

Abstract
The poxviruses (family Poxviridae) are a family of double-stranded viruses including several species that infect humans and their domestic animals, most notably Variola virus (VARV), the causative agent of smallpox. The evolutionary biology of these viruses poses numerous questions, for which we have only partial answers at present. Here we review evidence regarding the origin of poxviruses, the frequency of host transfer in poxvirus history, horizontal transfer of host genes to poxviruses, and the population processes accounting for patterns of nucleotide sequence polymorphism.
1. Introduction
The poxviruses (family Poxviridae) are a family of double-stranded DNA (dsDNA) viruses with very large genomes (130–360 kb in length), usually encoding more than 150 genes per genome (Lefkowitz et al. 2006). The Poxviridae are divided into two subfamilies: Entomopoxvirinae, infecting insects; and Chordopoxvirinae, infecting vertebrates (van Regenmortel et al. 2000). Poxvirus replication occurs in the cytoplasm, thus preventing the virus from using nuclear enzymes of the host and requiring it to encode its own enzymes for DNA replication. DNA replication is necessary to provide a DNA template from which intermediate gene products can be expressed and in turn regulate the late transcription process yielding the virion proteins (Lefkowitz et al. 2006). The occurrence of at least a portion of the viral life cycle in the host cytoplasm is a characteristic poxviruses share with all members of a proposed clade of large DNA viruses of eukaryotes, the Nucleo-Cytoplasmic Large DNA Viruses (NCLDV; Iyer et al. 2001, 2006).
Several species in the poxvirus family infect humans and their domestic animals, most notably Variola virus (VARV), the causative agent of smallpox, for which humans are the only known natural host. Smallpox epidemics have had major impacts on human history; and the discovery and promotion of vaccination against smallpox by Edward Jenner was a significant landmark in the development of Western medical science (Smith 1990). Interestingly, it is not known for certain which virus Jenner used for the first vaccinations (Baxby 1981). The modern vaccine is based on Vaccinia virus (VACV), but this virus is quite distinct from Cowpox virus (CPXV), traditionally assumed to be the virus used by Jenner to create the first vaccine. If Jenner really did use CPXV, how and when VACV replaced CPXV in vaccine cultures is unknown. Although VARV was eradicated as a naturally occurring human pathogen 1980, the threat of smallpox as a potential weapon of bioterrorism maintains active interest in the mechanisms of infection and pathogenesis of this virus (Artenstein and Grabenstein 2008).
The tools of molecular biology have provided numerous advances in understanding the basic biology and immunology of poxviruses (Moss and Shisler 2001; Shisler and Moss 2001; Everett and McFadden 2002; Smith and Kotwal 2002; Dunlop et al. 2003; Seet et al. 2003; Moss 2006; Zhang et al. 2009); yet there remains much that we do not understand (Lefkowitz et al. 2006). A number of unanswered questions relate to evolutionary biology, and an evolutionary perspective may provide important insights into the functional biology of these viruses, particularly in the area of immune evasion. Here we review the evidence in three areas of active research in poxvirus evolutionary biology: (1) phylogeny, with particular emphasis on transfer of poxviruses across host species boundaries and the origin of major human pathogens such as VARV; (2) horizontal transfer (or “capture”) of host genes and the role of transferred host genes in the evolution of immune evasion; and (3) the population biology of poxviruses, including the roles of mutation and selection in accounting for observed patterns of sequence diversity. We include analyses of published sequence data in order to illustrate some of the many interesting but unanswered questions regarding poxvirus evolutionary biology. In addition, we preface our review with some general considerations regarding the evolutionary biology of viruses; this field has given rise to an extensive and active literature, but one that has been plagued by a number of fairly widely held misconceptions.
