj.ki.5002380. [DOI] [PubMed] [Google Scholar]
- 235.Jonsson TJ, Murray MS, Johnson LC, Lowther WT. Reduction of cysteine sulfinic acid in peroxiredoxin by sulfiredoxin proceeds directly through a sulfinic phosphoryl ester intermediate. J Biol Chem. 2008;283:23846–23851. doi: 10.1074/jbc.M803244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Noh YH, Baek JY, Jeong W, Rhee SG, Chang T-S. Sulfiredoxin Translocation into Mitochondria Plays a Crucial Role in Reducing Hyperoxidized Peroxiredoxin III. J Biol Chem. 2009;284:8470–8477. doi: 10.1074/jbc.M808981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s- glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 238.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Current Opinion in Pharmacology. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 239.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular Mechanisms and Clinical Implications of Reversible Protein S-Glutathionylation. Antioxidants & Redox Signaling. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem. 2009 doi: 10.1074/jbc.M109.021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein- tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 243.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 244.Humphries KM, Juliano C, Taylor SS. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem. 2002;277:43505–43511. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- 245.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 246.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rahman I, Biswas SK, Jimenez LA, Torres M, Forman HJ. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid Redox Signal. 2005;7:42–59. doi: 10.1089/ars.2005.7.42. [DOI] [PubMed] [Google Scholar]
- 248.Vogelmann R, Nguyen-Tat MD, Giehl K, Adler G, Wedlich D, Menke A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J Cell Sci. 2005;118:4901–4912. doi: 10.1242/jcs.02594. [DOI] [PubMed] [Google Scholar]
- 249.Okamoto T, Akaike T, Sawa T, Miyamoto Y, van der Vliet A, Maeda H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J Biol Chem. 2001;276:29596–29602. doi: 10.1074/jbc.M102417200. [DOI] [PubMed] [Google Scholar]
- 250.Davis DA, Dorsey K, Wingfield PT, Stahl SJ, Kaufman J, Fales HM, Levine RL. Regulation of HIV-1 protease activity through cysteine modification. Biochemistry. 1996;35:2482–2488. doi: 10.1021/bi951525k. [DOI] [PubMed] [Google Scholar]
- 251.Davis DA, Newcomb FM, Starke DW, Ott DE, Mieyal JJ, Yarchoan R. Thioltransferase (glutaredoxin) is detected within HIV-1 and can regulate the activity of glutathionylated HIV-1 protease in vitro. J Biol Chem. 1997;272:25935–25940. doi: 10.1074/jbc.272.41.25935. [DOI] [PubMed] [Google Scholar]
- 252.Dafre AL, Sies H, Akerboom T. Protein S-thiolation and regulation of microsomal glutathione transferase activity by the glutathione redox couple. Arch Biochem Biophys. 1996;332:288–294. doi: 10.1006/abbi.1996.0344. [DOI] [PubMed] [Google Scholar]
- 253.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 254.Thomas JA, Poland B, Honzatko R. Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch Biochem Biophys. 1995;319:1–9. doi: 10.1006/abbi.1995.1261. [DOI] [PubMed] [Google Scholar]
- 255.Cotgreave IA, Gerdes RG. Recent trends in glutathione biochemistry--glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem Biophys Res Commun. 1998;242:1–9. doi: 10.1006/bbrc.1997.7812. [DOI] [PubMed] [Google Scholar]
- 256.Ghezzi P. Regulation of protein function by glutathionylation. Free Radic Res. 2005;39:573–580. doi: 10.1080/10715760500072172. [DOI] [PubMed] [Google Scholar]
