

Abstract
Spontaneous mitotic recombination is a potential source of genetic changes such as loss of heterozygosity and chromosome translocations, which may lead to genetic disease. In this study we have used a rad52 hyper-recombination mutant, rad52-Y66A, to investigate the process of spontaneous heteroallelic recombination in the yeast Saccharomyces cerevisiae. We find that spontaneous recombination has different genetic requirements, depending on whether the recombination event occurs between chromosomes or between chromosome and plasmid sequences. The hyper-recombination phenotype of the rad52-Y66A mutation is epistatic with deletion of MRE11, which is required for establishment of DNA damage-induced cohesion. Moreover, single-cell analysis of strains expressing YFP-tagged Rad52-Y66A reveals a close to wild-type frequency of focus formation, but with foci lasting 6 times longer. This result suggests that spontaneous DNA lesions that require recombinational repair occur at the same frequency in wild-type and rad52-Y66A cells, but that the recombination process is slow in rad52-Y66A cells. Taken together, we propose that the slow recombinational DNA repair in the rad52-Y66A mutant leads to a by-pass of the window-of-opportunity for sister chromatid recombination normally promoted by MRE11-dependent damage-induced cohesion thereby causing a shift towards interchromosomal recombination.
1. Introduction
In living cells, DNA damage occurs as a result of cell metabolism, developmental processes and exogenous sources such as chemical agents or radiation. Repair of DNA damage is essential to prevent chromosome loss and cell death. In the budding yeast Saccharomyces cerevisiae, homologous recombination (HR) is the major pathway for repair of DNA double-strand breaks (DSBs). However, although DSBs are recombinogenic, they do not appear to be the main source of spontaneous mitotic HR [1,2]. Hence, mutants exist that recombine at wild-type or higher levels despite the fact that they are defective in DNA DSB repair. The nature of the lesions provoking HR is still poorly defined, but understanding the phenotype of mutants that separate DNA DSB repair from spontaneous HR will likely provide clues to the mechanisms of spontaneous HR. Many of the genes involved in this process were identified in yeast by screening for mutants sensitive to ionizing radiation [3]. These mutants constitute the RAD52 epistasis group and include RAD50, RAD51, RAD52, RAD54, RAD55, RAD57, RAD59, RDH54, RFA1, MRE11 and XRS2 [4,5]. Amongst these genes in S. cerevisiae, disruption of RAD52 causes the most severe recombination defect.
The Mre11-Rad50-Xrs2 complex (MRX) is one of the earliest proteins detected at a DSB [6]. The recruitment of MRX to a DSB results in a high local concentration of the complex as visualized by fluorescence microscopy as Mre11 focus formation [6]. The MRX complex contributes to the initial processing of DSB ends into 3' single-stranded DNA (ssDNA) tails [7–10], which are essential for copying genetic information from an intact donor sequence during homologous recombination. Furthermore, recent studies have shown that the MRX complex is required for the postreplicative reestablishment of cohesion in response to genotoxic stress [11–14]. Notably, mre11 results in hyper-recombination between interchromosomal heteroalleles, but not between sister chromatids [15–18]. Importantly, the association of MRX with a DSB is transient and the dissociation of MRX from the site of DNA damage is concurrent with the appearance of ssDNA and recruitment of the Rad52 mediator protein [6], which in turn recruits the Rad51 recombinase to catalyze strand-invasion.
DSBs promote mitotic recombination and result in reciprocal exchange or gene conversion events [19,20]. Frequencies of gene conversion are highest near DSBs [21]. Moreover, conversion tracts are usually continuous and if multiple markers at a DNA DSB are involved, a central marker is almost always co-converted if the flanking markers are converted [21–26]. Gene conversion in yeast involves mismatch repair (MMR) of heteroduplex DNA (hDNA) for both meiotic [27,28] and mitotic events [29–33]. Thus, the amount of homology at the DSB-ends, the direction of mismatch repair and the length of hDNA greatly influence recombinational repair.
The RAD52 epistasis group is also important for spontaneous mitotic recombination although this process is less well characterized and the requirements for individual genes depend on the assay suggesting the existence of multiple pathways [4,5]. Importantly, Rad52 is essential for all types of spontaneous mitotic recombination, whereas Rad51 function is required only for some types of recombination. Finally, genes outside of the RAD52 epistasis group also affect spontaneous mitotic recombination as illustrated by a recent genome-wide analysis of the genetic control of Rad52 foci [34]. Some of these genes that affect recombination include factors that contribute to chromosome integrity by maintaining chromatin architecture and organization, regulating cell cycle and spindle checkpoints, and repairing DNA lesions via other pathways.
To gain insight into the mechanism(s) of spontaneous mitotic recombination, we analyzed the phenotype of a rad52-Y66A mutant that blocks the repair DNA damage induced by γ-irradiation, but is proficient for spontaneous mitotic recombination at a rate higher than wild type. This allele was generated by site-directed mutagenesis in an alanine scan of the conserved N terminus of Rad52 [35]. It was subsequently shown that rad52-Y66A cells are deficient in the repair of a single DSB induced during mating-type switching and are sensitive to a top1-T722A mutation, which causes the accumulation of covalent topoisomerase-DNA intermediates that are frequently converted to DSBs [1]. Further, the rad52-Y66A mutant is proficient for UV-induced heteroallelic recombination. The data presented here suggest that the rad52-Y66A hyper-recombination phenotype may result from a slowdown in DNA repair that leads to a loss of damage-induced cohesion prior to completion of repair, causing a shift from sister chromatid to interchromosomal recombination.
2. Materials and Methods
2.1. Genetic methods, yeast strains and plasmids
Yeast strains were manipulated using standard genetic techniques and media was prepared as described previously except that twice the amount of leucine was used (60mg/L) [36]. All strains used in this study are listed in Table 4 and all are RAD5 derivatives of W303 [37,38]. Other genetic markers have been described previously [39].
Table 4.
List of strains used in this study.