2. Evolutionary Biology of Viruses: General Considerations
Both evolutionary biologists and virologists have increasingly come to realize the importance of viruses as models for studying evolutionary processes. Because of their short generation times and high mutations rates – particularly in the case of viruses with RNA genomes – viruses provide an ideal system for studying evolution at the population level. In addition, because of the small genome sizes of many viruses, it is possible to sequence substantial numbers of complete genomes relatively inexpensively and thus to examine responses to a variety of natural and artificial selective pressures. Poxviruses, because of their large genome sizes, have not so far been used extensively for population studies; but, as the cost of sequencing goes down, population studies of complete genomes of poxviruses are likely to be increasingly feasible.
Many studies of virus evolution have been hampered by a speculative mindset inherited from the early, pre-molecular era in Neo-Darwinism. According to this point of view, we can study the evolution of phenotypes by predicting what natural selection “should” favor under a given set of circumstances and then comparing observed phenotypes to our predictions. The problem with such an approach is that it assumes that mutation will invariably supply the variation needed for a response to selection, which is by no means certain. For example, it is often assumed that viral mutation rates are “optimized,” representing some sort of balance between the introduction of deleterious and favorable mutations (Nowak and May 2000). But in fact, in most cases, it may not be possible for any mutation to occur that will globally change a virus’s mutation rate in the optimal direction. Thus a theoretically predicted optimum value for the mutation rate (or for any other phenotype) may have no biological meaning. Indeed, if favorable mutations are rare in comparison to deleterious mutations, the optimum mutation rate for any organism (viruses included) is likely to be zero; and one might expect natural selection to keep the mutation rate as low as possible (Williams 1966).
Viruses have provided strong evidence in support of the hypothesis that deleterious mutations greatly outnumber favorable mutations. Coding regions of viruses, like those of other organisms, provide evidence that most nonsynonymous (amino acid-altering) mutations are deleterious and have been subject to natural selection (“purifying selection”) acting to remove them from populations. In most viral protein-coding genes, the number of synonymous substitutions per synonymous site (dS) typically exceeds the number of nonsynonymous substitutions per nonsynonymous site (dN), indicating that purifying selection has acted against the latter (Hughes 1999). This pattern has been found in the protein-coding genes of many virus taxa (e.g., Hughes 2007a; Hughes and Hughes 2007; Hughes et al. 2007; Hughes and Piontkivska 2008; Irausquin and Hughes 2008). Moreover, in many viruses the rate of nucleotide substitution in non-coding regions is substantially less than dS (though usually not lower than dN), providing evidence of purifying selection on viral genomes outside protein-coding genes (Hughes 2007a, 2009; Hughes and Piontkivska 2008).
Sequence comparisons provide evidence of two distinct kinds of purifying selection: (1) past purifying selection, which has acted to eliminate deleterious mutations from the population, including highly deleterious mutations, which are eliminated very quickly or even immediately; and (2) ongoing purifying selection, acting against slightly deleterious variants, which may be present in the population at substantial allelic frequencies (Hughes 2008). Evidence of ongoing purifying selection against slightly deleterious nonsynonymous variants is provided by the occurrence of numerous relatively rare nonsynonymous variants (Hughes 2005; Hughes et al. 2003); and such a pattern has been seen in many viruses as well as in cellular organisms (Hughes 2007a; Hughes and Hughes 2007; Hughes et al. 2007; Hughes and Piontkivska 2008; Irausquin and Hughes 2008).
Escape from recognition by the host class I major histocompatibility complex and CD8+ T-cell recognition system has provided some of the most convincing evidence of positive Darwinian selection favoring amino acid replacements (O’Connor et al. 2004; Hughes et al. 2005). However, many papers have been published claiming evidence for positive selection in viruses based on the use of invalid statistical methods. Particularly egregious are the so-called “codon-based” tests for positive selection; these methods depend on the assumption that the existence of even one codon with dN/dS > 1 implies positive selection. But this assumption is false, since such codons occur in many sequence datasets even under strong purifying selection as a result of the stochastic nature of the mutational process (Hughes 2007a; Hughes and Friedman 2005a, 2008). Moreover, because these methods identify as “positively selected” any codon with one or more nonsynonymous changes and no synonymous changes (Hughes and Friedman 2008), they are highly sensitive to sequencing and alignment errors (Schneider et al. 2009). Claims of positive selection based on these methods should be treated with skepticism.