- 257.Biswas S, Chida AS, Rahman I. Redox modifications of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol. 2006;71:551–564. doi: 10.1016/j.bcp.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 258.Meister A. On the cycles of glutathione metabolism and transport. Curr Top Cell Regul. 1981;18:21–58. doi: 10.1016/b978-0-12-152818-8.50009-8. [DOI] [PubMed] [Google Scholar]
- 259.Roum JH, Buhl R, McElvany NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. Journal of Applied Physiology: Respiration, Environmental and Exercise Physiology. 1993;75:2419–2424. doi: 10.1152/jappl.1993.75.6.2419. [DOI] [PubMed] [Google Scholar]
- 260.Tirouvanziam R, Conrad CK, Bottiglieri T, Herzenberg LA, Moss RB. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc Natl Acad Sci U S A. 2006;103:4628–4633. doi: 10.1073/pnas.0511304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Rahman I, Skwarska E, Henry M, Davis M, O’Connor CM, FitzGerald MX, Greening A, MacNee W. Systemic and pulmonary oxidative stress in idiopathic pulmonary fibrosis. Free Radic Biol Med. 1999;27:60–68. doi: 10.1016/s0891-5849(99)00035-0. [DOI] [PubMed] [Google Scholar]
- 262.Pacht ER, Timerman AP, Lykens MG, Merola AJ. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. 1991;100:1397–1403. doi: 10.1378/chest.100.5.1397. [DOI] [PubMed] [Google Scholar]
- 263.Bunnell E, Pacht ER. Oxidized glutathione is increased in the alveolar fluid of patients with the adult respiratory distress syndrome. American Review of Respiratory Disease. 1993;148:1174–1178. doi: 10.1164/ajrccm/148.5.1174. [DOI] [PubMed] [Google Scholar]
- 264.Laurent T, Markert M, Feihl F, Schaller MD, Perret C. Oxidant-antioxidant balance in granulocytes during ARDS. Effect of N-acetylcysteine. Chest. 1996;109:163–166. doi: 10.1378/chest.109.1.163. [DOI] [PubMed] [Google Scholar]
- 265.Bernard GR, Wheeler AP, Arons MM, Morris PE, Paz HL, Russell JA, Wright PE. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest. 1997;112:164–172. doi: 10.1378/chest.112.1.164. [DOI] [PubMed] [Google Scholar]
- 266.Soltan-Sharifi MS, Mojtahedzadeh M, Najafi A, Reza Khajavi M, Reza Rouini M, Moradi M, Mohammadirad A, Abdollahi M. Improvement by N-acetylcysteine of acute respiratory distress syndrome through increasing intracellular glutathione, and extracellular thiol molecules and anti-oxidant power: evidence for underlying toxicological mechanisms. Hum Exp Toxicol. 2007;26:697–703. doi: 10.1177/0960327107083452. [DOI] [PubMed] [Google Scholar]
- 267.Cantin AM, Hubbard RC, Crystal RG. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. American Review of Respiratory Disease. 1989;139:370–372. doi: 10.1164/ajrccm/139.2.370. [DOI] [PubMed] [Google Scholar]
- 268.Borok Z, Grimes GJ, Buhl R, Bokser A, Hubbard RC, Holroyd K, Roum JH, Czerski DB, Cantin AM, Crystal RG. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet. 1991;338:215–216. doi: 10.1016/0140-6736(91)90350-x. [DOI] [PubMed] [Google Scholar]
- 269.Meyer A, Buhl R, Magnussen H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur Respir J. 1994;7:431–436. doi: 10.1183/09031936.94.07030431. [DOI] [PubMed] [Google Scholar]
- 270.Behr J, Maier K, Degenkolb B, Krombach F, Vogelmeier C. Antioxidative and clinical effects of high-dose N-acetylcysteine in fibrosing alveolitis. Adjunctive therapy to maintenance immunosuppression. Am J Respir Crit Care Med. 1997;156:1897–1901. doi: 10.1164/ajrccm.156.6.9706065. [DOI] [PubMed] [Google Scholar]