| Straina | Genotype |
|---|---|
| J1173 | MATα ade2–1 leu2ΔEcoRI trp1–1 |
| W1588-4Cb | MATa ade2–1 can1–100 his3–11,15 Ieu2–3,112 trp1–1 ura3–1 |
| W3749-14C | MATa ADE2 bar1∷LEU2 RAD52-YFP |
| W4091-13C | MATα rad52-Y66A ade2-n leu2ΔBstEII trp1–1 |
| W4091-4C | MATα ade2-n leu2ΔBstEII trp1–1 |
| W4092-13B | MATα ade2-a leu2ΔBstEII trp1–1 |
| W4092-4A | MATα rad52-Y66A ade2-a leu2ΔBstEII trp1–1 |
| W4138-17D | MATa ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4138-8B | MATa rad52-Y66A ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4373-1A | MATα mre11∷LEU2 ade2-n trp1–1 |
| W4373-28B | MATα rad52-Y66A mre11∷LEU2 ade2-n trp1–1 |
| W4374-19C | MATa rad52-Y66A mre11∷LEU2 ade2-a TRP1 lys2Δ |
| W4374-27A | MATa mre11∷LEU2 ade2-a TRP1 lys2Δ |
| W4377-14A | MATα rad52-Y66A msh2∷URA3 ade2-n leu2ΔBstEII trp1–1 |
| W4377-8A | MATα msh2∷URA3 ade2-n leu2ΔBstEIl trp1–1 |
| W4378-17D | MATa rad52-Y66A rad51Δ ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4378-34C | MATa rad51Δ ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4379-16D | MATα rad52-Y66A rad51Δ ade2-n leu2ΔBstEII trp1–1 |
| W4379-5C | MATα rad51Δ ade2-n leu2ΔBstEII trp1–1 |
| W4496-1B | MATα rad52∷HIS5 ade2-n leu2ΔBstEII trp1–1 |
| W4497-15A | MATa rad52∷HIS5 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4500-22A | MATa ctf4∷kanMX6 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4501-10A | MATα ctf4∷kanMX6 ade2-n leu2ΔBstEIl trp1–1 |
| W4501-2B | MATα rad52-Y66A ctf4∷kanMX6 ade2-n leu2ΔBstEII trp1–1 |
| W4568-20C | MATα rad52-Y66A msh2∷URA3 ade2-a leu2ΔBstEII trp1–1 |
| W4568-3B | MATa rad52-Y66A msh2∷URA3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4569-46C | MATa rad52-Y66A ctf4∷kanMX6 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4652-12D | MATa msh2∷URA3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4766-6A | MATa rad52∷HIS5 msh2∷URA3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W4766-6C | MATα rad52∷HIS5 msh2∷URA3 ade2-n leu2ΔBstEII trp1–1 |
| W4767-26B | MATa mre11∷LEU2 rad51Δ ade2-a TRP1 lys2Δ |
| W5010-30D | MATα mre11∷LEU2 rad51Δ ade2-n trp1–1 |
| W5176-15D | MATa msh2∷URA3 ade2–1 leu2ΔBstEII TRP1 lys2Δ |
| W5176-18C | MATα rad52-Y66A ade2–1 trp1–1 |
| W5176-19C | MATa rad52-Y66A ade2–1 leu2ΔBstEII TRP1 lys2Δ |
| W5176-24B | MATa rad52-Y66A msh2∷URA3 ade2–1 leu2ΔBstEII TRP1 lys2Δ |
| W5176-26C | MATα msh2∷URA3 ade2–1 leu2ΔBstEII trp1–1 |
| W5176-29D | MATα rad52-Y66A msh2∷URA3 ade2–1 leu2ΔBstEII trp1–1 |
| W5176-53C | MATα msh2∷URA3 ade2-a leu2ΔBstEII trp1–1 |
| W5176-8D | MATa ade2–1 leu2ΔBstEII TRP1 lys2Δ |
| W5461-19D | MATa rad52-Y66A mre11∷LEU2 msh2∷URA3 ade2-n trp1–1 |
| W5461-2D | MATα mre11∷LEU2 msh2∷URA3 ade2-a trp1–1 |
| W5461-35B | MATα rad52-Y66A mre11∷LEU2 msh2∷URA3 ade2-a TRP1 lys2Δ |
| W5461-4A | MATa mre11∷LEU2 msh2∷URA3 ade2-n TRP1 lys2Δ |
| W5507-11B | MATa msh2∷ura3∷HIS3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W5507-3B | MATa rad52-Y66A msh2∷ura3∷HIS3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W5508-1D | MATa rad52-Y66A msh2∷ura3∷HIS3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W5508-2C | MATa msh2∷ura3∷HIS3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W5509-6B | MATa msh2∷ura3∷HIS3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
| W5509-8B | MATa rad52-Y66A msh2∷ura3∷HIS3 ade2-a leu2ΔBstEII TRP1 lys2Δ |
All strains used in the study are derivatives of W1588-4C.
[37].
The ctf4∷kanMX6 allele was amplified from the available yeast gene deletion library strain [40] using primers 5'-CATCCTCTTCATGTACTACTTATGTCCA and 5'- AAGAAATAAAGAACTTTGAATTGATGC. The resulting PCR was transformed into W1588-4C [41]. The strategy for making a marker-free rad51 null (rad51Δ) strain was described previously [42]. The mre11∷LEU2 and msh2∷URA3 alleles were kindly provided by Dr. Lorraine Symington. The msh2∷ura3∷HIS3 allele was made by transforming the msh2∷URA3 strain with the “marker swap” pUH7 plasmid [43].
A CEN-based plasmid harboring the ade2-n allele was constructed by first cloning an ADE2-containing BglII fragment from pRS417 [44] into BamHI-digested YCp50 resulting in pWJ1190. Next, the ade2-n allele was gap-repaired onto AflII-digested pWJ1190 by transformation into W4091-4C resulting in plasmid pWJ1441.
To construct the single-copy pRS413-RAD52-YFP and pRS413-rad52-Y66A-YFP plasmids marked with HIS3, the RAD52 and rad52-Y66A alleles were first subcloned from pWJ646 [35] and its Y66A derivative to the pRS413 vector [44] using restriction enzymes XhoI and XmaI. Next, gap-repair was used to move the yellow fluorescent protein (YFP) encoding sequence onto the linearized plasmids by transformation into strain W3749-14C to produce pRS413-RAD52-YFP and pRS413-rad52-Y66A-YFP.
2.2. ADE2 heteroallelic recombination
Interchromosomal (C × C) recombination between non-functional ade2 heteroalleles was measured in diploid strains. The pair-wise combinations used for the analysis were ade2-n × ade2-a, ade2-1 × ade2-a and ade2-1 × ade2-n. The ade2 heteroalleles were previously described [45]. Cells were plated on SC and SC-Ade plates.
Chromosome-plasmid (C × P) recombination was measured in a haploid strain carrying the ade2-a heteroallele at the endogenous site and the ade2-n heteroallele on a single-copy plasmid (pWJ1441). Cells were plated on SC-Ura and SC-Ura-Ade plates.
The procedure for measuring the rate of heteroallelic recombination was as described previously [46], except that the single colonies were inoculated into liquid media. The plating efficiency and the number of recombinants were determined by plating an appropriate number of cells. For each strain, eight to 20 independent trials were carried out and the corresponding recombination rates and their standard deviations were calculated according to the median method [47].
Recombination was also analyzed in patches of cells grown at 30°C for two days and replica-plated onto the appropriate selective media. The plates were scanned after a four-day incubation. RAD52/RAD52, RAD52/rad52-Y66A and rad52-Y66A/rad52-Y66A diploid cells were patched onto SC plates and replica-plated onto SC-Ade plates. Haploid and diploid strains transformed with pWJ1441 were patched onto SC-Ura plates and replica-plated onto SC-Ura-Ade.
2.3. Analysis of sensitivity to UV- and γ-irradiation
Three to five trials were performed to measure γ-ray sensitivity of each haploid and diploid strain. Cells were grown in liquid YPD medium to a cell density of 107 cells/ml and briefly sonicated. Unless otherwise noted, the plates were incubated at 30°C for 3 days and the surviving colonies counted. Survival curves were produced as previously described [46].
Survival after UV-irradiation was measured in haploid and diploid strains by spot assays. Cells were grown in liquid SC-Leu media and exponential cultures were sonicated before plating. Ten-fold serial dilutions were made and 5 μl from each concentration was spotted onto SC-Leu plates prior to irradiation.