For example, McLysaght et al. (2003) tested for positive selection in poxviruses using a codon-based method subsequently shown to be highly non-conservative (Zhang 2004) as well as based on questionable assumptions (Hughes and Friedman 2008). The genes that they identified as subject to positive selection probably represent mainly cases of relaxation of purifying selection. This may be particularly true in the case of genes horizontally transferred from the host. There is evidence that such genes may undergo substantial change at the amino acid level, due to the higher mutation rate in viruses and the absence in the virus of certain functional constraints on the protein sequence that were present in the host (Hughes 2002).
Whatever the truth regarding the prevalence of positive selection on viral genomes, studying patterns of purifying selection is arguably of much greater biological and practical interest. Sequence motifs that show little polymorphism or long-term evolutionary change are in general likely to be subject to strong purifying selection and thus to play an important role in the viral life cycle. It is not rapidly changing sequences but unchanging sequence motifs – whether amino acid residues in virally encoded proteins or regulatory regions in non-coding portions of the viral genome – that are most likely to be adaptively important to the virus. Moreover, highly conserved sequences are likely to represent the most promising targets for both drugs and vaccines because they are not free to change without substantial fitness cost to the virus.
3. Origin and Phylogeny of Poxviruses
3.1. Origin of Poxviruses
The origin of viruses, like that of cellular life forms, is shrouded in mystery. A number of hypotheses have been proposed for the origin of viruses in general or that of certain viral groups, as well as for the related question of the monophyly of viruses as a whole or of major groups of viruses (Morse 1994; Koonin et al. 2006). Although it seems difficult to imagine that all viruses – including both DNA and RNA viruses – have a single evolutionary origin, it is still possible that certain major viral groups are monophyletic. One candidate for monophyly is the NCLDV group, which includes the family Poxviridae as well other large dsDNA viruses infecting eukaryotes (Iyer et al. 2001, 2006).
There are three major hypotheses for the origin of viruses (Koonin et al. 2006): (1) that viruses antedate cellular life forms, representing survivals from an ancient “virus world”; (2) that viruses arose as genes of cellular organisms that somehow “escaped” and became transmissible parasites of cells; and (3) that viruses arose from cells that underwent extensive genome reduction after becoming parasitic on other cells. These hypotheses are not necessarily mutually exclusive. If different groups of viruses arose independently, it is possible that more than one of these scenarios may have been involved in the origin of distinct viral groups.
Koonin and colleagues (2006) have drawn attention to the existence of certain virus-specific genes involved in fundamental processes across diverse groups of viruses (but not all viruses) but lacking homologs in cellular organisms. An example is the superfamily 3 helicase, found in both certain RNA viruses (such as picornaviruses as comoviruses) as well as in the NCLDV group, including poxviruses (Koonin et al. 2006). Koonin et al. (2006) argue that such genes provide evidence of an ancient pre-cellular virus world, and certainly this is one hypothesis that might explain their occurrence. Alternatively, it might also be hypothesized that such genes arose in cellular lineages that have since become extinct. The sharing of such genes across distinct viral groups might in turn be explained by horizontal transfer. Since viruses are able to pick up host genes (see below), it seems possible that a virus might obtain genes from an unrelated virus simultaneously infecting the same host.