- 271.Behr J, Degenkolb B, Krombach F, Vogelmeier C. Intracellular glutathione and bronchoalveolar cells in fibrosing alveolitis: effects of N-acetylcysteine. Eur Respir J. 2002;19:906–911. doi: 10.1183/09031936.02.00204902. [DOI] [PubMed] [Google Scholar]
- 272.Montaldo C, Cannas E, Ledda M, Rosetti L, Congiu L, Atzori L. Bronchoalveolar glutathione and nitrite/nitrate in idiopathic pulmonary fibrosis and sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19:54–58. [PubMed] [Google Scholar]
- 273.Beeh KM, Beier J, Haas IC, Kornmann O, Micke P, Buhl R. Glutathione deficiency of the lower respiratory tract in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2002;19:1119–1123. doi: 10.1183/09031936.02.00262402. [DOI] [PubMed] [Google Scholar]
- 274.Boots AW, Drent M, Swennen ELR, Moonen HJJ, Bast A, Haenen GRMM. Antioxidant status associated with inflammation in sarcoidosis: A potential role for antioxidants. Respiratory Medicine. 2009;103:364–372. doi: 10.1016/j.rmed.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 275.Brown DM, Beswick PH, Bell KS, Donaldson K. Depletion of glutathione and ascorbate in lung lining fluid by respirable fibres. Ann Occup Hyg. 2000;44:101–108. [PubMed] [Google Scholar]
- 276.Orman A, Kahraman A, Çakar H, Ellidokuz H, Serteser M. Plasma malondialdehyde and erythrocyte glutathione levels in workers with cement dust-exposure silicosis. Toxicology. 2005;207:15–20. doi: 10.1016/j.tox.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 277.Bianchi G, Bugianesi E, Ronchi M, Fabbri A, Zoli M, Marchesini G. Glutathione kinetics in normal man and in patients with liver cirrhosis. J Hepatol. 1997;26:606–613. doi: 10.1016/s0168-8278(97)80426-6. [DOI] [PubMed] [Google Scholar]
- 278.Aboutwerat A, Pemberton PW, Smith A, Burrows PC, McMahon RFT, Jain SK, Warnes TW. Oxidant stress is a significant feature of primary biliary cirrhosis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2003;1637:142–150. doi: 10.1016/s0925-4439(02)00225-9. [DOI] [PubMed] [Google Scholar]
- 279.Loguercio C, Federico A. Oxidative stress in viral and alcoholic hepatitis. Free Radical Biology and Medicine. 2003;34:1–10. doi: 10.1016/s0891-5849(02)01167-x. [DOI] [PubMed] [Google Scholar]
- 280.Czeczot H, Scibior D, Skrzycki M, Podsiad M. Glutathione and GSH-dependent enzymes in patients with liver cirrhosis and hepatocellular carcinoma. Acta Biochim Pol. 2006;53:237–242. [PubMed] [Google Scholar]
- 281.Priyanka Bandara JG, McCaughan Geoffrey, Naidoo Daya, Lux Ora, Salonikas Chris, Kench James, Byth Karen, Farrell Geoffrey C. Antioxidant levels in peripheral blood, disease activity and fibrotic stage in chronic hepatitis C. Liver International. 2005;25:518–526. doi: 10.1111/j.1478-3231.2005.01049.x. [DOI] [PubMed] [Google Scholar]
- 282.Marotta F, Yoshida C, Barreto R, Naito Y, Packer L. Oxidative-inflammatory damage in cirrhosis: effect of vitamin E and a fermented papaya preparation. J Gastroenterol Hepatol. 2007;22:697–703. doi: 10.1111/j.1440-1746.2007.04937.x. [DOI] [PubMed] [Google Scholar]
- 283.Osman H, Gabr O, Lotfy S, Gabr S. Serum levels of bcl-2 and cellular oxidative stress in patients with viral hepatitis. Indian Journal of Medical Microbiology. 2007;25:323–329. doi: 10.4103/0255-0857.37333. [DOI] [PubMed] [Google Scholar]
- 284.Joshi PC, Guidot DM. The alcoholic lung: epidemiology, pathophysiology, and potential therapies. Am J Physiol Lung Cell Mol Physiol. 2007;292:L813–823. doi: 10.1152/ajplung.00348.2006. [DOI] [PubMed] [Google Scholar]
- 285.Ma JY, Barger MW, Hubbs AF, Castranova V, Weber SL, Ma JK. Use of tetrandrine to differentiate between mechanisms involved in silica-versus bleomycin-induced fibrosis. J Toxicol Environ Health A. 1999;57:247–266. doi: 10.1080/009841099157692. [DOI] [PubMed] [Google Scholar]