2.4. Cell imaging and fluorescence microscopy
Cells were grown in liquid medium and analyzed by fluorescence microscopy as described [48]. Live cell images were captured with a cooled Orca-ER CCD camera (Hamamatsu, Japan) mounted on a Zeiss Axioplan II microscope (Carl Zeiss, Thornwood, NY). All images were captured at 100-fold magnification using a Plan-Apochromat 100x, 1.4 NA objective lens. The illumination source was a 100W mercury arc lamp (Osram, Munich, Germany). For each field of cells, eleven fluorescent images at the relevant wavelength were obtained at 0.3 μm intervals along the Z-axis to allow inspection of all focal planes of nuclei. YFP was visualized using a band-pass filter set (Cat. # 41028, Chroma, Brattleboro, VT). Images were acquired with a 2% (time-lapse experiments) or 10% neutral density filter in place to reduce photobleaching. Image acquisition time for Rad52-YFP and Rad52-Y66A-YFP were 1000ms. Images were acquired using OpenLab software (Improvision, Lexington, MA).
2.5. Statistical analyses
A t-test was used to determine the significance of differences for the recombination rates between the different strains and for γ-ray survival differences between haploid and diploid strains. For the percentage of cells with a Rad52-YFP focus, the significance was determined by chi-square analysis.
3. Results
3.1. A rad52-Y66A mutant displays context-dependent hyper-recombination
To explore further the mechanism of spontaneous mitotic recombination, we analyzed the hyper-recombination phenotype of a γ-ray sensitive rad52-Y66A mutant [1]. Heteroallelic recombination was measured for diploid wild-type, rad52-Y66A and rad52Δ strains using two previously described non-functional ade2 heteroalleles, ade2-a and ade2-n [45]. Two assays were used to measure the rate of prototroph formation due to recombination between two interchromosomal heteroalleles (C × C) in diploid strains (Fig. 1A) and between chromosomal and plasmid sequences (C × P) in haploid strains (Fig. 1B), respectively. Compared to a wild-type strain, a rad52Δ strain has about a 100-fold lower rate of C × C recombination. In contrast, rad52-Y66A has an eight-fold increase in C × C recombination compared to wild type (Table 1). The hyper-recombination phenotype of rad52-Y66A is recessive since recombination in a heterozygous RAD52/rad52-Y66A diploid is comparable to that of the wild-type strain (Fig. 1C(I)). For the wild-type strain, the rate of spontaneous C × P recombination is ten-fold lower than for C × C recombination indicating that the genomic context of the heteroalleles is important for their availability during mitotic recombination (Tables 1 and 2). Spontaneous recombination in the C × P assay is reduced by more than 20-fold in a rad52Δ strain compared to wild type indicating that Rad52 function is required for C × P recombination (Table 2). Interestingly, the C × P recombination rate of rad52-Y66A is not significantly different from the wild type indicating that the rad52-Y66A hyper-recombination phenotype is specific to the C × C recombination assay.
Fig. 1.

Heteroallelic recombination and DNA repair. (A) Assay for interchromosomal recombination between the ade2-a and ade2-n heteroalleles in diploid cells. -a and -n denote the deletion of the AatII and NdeI enzyme restriction sites, respectively. Three outcomes resulting in adenine prototrophs are illustrated: (i) reciprocal exchange, (ii) conversion of the ade-n allele, and (iii) conversion of the ade2-a allele. (B) Spontaneous recombination between heteroalleles located at the endogenous ADE2 locus (ade2-a) and on a single-copy plasmid (ade2-n). (C) Genomic context of heteroallelic recombination. Patches of prototroph recombinants after replica plating onto SC-Ade plates. (i) MATa RAD52 ade2-a/MATα RAD52 ade2-n, MATa rad52-Y66A ade2-a/MATα rad52-Y66A ade2-n, and MATa RAD52 ade2-a/MATα rad52-Y66A ade2-n diploid strains. (ii) MATa rad52-Y66A ade2-a haploid, MATα rad52-Y66A ade2-a haploid, and MATa rad52-Y66A ade2-a/MATα rad52-Y66A ade2-a transformed with single-copy plasmid carrying the ade2-n heteroallele. (D) Survival after γ-irradiation. Haploid (1n) and diploid (2n) strains were analyzed by counting colonies of surviving cells after 3 days unless otherwise stated. □ RAD52 diploid; ■ RAD52 haploid; ◯ rad52-Y66A diploid; ● rad52-Y66A haploid; △ rad52 null (rad52Δ) diploid; ◇ rad52 null (rad52Δ) diploid after 6 days; and ▲ rad52 null (rad52Δ) haploid. In the rad52 null diploid after 6 days many survivors exhibit loss of the TRP1, LYS2 and/or MAT markers either independently or in combination, indicating that >60% of the survivors are aneuploid (2n-1, 2n-2 and 2n-3), whereas <5% of wild-type survivors are aneuploid. Curves represent the average of 3 trials and error-bars indicate the standard error of the mean.
Table 1.
Spontaneous mitotic recombination between chromosome-chromosome (C x C) heteroalleles.
| Genotype | Spontaneous heteroallelic recombination |
||
|---|---|---|---|
| Rate χ 10−7 |
Fold changeb | ||
| RAD52 | rad52-Y66A | ||
| wild type | 10 ± 1.3** | 82 ± 7.1*,** | 8.2 |
| rad52Δ | 0.1 ± 0.05* | nac | na |
| mre11Δ | 79 ± 11*,** | 60 ± 8.1*,** | 0.8 |
| rad51Δ | 0.3 ± 0.07* | 0.4 ± 0.1*,** | 1.3 |
| mre11Δ rad51Δ | 0.4 ± 0.08* | ndd | na |
| ctf4Δ | 75 ± 10*,** | 100 ± 15*,** | 1.3 |
| msh2Δ | 20 ± 2.1*,** | 238 ± 40*,** | 12 |
| rad52Δ msh2Δ | 0.2 ± 0.1* | na | na |
| mre11Δ msh2Δ | 240 ± 32*,** | 418 ± 53*,** | 1.7 |
P < 0.05 compared to the wild type.
P < 0.05 compared to the rad52Δ.
Recombination rate (events per cell per generation) is presented as the mean ± SD as described in Materials and Methods.
Fold change for the rad52-Y66A allele compared to RAD52.
Not applicable.
Not determined.
Table 2.
Spontaneous mitotic recombination between chromosome-plasmid (C × P) heteroalleles in haploid strains
| Genotype | Spontaneous heteroallelic recombination |
||
|---|---|---|---|
| Rate × 10−7 |
Fold changeb | ||
| RAD52 | rad52-Y66A | ||
| wild type | 0.9 ± 0.2** | 2.1 ± 0.3** | 2.3 |
| rad52Δ | 0.04 ± 0.02* | nac | na |
| mre11Δ | 2.2 ± 0.6** | 0.5 ± 0.1** | 0.2 |
| rad51Δ | 0.3 ± 0.1* | 0.2 ± 0.06* | 0.7 |
| mre11Δ rad51Δ | 0.2 ± 0.05◆ | ndd | na |
| ctf4Δ | 8.1 ± 2.1*,** | 3.5 ± 1.3*,** | 0.4 |
| msh2Δ | 1.4 ± 0.2** | 0.7 ± 0.1** | 0.5 |
P < 0.05 compared to the wild type.
P < 0.05 compared to the rad52A.
P = 0.05 compared to the wild type.
Recombination rate (events per cell per generation) is presented as the mean ± SD as described in Materials and Methods.
Fold change of the rad52-Y66A allele compared to the RAD52 on the different genetic backgrounds.
Not applicable.
Not determined.