3.2. Phylogeny of Poxviridae
The availability of complete genome sequences for a number of poxviruses has inspired researchers to undertake phylogenetic analyses of this family using a variety of methods and approaches. Genomes of any two poxvirus species will share a certain proportion of their genes, but there are likely to be a number of genes in each genome that lack orthologs in the other. This observation has suggested that a phylogeny might be constructed based on the presence/absence of gene families (Gubser et al. 2004; Hughes and Friedman 2005b; Xing et al. 2006). Another alternative has been to use gene order (Xing et al. 2006). Finally, researchers have used sequence similarity searches to identify a set of conserved genes shared by all of the genomes to be analyzed, and have then based a phylogeny on these conserved genes (McLysacht et al. 2003; Gubser et al. 2004; Hughes and Friedman 2005b; Xing et al. 2006; Bratke and McLysaght 2008).
In general, these different approaches have yielded similar results, although details may differ among different analyses, depending on the methods used and the sequences included. Figure 1 shows a phylogenetic tree based on concatenated amino acid sequences of 29 conserved orthologous proteins. This tree was constructed by the neighbor-joining (NJ) method (Saitou and Nei 1987) on the basis of the JTT amino acid distance, assuming that rate variation among sites follows a gamma distribution. The shape parameter of the gamma distribution (a = 0.86) was estimated by the TREE-PUZZLE program (Schmidt et al. 2002). Reliability of branching patterns in the tree was assessed by bootstrapping (Felsenstein 1985); 1000 bootstrap samples were used. The phylogenetic tree of Chordopoxvirinae was rooted with two viruses belonging to Entomopoxvirinae (Figure 1). The phylogenetic tree (Figure 1) was similar to previously published phylogenies (McLysacht et al. 2003; Gubser et al. 2004; Hughes and Friedman 2005b; Xing et al. 2006). Note that this is only a preliminary analysis. Complete genome sequences are now available for several isolates of a number of poxvirus species, but in this analysis only one genome per species was included.
Figure 1.

NJ tree (topology only) of poxviruses based on concatenated amino acid sequences from 29 conserved orthologous proteins (13, 475 aligned sites). The tree was constructed on the basis of the JTT amino acid distance, assuming that rate variation among sites follows a gamma distribution (shape parameter a = 0.86). Numbers on the branches represent percentages of 1000 bootstrap samples supporting each branch; only values ≥ 50 % are shown. For accession numbers of genome sequences, see Supplementary Table S1.
Before genomic sequences were available, it was often speculated that the association of poxviruses with their vertebrate hosts is very ancient, and that host and parasite have co-evolved throughout the history of amniotes (Fenner and Kerr 1994). Phylogenetic analyses have generally not supported this hypothesis. It is true that certain clades of poxviruses infect members of specific mammalian taxa. For example, our phylogenetic analysis provided 100% bootstrap support for a clade including five viruses infecting members of the Artiodactyla (even-toed hoofed mammals): Deerpox virus, Swinepox virus, Goatpox virus, Sheeppox virus, and Lumpy skin disease virus (Figure 1). On the other hand, two other viruses of artiodactyls, Orf virus and Bovine papular stomatitis virus, did not cluster with the former group (Figure 1). Likewise, our phylogenetic analysis provided 100% bootstrap support for a clade including viruses of artiodactyls (CPXV and Camelpox virus), rodents (Ectromelia virus and Taterapox virus), and primates (VARV), as well as Monkeypox virus, which infects both rodents and primates, and VACV, whose natural host is unknown (Figure 1). The latter clade corresponds to the genus Orthopoxvirus (van Regenmortel et al. 2000). Overall, phylogenetic analyses provide strong support for the hypothesis that transfers from one host species to another have been a recurrent feature of the evolution of the Chordopoxvirinae. Thus, in general, host lineages and poxvirus lineages have not co-evolved.
3.3. The Origin of VARV
Because VARV lacks a known non-human animal reservoir, its origin as a human pathogen has been mysterious. Based on Jenner’s reported use of CPXV in vaccination for VARV, it has often been speculated that VARV arose as a virulent form of CPXV transferred to humans from domestic cattle. For example, Diamond (1997) listed VARV as one of several cases of human pathogens derived from domestic animals and developed a fanciful interpretation of human history based on the competitive advantage cultures with livestock are supposed to have gained from exposure to these pathogens. However, sequence analyses provide strong evidence against the domestic animal origin of several of the pathogens listed by Diamond (1997); e.g., Mycobacterium tuberculosis (Hughes et al. 2002) and Plasmodium falciparum (Hughes and Verra 2002).