- 286.Candan F, Alagozlu H. Captopril inhibits the pulmonary toxicity of paraquat in rats. Hum Exp Toxicol. 2001;20:637–641. doi: 10.1191/096032701718890540. [DOI] [PubMed] [Google Scholar]
- 287.Arslan SO, Zerin M, Vural H, Coskun A. The effect of melatonin on bleomycin- induced pulmonary fibrosis in rats. J Pineal Res. 2002;32:21–25. doi: 10.1034/j.1600-079x.2002.10796.x. [DOI] [PubMed] [Google Scholar]
- 288.Nieto N, Cederbaum AI. S-Adenosylmethionine Blocks Collagen I Production by Preventing Transforming Growth Factor-{beta} Induction of the COL1A2 Promoter. J Biol Chem. 2005;280:30963–30974. doi: 10.1074/jbc.M503569200. [DOI] [PubMed] [Google Scholar]
- 289.Atzori L, Chua F, Dunsmore SE, Willis D, Barbarisi M, McAnulty RJ, Laurent GJ. Attenuation of bleomycin induced pulmonary fibrosis in mice using the heme oxygenase inhibitor Zn-deuteroporphyrin IX-2,4-bisethylene glycol. Thorax. 2004;59:217–223. doi: 10.1136/thx.2003.008979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.El-Medany A, Hagar HH, Moursi M, At Muhammed R, El-Rakhawy FI, El-Medany G. Attenuation of bleomycin-induced lung fibrosis in rats by mesna. European Journal of Pharmacology. 2005;509:61–70. doi: 10.1016/j.ejphar.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 291.Boyacı H, Maral H, Turan G, Başyiğit İ, Dillioğlugil M, Yıldız F, Tugay M, Pala A, Erçin C. Effects of erdosteine on bleomycin-induced lung fibrosis in rats. Molecular and Cellular Biochemistry. 2006;281:129–137. doi: 10.1007/s11010-006-0640-3. [DOI] [PubMed] [Google Scholar]
- 292.Yeter Deger FY, Ertekin Ali, Mert Nihat, Dede Semiha, Mert Handan. Protective effect of alpha-tocopherol on oxidative stress in experimental pulmonary fibrosis in rats. Cell Biochemistry and Function. 2007;25:633–637. doi: 10.1002/cbf.1362. [DOI] [PubMed] [Google Scholar]
- 293.Arafa HMM, Abdel-Wahab MH, El-Shafeey MF, Badary OA, Hamada FMA. Anti-fibrotic effect of meloxicam in a murine lung fibrosis model. European Journal of Pharmacology. 2007;564:181–189. doi: 10.1016/j.ejphar.2007.02.065. [DOI] [PubMed] [Google Scholar]
- 294.Hettiarachchi J, Fernando SS. Pulmonary fibrosis following paraquat poisoning. Ceylon Med J. 1988;33:141–142. [PubMed] [Google Scholar]
- 295.Ghazi-Khansari M, Mohammadi-Karakani A, Sotoudeh M, Mokhtary P, Pour-Esmaeil E, Maghsoud S. Antifibrotic effect of captopril and enalapril on paraquat-induced lung fibrosis in rats. J Appl Toxicol. 2007;27:342–349. doi: 10.1002/jat.1212. [DOI] [PubMed] [Google Scholar]
- 296.Abidi P, Afaq F, Arif JM, Lohani M, Rahman Q. Chrysotile-mediated imbalance in the glutathione redox system in the development of pulmonary injury. Toxicol Lett. 1999;106:31–39. doi: 10.1016/s0378-4274(99)00013-2. [DOI] [PubMed] [Google Scholar]
- 297.Lukivskaya O, Zavodnik L, Knas M, Buko V. Antioxidant mechanism of hepatoprotection by ursodeoxycholic acid in experimental alcoholic steatohepatitis. Adv Med Sci. 2006;51:54–59. [PubMed] [Google Scholar]
- 298.Fernandez-Checa JC, Colell A, Garcia-Ruiz C. S-Adenosyl-L-methionine and mitochondrial reduced glutathione depletion in alcoholic liver disease. Alcohol. 2002;27:179–183. doi: 10.1016/s0741-8329(02)00229-x. [DOI] [PubMed] [Google Scholar]
- 299.Purucker E, Wernze W, Krandik G. Glutathione in plasma, liver, and kidney in the development of CCl4-induced cirrhosis of the rat. Res Exp Med (Berl) 1995;195:193–199. doi: 10.1007/BF02576788. [DOI] [PubMed] [Google Scholar]