It is possible that the difference between C × C and C × P recombination may be due to ploidy or heterozygosity at the mating-type locus. C × C recombination was measured in a MATa/MATα diploid strain whereas C × P recombination was determined in a MATa haploid strain. Both MATα rad52-Y66A ade2-a haploid and MATa/MATα rad52-Y66A/rad52-Y66A ade2-a/ade2-a diploid strains were transformed with a plasmid containing the ade2-n heteroallele and spontaneous recombination assayed in patches of cells. This experiment shows that the recombination levels in both strains are comparable to that of the MATa rad52-Y66A ade2-a haploid transformed with the same plasmid (Fig. 1C(II)). This result indicates that the lower rate of recombination between the C × P heteroalleles in a haploid strain is not mating-type specific nor due to ploidy.
Further, homozygous RAD52, rad52-Y66A and rad52Δ diploid and haploid strains were analyzed for their ability to repair γ-ray induced DNA damage (Fig. 1D). Survival curves show that both haploid and diploid rad52-Y66A strains are more sensitive to ionizing radiation than the wild type. In addition, both wild-type and rad52-Y66A diploids are significantly more resistant to γ-ray induced damage than the respective haploid strains, which is in contrast to the rad52Δ diploid that is more sensitive than the haploid (Fig. 1D). Further, we found that additional slow growing survivors of the irradiated rad52Δ diploid cells appear after 6 days. However, based on marker loss analysis these colonies are likely to have lost whole chromosomes as a result of the induced DNA damage rather than reflecting its repair. Although recombinational repair of DSBs occurs preferentially from the sister chromatid [49], a reasonable explanation for the increased resistance to γ-rays in the wild-type and rad52-Y66A diploid strains is that the repair of induced DSBs is additionally aided by the presence of the homologue. Taken together, the hyper-recombination phenotype of rad52-Y66A is independent of ploidy and is specific for heteroallelic recombination between chromosomes (C × C assay). A possibility is that recombination between C × C and C × P heteroalleles may have distinct requirements for RAD52.
3.2. The rad52-Y66A and mre11Δ mutants are in the same epistasis group for hyper-recombination
To determine whether the difference between C × C and C × P recombination is specific to rad52-Y66A, we tested another hyper-recombination mutant from the RAD52 epistasis group. Compared to a wild-type strain, spontaneous C × C recombination in an mre11Δ mutant is induced eight- to ten-fold [16]. This recombination rate is not increased further by the presence of the rad52-Y66A mutation (Table 1). Interestingly, C × P recombination in mre11Δ or rad52-Y66Amre11Δ is not significantly different from wild-type levels (Table 2). These data indicate that mre11Δ and rad52-Y66A display similar context-dependent hyper-recombination.
Since the difference between the two assays is not specific to rad52-Y66A, we decided to determine the requirements of heteroallelic recombination for Rad51, a RecA homologue and another member of the RAD52 epistasis group. Unlike mre11Δ and rad52-Y66A strains, the lack of Rad51 function significantly reduces C × C recombination by 30 to 50-fold compared to the wild type [50]. rad52-Y66Arad51Δ and mre11Δ rad51Δ double mutants are as defective for recombination as the rad51Δ single mutant (Table 2). Therefore, the C × C events leading to the rad52-Y66A and mre11Δ hyper-recombination phenotype require Rad51 function. In addition, the rate of C × P recombination in rad51Δ, rad52-Y66Arad51Δ and mre11Δ rad51Δ strains is significantly reduced demonstrating that this type of recombination also requires Rad51 activity (Table 2).
3.3. ctf4Δ and msh2Δ cause hyper-recombination by different mechanisms compared to rad52-Y66A
To determine if the difference between C × C and C × P recombination assays extends outside of the RAD52 epistasis group, we measured recombination rates in two other previously reported hyper-recombination mutants, ctf4 and msh2 [51,52].
Msh2 is a key factor in mismatch repair (MMR). In addition, Msh2 helps to prevent genetic recombination between divergent sequences likely by binding mismatches and interacting with the recombination machinery to block, reverse and/or destroy mismatched intermediates [28,52–54]. C × C recombination in a msh2Δ strain shows a slight, but significant increase in the rate of recombination compared to wild type (Table 1). To determine if the C × C hyper-recombination phenotype of rad52-Y66A requires Msh2 function, the rad52-Y66Amsh2Δ double mutant was analyzed. The rate of prototroph formation between C × C heteroalleles in the rad52-Y66Amsh2Δ double mutant is synergistically increased over the rates of either single mutant to 24-fold over the wild-type rate (Table 1). The synergistic effect is specific for C × C recombination as it was not observed for C × P recombination (Table 2). The rad52-Y66Amsh2Δ hyper-recombination phenotype could be due to lack of functional Rad52 activity in a msh2Δ background. To assess this possibility, a rad52Δ msh2Δ strain was analyzed in the C × C assay. Whereas the recombination rate for msh2Δ is 20 × 10−7, the rate for rad52Δ msh2Δ is only 0.2 × 10−7 (Table 1). This result indicates that recombination in a msh2Δ strain requires Rad52 function. Moreover, the synergistic effect is not caused by an additional overall defect in DNA repair efficiency as the MSH2 deletion does not significantly increase the UV- or γ-ray sensitivity of rad52-Y66A (Fig. 2). Taken together, these data indicate that rad52-Y66A and msh2Δ stimulate recombination by different mechanisms.
Fig. 2.

UV- and γ-ray sensitivity of rad52-Y66A and msh2Δ mutants. (A) Survival of diploid strains after γ-irradiation. □ RAD52; ◆ msh2 null (msh2Δ); ◯ rad52-Y66A; ◇ rad52-Y66A msh2Δ; and △ rad52Δ. (B) Survival of diploid strains after UV-irradiation. Ten-fold serial dilutions were plated for each strain onto YPD plates and irradiated with 30 or 60 J/m2. The unirradiated control (0 J/m2) shows the starting concentration for each strain. Curves represent the average of 3 trials and error-bars indicate the standard error of the mean. (C) Hydroxyurea sensitivity of rad52-Y66A. Ten-fold serial dilutions for each strain were used to test survival after DNA damage induced by hydroxyurea (HU, 25 mM and 50 mM). Plates were scanned after 3 days at 30°C.
CTF4 is required for sister chromatid cohesion and fidelity of chromosome segregation and was recently isolated in a genome-wide screen for mutations that increase spontaneous Rad52 foci [34,55–57]. Ctf4 is also a chromatin-associated protein that interacts with DNA polymerase α and may link sister chromatid cohesion to DNA synthesis [58]. Cohesin is assembled at the time of DNA replication at centromeres and other cohesin binding sites [59–62]. Accordingly, ctf4 mutants display increased loss rates for chromosome III as well as for an artificial centromere-based circular chromosome [55]. Similar to rad52-Y66A and mre11Δ, deletion of CTF4 increases recombination eight-fold for the C × C assay, which is consistent with the previously reported hyper-recombination phenotype of this mutant (Table 1) [51]. To determine whether the hyper-recombination phenotype of rad52-Y66A is affected in a ctf4Δ background, the double mutant was analyzed. The C × C recombination rate of rad52-Y66A ctf4Δ is not significantly different from the rad52-Y66A rate. In addition, unlike the results obtained for rad52-Y66A and mre11Δ, ctf4Δ has an eight-fold increase over wild type levels for C × P recombination. This rate is significantly reduced two-fold in a rad52-Y66Actf4Δ strain (Table 2). These data suggest that ctf4Δ causes hyper-recombination by a different mechanism from rad52-Y66A likely by affecting both centromere function and global sister chromatid cohesion [55–57].