Like previous phylogenetic analyses, our tree showed VARV to be a sister group of Camelpox virus and Taterapox virus (Figure 1). Although several branching patterns within the Orthopoxvirus clade were not well resolved, the clustering of VARV with Camelpox virus and Taterapox virus received 98% bootstrap support (Figure 1). This pattern provides evidence against the hypothesis that CPXV was the ancestor of VARV. Taterapox was isolated from a West African rodent, Kemp’s gerbil (formerly Tatera kempi, now Gerbilliscus kempi; Lourie et al. 1975). Thus, it has been suggested that VARV was transferred to the human population from an African rodent host (Esposito et al. 2006).
Several authors have used examined historical and archaeological evidence in order to estimate the time of the first VARV infection of humans. Dixon (1962) invoked the fact that smallpox is not mentioned in the Old and New Testament writings or in any of the surviving literature of classical Greece and Rome as evidence for a recent origin, at least in the Mediterranean region. The earliest unmistakable descriptions of smallpox in historical records are said to be from the 4th Century A.D. in China and the 7th Century A.D. in India and the Mediterranean (Li et al. 2007). On the other hand, smallpox-like lesions have been described from Egyptian mummies dating from 1580-1100 B.C., and ancient Chinese and Indian medical treatises from the 2nd Millennium B.C. describe smallpox-like symptoms (Li et al. 2007).
A number of recent studies have used molecular sequence data to estimate divergence times within the phylogeny of VARV and related viruses, but these are highly dependent on the assumptions used in calibration. Figure 2 shows the NJ tree of VARV isolates, rooted with a Taterapox isolate. The tree was constructed on the basis of the maximum composite likelihood distance (Tamura et al. 2007) in 132 aligned protein-coding genes. Like other phylogenetic analyses of VARV sequences (Esposito et al. 2006; Li et al. 2007), the tree shows two major clades of VARV sequences: primary clade I (P-I) and primary clade II (P-II), both of which received 100% bootstrap support (Figure 2). P-II includes two subclades: (1) sequences from South America, traditionally designated alastrim minor, and characterized by a milder disease phenotype; and (2) sequences from West Africa (Figure 2).
Figure 2.

NJ tree (topology only) of smallpox virus isolates based on maximum composite likelihood distance at 132 concatenated orthologous protein-coding genes (118, 992 aligned sites). The tree was constructed on the basis of the maximum composite likelihood distance. Numbers on the branches represent percentages of 1000 bootstrap samples supporting each branch; only values ≥ 95 % are shown.
A key component of estimating divergence times is providing an estimate of the neutral substitution rate, which is expected to equal the mutation rate. Synonymous sites in coding regions provide the best estimate of the neutral substitution rate. Although there may be some purifying selection at synonymous sites because of constraints on codon usage or other such factors, synonymous sites are much less subject to purifying selection than are nonsynonymous sites. Moreover, in viruses, sites outside coding regions are often much more constrained than are synonymous sites in coding regions, apparently because the former play important roles in regulating gene expression (Hughes 2007a, 2009; Hughes and Piontkivska 2008).
Experimentally derived estimates of the mutation rates of DNA viruses are generally much lower than those of RNA viruses (Drake et al. 1998; Drake and Holland 1999). For example, the spontaneous mutation rate per base pair per generation of human immunodeficiency virus 1 (HIV-1) has been estimated at about 2 × 10−5 mutations per base pair per replication, whereas estimates for three bacteriophages with DNA genomes ranged from 2 × 10−8 to 7 × 10−7 mutations per base pairs per replication (Drake et al. 1998). In order to use experimentally derived estimates in molecular clock studies, it is necessary to have an estimate of the average number of replications per unit time, a parameter that may be difficult to estimate for many viruses.