- 300.Cabre M, Camps J, Paternain JL, Ferre N, Joven J. Time-course of changes in hepatic lipid peroxidation and glutathione metabolism in rats with carbon tetrachloride-induced cirrhosis. Clin Exp Pharmacol Physiol. 2000;27:694–699. doi: 10.1046/j.1440-1681.2000.03322.x. [DOI] [PubMed] [Google Scholar]
- 301.Kondou H, Mushiake S, Etani Y, Miyoshi Y, Michigami T, Ozono K. A blocking peptide for transforming growth factor-beta1 activation prevents hepatic fibrosis in vivo. J Hepatol. 2003;39:742–748. doi: 10.1016/s0168-8278(03)00377-5. [DOI] [PubMed] [Google Scholar]
- 302.Tahan V, Ozaras R, Canbakan B, Uzun H, Aydin S, Yildirim B, Aytekin H, Ozbay G, Mert A, Senturk H. Melatonin reduces dimethylnitrosamine-induced liver fibrosis in rats. J Pineal Res. 2004;37:78–84. doi: 10.1111/j.1600-079X.2004.00137.x. [DOI] [PubMed] [Google Scholar]
- 303.Vendemiale G, Grattagliano I, Altomare E, Serviddio G, Portincasa P, Prigigallo F, Palasciano G. Mitochondrial oxidative damage and myocardial fibrosis in rats chronically intoxicated with moderate doses of ethanol. Toxicology Letters. 2001;123:209–216. doi: 10.1016/s0378-4274(01)00401-5. [DOI] [PubMed] [Google Scholar]
- 304.White AC, Das SK, Fanburg BL. Reduction of glutathione is associated with growth restriction and enlargement of bovine pulmonary artery endothelial cells produced by transforming growth factor-beta 1. Am J Respir Cell Mol Biol. 1992;6:364–368. doi: 10.1165/ajrcmb/6.4.364. [DOI] [PubMed] [Google Scholar]
- 305.Sanchez A, Alvarez AM, Benito M, Fabregat I. Apoptosis induced by transforming growth factor-beta in fetal hepatocyte primary cultures: involvement of reactive oxygen intermediates. J Biol Chem. 1996;271:7416–7422. doi: 10.1074/jbc.271.13.7416. [DOI] [PubMed] [Google Scholar]
- 306.Sanchez A, Alvarez AM, Benito M, Fabregat I. Cycloheximide prevents apoptosis, reactive oxygen species production, and glutathione depletion induced by transforming growth factor beta in fetal rat hepatocytes in primary culture. Hepatology. 1997;26:935–943. doi: 10.1002/hep.510260420. [DOI] [PubMed] [Google Scholar]
- 307.Arsalane K, Dubois CM, Muanza T, Begin R, Boudreau F, Asselin C, Cantin AM. Transforming growth factor-beta1 is a potent inhibitor of glutathione synthesis in the lung epithelial cell line A549: transcriptional effect on the GSH rate-limiting enzyme gamma-glutamylcysteine synthetase. Am J Respir Cell Mol Biol. 1997;17:599–607. doi: 10.1165/ajrcmb.17.5.2833. [DOI] [PubMed] [Google Scholar]
- 308.Pittet JF, Griffiths MJ, Geiser T, Kaminski N, Dalton SL, Huang X, Brown LA, Gotwals PJ, Koteliansky VE, Matthay MA, Sheppard D. TGF-beta is a critical mediator of acute lung injury. J Clin Invest. 2001;107:1537–1544. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Jardine H, MacNee W, Donaldson K, Rahman I. Molecular mechanism of transforming growth factor (TGF)-beta1-induced glutathione depletion in alveolar epithelial cells. Involvement of AP- 1/ARE and Fra-1. J Biol Chem. 2002;277:21158–21166. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- 310.Franklin CC, Rosenfeld-Franklin ME, White C, Kavanagh TJ, Fausto N. TGFbeta1-induced suppression of glutathione antioxidant defenses in hepatocytes: caspase-dependent post-translational and caspase-independent transcriptional regulatory mechanisms. Faseb J. 2003;17:1535–1537. doi: 10.1096/fj.02-0867fje. [DOI] [PubMed] [Google Scholar]