3.4. rad52-Y66A results in longer lasting Rad52 foci
Previous studies have shown that fluorescently tagged Rad52 relocalizes from a diffuse nuclear pattern to distinct foci upon induction of DSBs. Focus formation occurs almost exclusively in S phase of mitotically growing cells [48]. To investigate the effect of rad52-Y66A on focus formation, strains expressing Rad52-YFP and Rad52-Y66A-YFP were analyzed (Fig. 3). The localization of these proteins was investigated in rad52Δ diploid cells transformed with a single-copy plasmid carrying either RAD52-YFP or rad52-Y66A-YFP. Fluorescence microscopy shows that the level of spontaneous foci is significantly elevated for rad52-Y66A-YFP in both unbudded (G1) and budded cells (S/G2/M) (Fig. 3A and 3B). The levels of spontaneous Rad52 foci were also measured in a msh2Δ background. The experiment was performed in a rad52Δ msh2Δ diploid strain transformed with either the single-copy plasmid carrying RAD52-YFP or rad52-Y66A-YFP. The level of spontaneous Rad52-YFP foci in msh2Δ budded cells is not significantly different from the levels in a MSH2 background. However, the levels of spontaneous Rad52-Y66A-YFP foci in a msh2Δ strain are significantly lower than in a MSH2 background (Fig. 3B).
Fig. 3.

Spontaneous Rad52 foci. Diploid strains rad52Δ/rad52Δ and rad52Δ msh2Δ/rad52Δ msh2Δ transformed with a single-copy plasmid carrying either RAD52-YFP or rad52-Y66A-YFP. (A) Microscopy of cells. Spontaneous Rad52 foci can be seen in the bottom three panels. (B) Percentage of cells carrying a spontaneous Rad52 focus in unbudded (G1, white box) and budded (S/G2/M, grey box) cells. RAD52-YFP MSH2 0/358 unbudded and 67/442 budded cells. rad52-Y66A-YFP MSH2 82/220 unbudded and 346/434 budded cells. RAD52-YFP msh2Δ 3/197 unbudded and 70/432 budded cells. rad52-Y66A-YFP msh2Δ 37/293 unbudded and 219/518 budded cells. (C) Duration of Rad52 foci determined by time-lapse microscopy. Each circle represents a focus. Black circles: focus lasted within the specified category. Grey circles: focus lasted at least this number of minutes. (D) Percentage of cells that formed at least one Rad52 focus per cell cycle. RAD52-YFP MSH2 43/61 cells. rad52-Y66A-YFP MSH2 59/72 cells. RAD52-YFP msh2Δ 32/56 cells. rad52-Y66A-YFP msh2Δ 12/19 cells.
It is possible that changes in the levels of spontaneous Rad52 foci are the result of changes in the number of DNA lesions. Alternatively, the number of DNA lesions remains the same, but the length of time that the repair proteins remain at foci changes. To distinguish between these possibilities, time-lapse experiments were performed to measure the duration of foci (Fig. 3C). These experiments show that the median Rad52-YFP focus lasts 15 minutes and the median Rad52-Y66A-YFP focus lasts 84 minutes. In a msh2Δ strain, a Rad52-YFP focus lasts 12 minutes, whereas a Rad52-Y66A-YFP focus lasts 60 minutes. Moreover, the cumulative percentage of rad52-Y66A, msh2Δ and rad52-Y66A msh2Δ cells that form at least one Rad52 focus per cell cycle in the time-lapse experiments is not significantly different from that of wild-type cells (Fig. 3D). These data show that the increased duration of foci in rad52-Y66A is largely responsible for the higher frequency of foci observed in both MSH2 and msh2Δ cells (Fig. 3B). Compared to the MSH2 strain, the decrease in duration time of Rad52-Y66A-YFP foci in msh2Δ may be responsible for the lower frequency of foci seen in msh2Δ (Fig. 3B). Thus, our results suggest that rad52-Y66A does not result in an increase in the frequency of DNA lesions, but rather that recombination is slower in the rad52-Y66A mutant.
Lastly, we did not observe RAD52-YFP and RAD52-YFPmsh2Δ cells progressing through the cell cycle until the Rad52 focus disassembles. However, rad52-Y66A-YFP and rad52-Y66A-YFPmsh2Δ cells occasionally divide disregarding the presence of a repair focus likely due to adaptation to the DNA damage checkpoint. This result explains the occurrence of Rad52-Y66A-YFP foci in G1 cells, which is not observed in wild-type cells.
3.5. rad52-Y66A alters the choice of donor template
Long-lasting Rad52-Y66A foci indicate that the DNA repair proteins are associated with the site of DNA damage for a longer time than in wild-type cells. Prolonging this association could affect several aspects of recombination including crossover frequency, gene conversion tract length, and choice of donor template thereby causing the hyper-recombination phenotype observed in rad52-Y66A cells. If the hyper-recombination phenotype of rad52-Y66A cells were due to a change in crossover frequency or conversion tract length, we would expect the degree of hyper-recombination to vary with the distance between the heteroalleles, while a change in the choice of donor template should not vary with the distance between the heteroalleles. To evaluate these alternatives, spontaneous C × C recombination was measured between heteroallelic polymorphisms separated by ~200 bp or ~1150 bp. In the original C × C recombination assay discussed above, the polymorphic sites are separated by ~950 bp (Table 3, top). This analysis shows that the rad52-Y66A strain is hyper-recombinogenic to approximately the same degree independent of the length separating the ade2 polymorphisms (Table 3). The same trend is observed in the rad52-Y66A msh2genetic background, where mismatch repair is decreased (Table 3). These result suggests that the rad52-Y66A hyper-recombination phenotype is due to a change in the choice of donor template rather than a change in crossover frequency and/or conversion tract length.
Table 3.
Spontaneous interchromosomal recombination between ade2 heteroalleles where the polymorphisms are separated by different lengths.
| Genotype | Spontaneous heteroallelic recombinationa |
|||||
|---|---|---|---|---|---|---|
| Rate × 10−7 | ||||||
| 200 bp | Fold changeb | 950 bp | Fold changeb | 1150 bp | Fold changeb | |
| wild type | 1.1±0.3 | na | 10±1.3 | na | 4.3±0.7 | na |
| rad52-Y66A | 11±1.8 | 10 | 82±7.1 | 8 | 60±7.7 | 14 |
| msh2Δ | 3.5±0.6 | 3 | 20±2.1 | 2 | 15±2.1 | 3 |
| rad52-Y66A msh2Δ | 34±4.7 | 31 | 238±40 | 24 | 342±50 | 80 |
Top: Assays for spontaneous interchromosomal recombination between ade2 heteroalleles where the polymorphisms are separated by different lengths. Dashed lines indicate the approximate number of base pairs separating each polymorphism.
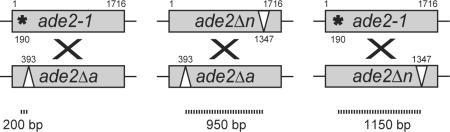
Recombination rate (events per cell per generation) is presented as the mean ± SD as described in Materials and Methods.
Fold change compared to wild type.
3.6. The rad52-Y66A mutant phenocopies mre11Δ
An mre11Δ strain is unable to repair DNA damage induced by hydroxyurea (HU) [63], which causes the formation of replication-associated DSBs. To further explore the epistatic relationship between rad52-Y66A and mre11Δ, we tested the sensitivity of rad52-Y66A to HU. Indeed, we find that HU strongly impairs growth and colony formation for the rad52-Y66A strain (Fig. 2C).