An alternative approach is to use some external calibration from epidemiological history or from the fossil record of host species. As an example of the former, Li et al. (2007) used time of isolation of various VARV isolates to calibrate major events in the VARV phylogeny. This approach led to what are almost certainly underestimates of the divergence times of major clades of VARV; alternatively, those authors proposed a calibration based on the separation of the South American VARV from West African P-II (Li et al. 2007). In a paper published in Russian, Babkin and Shchelkunov (2006) suggested that the separation of alastrim from West African P-II could be dated from the beginning of the African slave trade in the 16th Century. Esposito and colleagues (2006) suggested that this event might be dated to the 18th Century, which saw importation to South America of Yoruba slaves from West Africa.
Using 132 protein-coding genes, we estimated the number of neutral substitutions per site (Table 1): (1) the rate of substitution at four-fold degenerate sites (Li 1993); and (2) the number of synonymous substitutions per synonymous site (dS; Nei and Gojobori 1986). The two methods yielded nearly identical results for mean evolutionary distances between the major clades of VARV sequences and between VARV and Taterapox virus (Table 1). Using 300 years for the divergence of alastrim from West African P-II, we obtained a substitution rate of 6 × 10−6 substitutions/site/year (Table 1). Assuming 500 years for the divergence of alastrim from West African P-II, we obtained a substitution rate of 4 × 10−6 substitutions/site/year (Table 1). These rate estimates place the separation of the P-I and P-II clades 700–1000 years ago and the divergence of VARV from Taterapox 3000–4000 years ago, times that are consistent with archaeological and historical evidence regarding the appearance of smallpox as a human disease.
Table 1.
Estimates of VARV divergence times.
| Divergence | Substitutions per site | Times estimates (years before present) | |||
|---|---|---|---|---|---|
| 4-fold degenerate sites | Synonymous sites | rate = 6 × 10−6 subs/site/year | rate = 4 × 10−6 subs/site/year | rate = 3.5 × 10−8 subs/site/year | |
| P-II W. Africa vs. alastrim | 0.00355 ± 0.00029 | 0.00374 ± 0.00029 | 300a | 500a | 5,000 |
| P-I vs. P-II | 0.00794 ± 0.0068 | 0.007970.± 0.00042 | 700 | 1,000 | 10,000 |
| VARV vs. Taterapox virus | 0.03476 ± 0.00146 | 0.03497 ± 0.00102 | 2900 | 4,000 | 50,000 |
Calibration
Among viruses with large dsDNA genomes, more attention has been devoted to estimating substitution rates in herpesviruses than in poxviruses. Sakaoka and colleagues (1994) obtained an estimate of 3.5 × 10−8 substitutions per site per years for Herpes simplex virus type 1 (HSV-1) based on restriction site polymorphisms. This value is surprisingly low in comparison to our estimates of the substitution rate in VARV. The analysis of restriction site polymorphism might be expected to produce a lower estimate than examination of synonymous sites, since the former method treats both synonymous and nonsynonymous sites equally. However, it is unclear whether the difference in methods can explain a difference of two orders of magnitude. It is also possible that the synonymous substitution rate of poxviruses is indeed substantially greater than that of herpesviruses as a result of shorter generation times in the former; but again it is unclear whether a generation effect is sufficient to explain a 100-fold difference in synonymous substitution rate.