- 311.Bakin AV, Stourman NV, Sekhar KR, Rinehart C, Yan X, Meredith MJ, Arteaga CL, Freeman ML. Smad3-ATF3 signaling mediates TGF-[beta] suppression of genes encoding Phase II detoxifying proteins. Free Radical Biology and Medicine. 2005;38:375–387. doi: 10.1016/j.freeradbiomed.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 312.Liu R-M, Liu Y, Forman HJ, Olman M, Tarpey MM. Glutathione regulates transforming growth factor-{beta}-stimulated collagen production in fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2004;286:L121–128. doi: 10.1152/ajplung.00231.2003. [DOI] [PubMed] [Google Scholar]
- 313.Tiitto LH, Peltoniemi MJ, Kaarteenaho-Wiik RL, Soini YM, Paakko PK, Sormunen RT, Kinnula VL. Cell-specific regulation of gamma-glutamylcysteine synthetase in human interstitial lung diseases. Hum Pathol. 2004;35:832–839. doi: 10.1016/j.humpath.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 314.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione Metabolism and Its Implications for Health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 315.Baumgardner JN, Shankar K, Hennings L, Albano E, Badger TM, Ronis MJJ. N-Acetylcysteine Attenuates Progression of Liver Pathology in a Rat Model of Nonalcoholic Steatohepatitis. J Nutr. 2008;138:1872–1879. doi: 10.1093/jn/138.10.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Yang YY, Lee KC, Huang YT, Wang YW, Hou MC, Lee FY, Lin HC, Lee SD. Effects of N-acetylcysteine administration in hepatic microcirculation of rats with biliary cirrhosis. J Hepatol. 2008;49:25–33. doi: 10.1016/j.jhep.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 317.Vendemiale G, Grattagliano I, Caruso ML, Serviddio G, Valentini AM, Pirrelli M, Altomare E. Increased oxidative stress in dimethylnitrosamine-induced liver fibrosis in the rat: effect of N-acetylcysteine and interferon-alpha. Toxicol Appl Pharmacol. 2001;175:130–139. doi: 10.1006/taap.2001.9234. [DOI] [PubMed] [Google Scholar]
- 318.Pereira-Filho G, Ferreira C, Schwengber A, Marroni C, Zettler C, Marroni N. Role of N-acetylcysteine on fibrosis and oxidative stress in cirrhotic rats. Arq Gastroenterol. 2008;45:156–162. doi: 10.1590/s0004-28032008000200013. [DOI] [PubMed] [Google Scholar]
- 319.Shahzeidi S, Sarnstrand B, Jeffery PK, McAnulty RJ, Laurent GJ. Oral N- acetylcysteine reduces bleomycin-induced collagen deposition in the lungs of mice. Eur Respir J. 1991;4:845–852. [PubMed] [Google Scholar]
- 320.Cortijo J, Cerda-Nicolas M, Serrano A, Bioque G, Estrela JM, Santangelo F, Esteras A, Llombart-Bosch A, Morcillo EJ. Attenuation by oral N-acetylcysteine of bleomycin-induced lung injury in rats. Eur Respir J. 2001;17:1228–1235. doi: 10.1183/09031936.01.00049701. [DOI] [PubMed] [Google Scholar]
- 321.Mata M, Ruiz A, Cerda M, Martinez-Losa M, Cortijo J, Santangelo F, Serrano-Mollar A, Llombart-Bosch A, Morcillo EJ. Oral N-acetylcysteine reduces bleomycin-induced lung damage and mucin Muc5ac expression in rats. Eur Respir J. 2003;22:900–905. doi: 10.1183/09031936.03.00018003. [DOI] [PubMed] [Google Scholar]
- 322.Hagiwara SI, Ishii Y, Kitamura S. Aerosolized administration of N-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am J Respir Crit Care Med. 2000;162:225–231. doi: 10.1164/ajrccm.162.1.9903129. [DOI] [PubMed] [Google Scholar]
- 323.Sime PJ, O’Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 324.Selman M, Thannickal VJ, Pardo A, Zisman DA, Martinez FJ, Lynch JP., 3rd Idiopathic pulmonary fibrosis: pathogenesis and therapeutic approaches. Drugs. 2004;64:405–430. doi: 10.2165/00003495-200464040-00005. [DOI] [PubMed] [Google Scholar]
- 325.Hunninghake GW. Antioxidant therapy for idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2285–2287. doi: 10.1056/NEJMe058210. [DOI] [PubMed] [Google Scholar]