Further, the similar phenotypes of rad52-Y66A and mre11Δ mutants led us to measure the rate of C × C recombination in an mre11Δ msh2Δ strain. If the mechanism by which rad52-Y66A and mre11Δ result in a hyper-recombination phenotype is similar, then the increase in recombination of a rad52-Y66A msh2Δ strain should be comparable to an mre11Δ msh2Δ strain. Indeed, the hyper-recombination rate of mre11Δ msh2Δ is the same as for rad52-Y66A msh2Δ (Table 1). In addition, the C × C recombination rate of the rad52-Y66A mre11Δ msh2Δ triple mutant is not significantly higher than either the rad52-Y66A msh2Δ or mre11Δ msh2 double mutants, further showing that rad52-Y66A and mre11Δ are in the same epistasis group (see section 3.2). Taken together with previous studies demonstrating that Mre11 is required for sister chromatid recombination [64,65], the results presented here support the conclusion that the hyper-recombination phenotypes of rad52-Y66A and mre11Δ could reflect the same underlying mechanism.
4. Discussion
In the budding yeast S. cerevisiae, recombinational repair during mitotic growth relies on several DNA repair proteins, including those encoded by genes of the RAD52 epistasis group. In this study, we measured spontaneous heteroallelic recombination between two chromosomes (C × C) or between a chromosome and a plasmid (C × P). Interestingly, we find that the two configurations display different requirements. The rate of wild-type C × P recombination is ten-fold lower than the rate of C × C recombination. Several factors that could contribute to this difference are outlined below.
One possible explanation for the ten-fold difference may relate to the timing of the replication of the two heteroalleles since spontaneous recombination is likely to occur primarily during S phase [48]. For example, some origins initiate DNA replication at the start of S phase [66], other origins fire slightly later during S [67], whereas still others fire during the second half of S phase [68,69]. Homologous chromosomes are under the control of identical replication origins which are likely to fire at the same time. On the other hand, DNA replication of the plasmid is controlled by a different origin which is likely to initiate at a different time. Thus, the differential timing of replication may make the plasmid and chromosome sequences available for recombination at different times thus lowering their potential to recombine. Another difference between chromosome and plasmid sequences is the length of homology. Chromosomes can potentially pair along their entire length, while the plasmid contains only 2.2 kb of homologous DNA with the chromosomal site. Thus, the shorter homology on the plasmid may be limiting. Finally, the difference between the two configurations may reflect the genomic context of the heteroalleles. For example, chromatin structure may be different between plasmid and chromosomal sequences, and the plasmid-borne ade2-n allele is likely to be spatially restricted due to its centromere-mediated anchoring at the spindle pole body [70]. These possibilities in turn may reduce the probability of an interaction with the chromosomal ade2-a locus (Fig. 4A, top row).
Fig. 4.

Model for rad52-Y66A hyper-recombination. (A) Genetic requirements for C × C versus C × P recombination. Centromere regions are tethered to the spindle-pole body (SPB) at the nuclear envelope (NE) thereby restricting the mobility of the plasmid-borne ade2-n allele and reducing its availability for heteroallelic recombination. Loss of centromere function in the ctf4Δ mutant mobilizes the plasmid to become available for heteroallelic recombination. The mre11Δ and rad52-Y66A mutants show defects in DNA damage-induced cohesion or a failure to complete recombination before cohesion is disassembled, respectively, leading to increased C × C recombination. The spatial restriction of the plasmid-borne heteroallele to the SPB is not releaved in mre11Δ and rad52-Y66A mutants. (B) Transient cohesion model. Mre11 facilitates damage-induced cohesion (dashed lines) which favors the sister chromatid as a template for DSB repair (in grey, i and ii). After approximately 20 min, Mre11 dissociates from the DSB, which may be followed by a gradual loss of cohesion. If repair is not accomplished before loss of cohesion, the DSB may be released to interact with the homologue (in black, iii and vi).
Several issues were considered to explain the hyper-recombination phenotype of the rad52-Y66A mutant. The epistasis analysis of rad52-Y66A relative to other hyper-recombination mutants, msh2Δ, ctf4Δ and mre11Δ, shows that the Y66A mutation most closely phenocopies the mre11Δ deletion, which is defective in sister chromatid recombination (Fig. 4). The phenotypic similarities were further supported by its mre11Δ-like HU sensitivity. The partial rescue of γ-ray sensitivity in rad52-Y66A diploids further supports the notion that these cells frequently utilize the homologous chromosome as a template for repair instead of the sister chromatid. In addition, changing the distance between the heteroalleles does not affect the degree of hyper-recombination, suggesting that rad52-Y66A alters the choice of donor template rather than recombination crossover frequency and/or conversion tract length, although these parameters were not measured directly. Taken together, our data support the hypothesis that the rad52-Y66A mutant phenotype may result from the failure to repair damage using the sister chromatid, thereby increasing interchromosomal interactions that lead to hyper-recombination.
Tyrosine at position 66 of yeast Rad52 is highly conserved through eukaryotic evolution [44]. No structural information is available for yeast Rad52, but in the X-ray crystal structure of human Rad52, the corresponding tyrosine 51 is burrowed below the DNA binding groove with little surface exposure [71,72]. Thus, the structural information indicates that this residue is unlikely to have direct contacts with DNA or other protein, but may rather be important for putative conformational changes during recombination. Consistent with this notion, the human Rad52-Y51A protein is proficient for DNA binding. Together with the separation-of-function phenotype of the yeast rad52-Y66A mutant, this information suggests that, rather than affecting the initial association with DNA, the Y66A mutation prevents a later step during homologous recombination. This step, which is dispensible for heteroallelic recombination, but important during the repair of IR-induced lesions, may be second-end capture [73,74].
In response to induced DNA damage, postreplicative cohesion is assembled in an MRE11-dependent fashion [11–14]. This assembly is likely to occur during the transient association of Mre11 with the site of DNA damage immediately upon its recognition. We previously showed that Mre11 dissociates from the site of DNA damage after approximately 20 minutes raising the possibility that damage-induced cohesion is not maintained in the event of persistent damage [6]. Here we show that Rad52-Y66A-YFP foci often persist for several hours. If DNA damage-induced cohesion is relaxed upon dissociation of Mre11, before repair from the sister chromatid takes place, survival would be enhanced by redirecting repair towards the homologue resulting in interchromosomal recombination (Fig. 4B). Such a scenario might be beneficial to promote survival, even in wild-type cells, in the event that sister chromatid recombination cannot be completed due to damage of both sister chromatids.
Acknowledgments
This work was supported by The Danish Agency for Science, Technology and Innovation and the Villum Kann Rasmussen Foundation (ML), the Danish Research Council for Technology and Production Sciences (UHM), and the National Institutes of Health (GM50237 and GM67055 to RR). We thank Marisa Wagner for constructing the pWJ1190 plasmid and Lorraine Symington for sharing strains.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest The authors declare that there are no conflicts of interest.
References
- [1].Lettier G, Feng Q, de Mayolo AA, Erdeniz N, Reid RJ, Lisby M, Mortensen UH, Rothstein R. The role of DNA double-strand breaks in spontaneous homologous recombination in S. cerevisiae. PLoS Genet. 2006;2:e194. doi: 10.1371/journal.pgen.0020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fabre F, Chan A, Heyer WD, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. USA. 2002;10:10. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Game JC, Cox BS. Allelism tests of mutants affecting sensitivity to radiation in yeast and a proposed nomenclature. Mutat. Res. 1971;6:37–55. [Google Scholar]
- [4].Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response; spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- [7].Johzuka K, Ogawa H. Interaction of Mre11 and Rad50: two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics. 1995;139:1521–1532. doi: 10.1093/genetics/139.4.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tsubouchi H, Ogawa H. A novel mre11 mutation impairs processing of double-strand breaks of DNA during both mitosis and meiosis. Mol. Cell. Biol. 1998;18:260–268. doi: 10.1128/mcb.18.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sugawara N, Haber JE. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol. Cell. Biol. 1992;12:563–575. doi: 10.1128/mcb.12.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ivanov EL, Sugawara N, White CI, Fabre F, Haber JE. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol. Cell. Biol. 1994;14:3414–3425. doi: 10.1128/mcb.14.5.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Strom L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- [12].Unal E, Heidinger-Pauli JM, Koshland D. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7) Science. 2007;317:245–248. doi: 10.1126/science.1140637. [DOI] [PubMed] [Google Scholar]
- [13].Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- [14].Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schiestl RH, Zhu J, Petes TD. Effect of mutations in genes affecting homologous recombination on restriction enzyme-mediated and illegitimate recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 1994;14 :4493–4500. doi: 10.1128/mcb.14.7.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ajimura M, Leem SH, Ogawa H. Identification of new genes required for meiotic recombination in Saccharomyces cerevisiae. Genetics. 1993;133:51–66. doi: 10.1093/genetics/133.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ivanov EL, Korolev VG, Fabre F. XRS2, a DNA repair gene of Saccharomyces cerevisiae, is needed for meiotic recombination. Genetics. 1992;132:651–664. doi: 10.1093/genetics/132.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Malone RE, Ward T, Lin S, Waring J. The RAD50 gene, a member of the double strand break repair epistasis group, is not required for spontaneous mitotic recombination in yeast. Curr. Genet. 1990;18:111–116. doi: 10.1007/BF00312598. [DOI] [PubMed] [Google Scholar]
- [19].Ray A, Siddiqi I, Kolodkin AL, Stahl FW. Intra-chromosomal gene conversion induced by a DNA double-strand break in Saccharomyces cerevisiae. J. Mol. Biol. 1988;201:247–260. doi: 10.1016/0022-2836(88)90136-2. [DOI] [PubMed] [Google Scholar]
- [20].Nickoloff JA, Singer JD, Hoekstra MF, Heffron F. Double-strand breaks stimulate alternative mechanisms of recombination repair. J. Mol. Biol. 1989;207:527–541. doi: 10.1016/0022-2836(89)90462-2. [DOI] [PubMed] [Google Scholar]
- [21].Sweetser DB, Hough H, Whelden JF, Arbuckle M, Nickoloff JA. Fine-resolution mapping of spontaneous and double-strand break-induced gene conversion tracts in Saccharomyces cerevisiae reveals reversible mitotic conversion polarity. Mol. Cell. Biol. 1994;14:3863–3875. doi: 10.1128/mcb.14.6.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Borts RH, Haber JE. Meiotic recombination in yeast: alteration by multiple heterozygosities. Science. 1987;237:1459–1465. doi: 10.1126/science.2820060. [DOI] [PubMed] [Google Scholar]
- [23].Borts RH, Haber JE. Length and distribution of meiotic gene conversion tracts and crossovers in Saccharomyces cerevisiae. Genetics. 1989;123:69–80. doi: 10.1093/genetics/123.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Willis KK, Klein HL. Intrachromosomal recombination in Saccharomyces cerevisiae: reciprocal exchange in an inverted repeat and associated gene conversion. Genetics. 1987;117:633–643. doi: 10.1093/genetics/117.4.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Aguilera A, Klein HL. Yeast intrachromosomal recombination: long gene conversion tracts are preferentially associated with reciprocal exchange and require the RAD1 and RAD3 gene products. Genetics. 1989;123:683–694. doi: 10.1093/genetics/123.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Judd SR, Petes TD. Physical lengths of meiotic and mitotic gene conversion tracts in Saccharomyces cerevisiae. Genetics. 1988;118:401–410. doi: 10.1093/genetics/118.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Petes TD, Malone RE. L.S. Symington Recombination in yeast. In: Broach JR, Pringle JR, Jones EW, editors. The Molecular & Cellular Biology of the Yeast Saccharomyces: Genome Dynamics, Protein Synthesis and Energetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1991. pp. 407–521. [Google Scholar]
- [28].Alani E, Reenan RA, Kolodner RD. Interaction between mismatch repair and genetic recombination in Saccharomyces cerevisiae. Genetics. 1994;137:19–39. doi: 10.1093/genetics/137.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].White CI, Haber JE. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J. 1990;9:663–673. doi: 10.1002/j.1460-2075.1990.tb08158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Haber JE, Ray BL, Kolb JM, White CI. Rapid kinetics of mismatch repair of heteroduplex DNA that is formed during recombination in yeast. Proc. Natl. Acad. Sci. USA. 1993;90:3363–3367. doi: 10.1073/pnas.90.8.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ray BL, White CI, Haber JE. Heteroduplex formation and mismatch repair of the “stuck” mutation during mating-type switching in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991;11:5372–5380. doi: 10.1128/mcb.11.10.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cross SH, Allshire RC, McKay SJ, McGill NI, Cooke HJ. Cloning of human telomeres by complementation in yeast. Nature. 1989;338:771–774. doi: 10.1038/338771a0. [DOI] [PubMed] [Google Scholar]
- [33].Weng YS, Nickoloff JA. Evidence for independent mismatch repair processing on opposite sides of a double-strand break in Saccharomyces cerevisiae. Genetics. 1998;148:59–70. doi: 10.1093/genetics/148.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Alvaro D, Lisby M, Rothstein R. Genome-wide analysis of Rad52 foci reveals diverse mechanisms impacting recombination. PLoS Genet. 2007;3:2439–2449. doi: 10.1371/journal.pgen.0030228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mortensen UH, Erdeniz N, Feng Q, Rothstein R. A molecular genetic dissection of the evolutionarily conserved N terminus of yeast Rad52. Genetics. 2002;161:549–562. doi: 10.1093/genetics/161.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sherman F, Fink GR, Hicks JB. Methods in Yeast Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1986. [Google Scholar]
- [37].Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- [38].Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- [39].Erdeniz N, Rothstein R. Rsp5, a ubiquitin-protein ligase, is involved in degradation of the single-stranded-DNA binding protein rfa1 in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000;20:224–232. doi: 10.1128/mcb.20.1.224-232.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- [41].Alvaro D, Sunjevaric I, Reid RJ, Lisby M, Stillman DJ, Rothstein R. Systematic hybrid LOH: a new method to reduce false positives and negatives during screening of yeast gene deletion libraries. Yeast. 2006;23:1097–1106. doi: 10.1002/yea.1423. [DOI] [PubMed] [Google Scholar]