- 326.King TE., Jr Clinical advances in the diagnosis and therapy of the interstitial lung diseases. Am J Respir Crit Care Med. 2005;172:268–279. doi: 10.1164/rccm.200503-483OE. [DOI] [PubMed] [Google Scholar]
- 327.Ziesche R, Hofbauer E, Wittmann K, Petkov V, Block LH. A preliminary study of long-term treatment with interferon gamma-1b and low-dose prednisolone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 1999;341:1264–1269. doi: 10.1056/NEJM199910213411703. [DOI] [PubMed] [Google Scholar]
- 328.Raghu G, Brown KK, Bradford WZ, Starko K, Noble PW, Schwartz DA, King TE., Jr A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350:125–133. doi: 10.1056/NEJMoa030511. [DOI] [PubMed] [Google Scholar]
- 329.Prasse A, Muller KM, Kurz C, Hamm H, Virchow JC., Jr Does interferon- gamma improve pulmonary function in idiopathic pulmonary fibrosis? Eur Respir J. 2003;22:906–911. doi: 10.1183/09031936.03.00091802. [DOI] [PubMed] [Google Scholar]
- 330.Selman M. A dark side of interferon-gamma in the treatment of idiopathic pulmonary fibrosis? Am J Respir Crit Care Med. 2003;167:945–946. doi: 10.1164/rccm.2301004. [DOI] [PubMed] [Google Scholar]
- 331.Tomioka H, Kuwata Y, Imanaka K, Hashimoto K, Ohnishi H, Tada K, Sakamoto H, Iwasaki H. A pilot study of aerosolized N-acetylcysteine for idiopathic pulmonary fibrosis. Respirology. 2005;10:449–455. doi: 10.1111/j.1440-1843.2005.00725.x. [DOI] [PubMed] [Google Scholar]
- 332.Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Capron F, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 333.Gao L, Broughman JR, Iwamoto T, Tomich JM, Venglarik CJ, Forman HJ. Synthetic chloride channel restores glutathione secretion in cystic fibrosis airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L24–30. doi: 10.1152/ajplung.2001.281.1.L24. [DOI] [PubMed] [Google Scholar]
- 334.Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, Cole SP, Bear CE. CFTR directly mediates nucleotide-regulated glutathione flux. Embo J. 2003;22:1981–1989. doi: 10.1093/emboj/cdg194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kariya C, Leitner H, Min E, van Heeckeren C, van Heeckeren A, Day BJ. A role for CFTR in the elevation of glutathione levels in the lung by oral glutathione administration. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1590–1597. doi: 10.1152/ajplung.00365.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Stafanger G, Garne S, Howitz P, Morkassel E, Koch C. The clinical effect and the effect on the ciliary motility of oral N-acetylcysteine in patients with cystic fibrosis and primary ciliary dyskinesia. Eur Respir J. 1988;1:161–167. [PubMed] [Google Scholar]
- 337.Griese M, Ramakers J, Krasselt A, Starosta V, Van Koningsbruggen S, Fischer R, Ratjen F, Mullinger B, Huber RM, Maier K, Rietschel E, Scheuch G. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am J Respir Crit Care Med. 2004;169:822–828. doi: 10.1164/rccm.200308-1104OC. [DOI] [PubMed] [Google Scholar]
- 338.Hartl D, Starosta V, Maier K, Beck-Speier I, Rebhan C, Becker BF, Latzin P, Fischer R, Ratjen F, Huber RM, Rietschel E, Krauss-Etschmann S, Griese M. Inhaled glutathione decreases PGE2 and increases lymphocytes in cystic fibrosis lungs. Free Radic Biol Med. 2005;39:463–472. doi: 10.1016/j.freeradbiomed.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 339.Bishop C, Hudson VM, Hilton SC, Wilde C. A pilot study of the effect of inhaled buffered reduced glutathione on the clinical status of patients with cystic fibrosis. Chest. 2005;127:308–317. doi: 10.1378/chest.127.1.308. [DOI] [PubMed] [Google Scholar]