- [42].Reid R, Lisby M, Rothstein R. Cloning-free genome alterations in Saccharomyce cerevisiae using adaptamer-mediated PCR. Methods in Enzymology. 2002;350:258–277. doi: 10.1016/s0076-6879(02)50968-x. [DOI] [PubMed] [Google Scholar]
- [43].Cross FR. 'Marker swap' plasmids: convenient tools for budding yeast molecular genetics. Yeast. 1997;13:647–653. doi: 10.1002/(SICI)1097-0061(19970615)13:7<647::AID-YEA115>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- [44].Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rattray AJ, Symington LS. Use of a chromosomal inverted repeat to demonstrate that the RAD51 and RAD52 genes of Saccharomyces cerevisiae have different roles in mitotic recombination. Genetics. 1994;138:587–595. doi: 10.1093/genetics/138.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Smith J, Rothstein R. A mutation in the gene encoding the Saccharomyces cerevisiae single-stranded DNA-binding protein Rfa1 stimulates a RAD52-independent pathway for direct-repeat recombination. Mol. Cell. Biol. 1995;15:1632–1641. doi: 10.1128/mcb.15.3.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lea DE, Coulson CA. The distribution in the numbers of mutants in bacterial populations. J. Genet. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- [48].Lisby M, Rothstein R, Mortensen UH. Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. USA. 2001;98:8276–8282. doi: 10.1073/pnas.121006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kadyk LC, Hartwell LH. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics. 1992;132:387–402. doi: 10.1093/genetics/132.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bai Y, Davis AP, Symington LS. A novel allele of RAD52 that causes severe DNA repair and recombination deficiencies only in the absence of RAD51 or RAD59. Genetics. 1999;153:1117–1130. doi: 10.1093/genetics/153.3.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kouprina N, Kroll E, Bannikov V, Bliskovsky V, Gizatullin R, Kirillov A, Shestopalov B, Zakharyev V, Hieter P, Spencer F, et al. CTF4 (CHL15) mutants exhibit defective DNA metabolism in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 1992;12:5736–5747. doi: 10.1128/mcb.12.12.5736. published erratum appears in Mol. Cell. Biol. 1993 Nov;13(11):7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Selva EM, Maderazo AB, Lahue RS. Differential effects of the mismatch repair genes MSH2 and MSH3 on homeologous recombination in Saccharomyces cerevisiae. Mol. Gen. Genet. 1997;257:71–82. doi: 10.1007/pl00008619. [DOI] [PubMed] [Google Scholar]
- [53].Selva EM, New L, Crouse GF, Lahue RS. Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae. Genetics. 1995;139:1175–1188. doi: 10.1093/genetics/139.3.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chambers SR, Hunter N, Louis EJ, Borts RH. The mismatch repair system reduces meiotic homeologous recombination and stimulates recombination-dependent chromosome loss. Mol. Cell. Biol. 1996;16:6110–6120. doi: 10.1128/mcb.16.11.6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kouprina N, Pashina OB, Nikolaishwili NT, Tsouladze AM, Larionov VL. Genetic control of chromosome stability in the yeast Saccharomyces cerevisiae. Yeast. 1988;4:257–269. doi: 10.1002/yea.320040404. [DOI] [PubMed] [Google Scholar]
- [56].Hanna JS, Kroll ES, Lundblad V, Spencer FA. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol. Cell. Biol. 2001;21:3144–3158. doi: 10.1128/MCB.21.9.3144-3158.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Spencer F, Gerring SL, Connelly C, Hieter P. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics. 1990;124:237–249. doi: 10.1093/genetics/124.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Miles J, Formosa T. Protein affinity chromatography with purified yeast DNA polymerase alpha detects proteins that bind to DNA polymerase. Proc. Natl. Acad. Sci. USA. 1992;89:1276–1280. doi: 10.1073/pnas.89.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Uhlmann F, Nasmyth K. Cohesion between sister chromatids must be established during DNA replication. Curr. Biol. 1998;8:1095–1101. doi: 10.1016/s0960-9822(98)70463-4. [DOI] [PubMed] [Google Scholar]
- [60].Blat Y, Kleckner N. Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell. 1999;98:249–259. doi: 10.1016/s0092-8674(00)81019-3. [DOI] [PubMed] [Google Scholar]
- [61].Tanaka T, Cosma MP, Wirth K, Nasmyth K. Identification of cohesin association sites at centromeres and along chromosome arms. Cell. 1999;98:847–858. doi: 10.1016/s0092-8674(00)81518-4. [DOI] [PubMed] [Google Scholar]
- [62].Laloraya S, Guacci V, Koshland D. Chromosomal addresses of the cohesin component Mcd1p. J. Cell. Biol. 2000;151:1047–1056. doi: 10.1083/jcb.151.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Krogh BO, Llorente B, Lam A, Symington LS. Mutations in Mre11 phosphoesterase motif I that impair Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex stability in addition to nuclease activity. Genetics. 2005;171:1561–1570. doi: 10.1534/genetics.105.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bressan DA, Baxter BK, Petrini JH. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999;19:7681–7687. doi: 10.1128/mcb.19.11.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gonzalez-Barrera S, Cortes-Ledesma F, Wellinger RE, Aguilera A. Equal sister chromatid exchange is a major mechanism of double-strand break repair in yeast. Mol. Cell. 2003;11:1661–1671. doi: 10.1016/s1097-2765(03)00183-7. [DOI] [PubMed] [Google Scholar]
- [66].Reynolds AE, McCarroll RM, Newlon CS, Fangman WL. Time of replication of ARS elements along yeast chromosome III. Mol. Cell. Biol. 1989;9:4488–4494. doi: 10.1128/mcb.9.10.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Brewer BJ, Fangman WL. Initiation at closely spaced replication origins in a yeast chromosome. Science. 1993;262:1728–1731. doi: 10.1126/science.8259517. [DOI] [PubMed] [Google Scholar]
- [68].Ferguson BM, Brewer BJ, Reynolds AE, Fangman WL. A yeast origin of replication is activated late in S phase. Cell. 1991;65:507–515. doi: 10.1016/0092-8674(91)90468-e. [DOI] [PubMed] [Google Scholar]
- [69].Friedman KL, Diller JD, Ferguson BM, Nyland SV, Brewer BJ, Fangman WL. Multiple determinants controlling activation of yeast replication origins late in S phase. Genes Dev. 1996;10:1595–1607. doi: 10.1101/gad.10.13.1595. [DOI] [PubMed] [Google Scholar]
- [70].Stoler S, Rogers K, Weitze S, Morey L, Fitzgerald-Hayes M, Baker RE. Scm3, an essential Saccharomyces cerevisiae centromere protein required for G2/M progression and Cse4 localization. Proc. Natl. Acad. Sci. USA. 2007;104:10571–10576. doi: 10.1073/pnas.0703178104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kagawa W, Kurumizaka H, Ishitani R, Fukai S, Nureki O, Shibata T, Yokoyama S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell. 2002;10:359–371. doi: 10.1016/s1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- [72].Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB, Kagawa W, Kurumizaka H, Ishitani R, Fukai S, Nureki O, et al. Structure of the single-strand annealing domain of human RAD52 protein crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Proc. Natl. Acad. Sci. USA. 2002;99:13492–13497. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nimonkar AV, Sica RA, Kowalczykowski SC. Rad52 promotes second-end DNA capture in double-stranded break repair to form complement-stabilized joint molecules. Proc. Natl. Acad. Sci. USA. 2009;106:3077–3082. doi: 10.1073/pnas.0813247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].McIlwraith MJ, West SC. DNA repair synthesis facilitates RAD52-mediated second-end capture during DSB repair. Mol. Cell. 2008;29:510–516. doi: 10.1016/j.molcel.2007.11.037. [DOI] [PubMed] [Google Scholar]