When the rate of 3.5 × 10−8 substitutions per site per year was applied to divergence events in the VARV phylogeny, it yielded very early estimates of divergence times, placing the VARV-Taterapox virus divergence 50,000 years ago (Table 1). Although these earlier times do not fit well with the appearance of smallpox in historical and archaeological records, one cannot definitively rule out this interpretation at present. It is possible that smallpox existed in non-literate human populations long before its appearance in recorded history. Thus, in our present state of knowledge, we are left with two alternatives: either the synonymous substitution rate (and possibly also the mutation rate) of poxviruses is much higher than that of herpesviruses, or the origin of VARV long predates its appearance in archaeological or historical records. Neither of these alternatives seems wholly satisfactory. Clearly we need a better understanding of mutation and synonymous substitution rates in DNA viruses as a whole, not only in poxviruses. For example, it is possible that the substitution rate in herpesviruses has been substantially underestimated. Only when we have a more complete understanding of mutation and synonymous substitution rates of DNA viruses can we provide a reasonable calibration for the molecular clock in the case of VARV.
4. Horizontal Gene Transfer
4.1. Poxvirus “Capture” of Host Genes
Interest in horizontal transfer of host genes to poxviruses was sparked by the discovery of homologs of vertebrate immune system signaling molecules in the genomes of poxviruses and herpesviruses (McFadden 1995). Hughes and Friedman (2005b) conducted a systematic search for horizontally transferred genes by phylogenetic methods, while Bratke and McLysaght (2008) added an examination of conserved syntenic relationships around putative horizontally transferred genes with the goal of distinguishing between single and multiple transfer events. The latter authors’ method depended on two implicit assumptions (1) that incorporation of the same host gene into the same location in the viral genome is unlikely to occur independently more than once; and (2) that, once horizontally transferred, genes are not likely to change locations in poxvirus genomes.
Some of the poxvirus genes with homologs to vertebrate genes clearly represent ancient genes that were probably present since the origin of the poxviruses or even the origin of the NCLDV clade (Bugert and Darai 2000; Hughes 2002; Hughes and Friedman 2005b). In phylogenetic analyses, evidence of an ancient origin is provided by a topology in which all poxvirus homologs cluster together, apart from those of cellular organisms (Hughes 2002; Hughes and Friedman 2005b). On the other hand, horizontal transfer is indicated by a topology in which one or more viral genes cluster with certain genes of cellular organisms but not others, particularly when the viral gene clusters with that of its host.
4.2. Interleukin-10 Family
Viral homologs of vertebrate host immune system genes include several members of the interleukin-10 (IL-10) family. This family of cytokines also includes human IL-19, IL-20, and IL-24. In humans, these four proteins are all encoded by genes mapping to a 195-kb region on chromosome 1q32 (Commins et al. 2008). All four of these cytokines are involved in regulating immune responses; but their roles are complex and so far not completely elucidated (Commins et al. 2008). The first viral IL-10 homolog to be discovered was in Human herpesvurus 4 (also known as Epstein-Barr virus or EBV), and this molecule is perhaps the best studied viral cytokine homolog (Kanai et al. 2007). The IL-10 molecule of EBV acts together with host IL-10 to down-regulate the host immune response early in EBV infection of host B cells (Kanai et al. 2007).
Figure 3 shows a phylogenetic tree of vertebrate IL-10, IL-19, IL-20, and IL-24 sequences, along with homolog from poxviruses of mammals. [This tree was based on the Poisson-corrected amino acid distance, as were the trees shown in Figures 4 and 5, below. This distance was used because the number of aligned amino acid sites available for analysis was small and thus more complicated models were not necessarily applicable. In each of the three datasets, trees based on the JTT model and the assumption that rates varied among sites following a gamma distribution showed a similar topology but sometimes poorer resolution (not shown). Estimation of the shape parameter of the gamma distribution yielded high values in all three data sets, indicating a low rate variation among sites and thus a close approximation to the Poisson distribution.]
Figure 3.

NJ tree of vertebrate interleukin-10 (IL-10) cytokine family members and homologs from poxviruses of mammals. The tree was based on the Poisson-corrected amino acid distance at 110 aligned sites. Numbers on the branches represent percentages of 1000 bootstrap samples supporting each branch; only values ≥ 50 % are shown.
Figure 4.

NJ tree of ankyrin repeat-containing proteins from BPSV and ORFV. The tree was based on the Poisson-corrected amino acid distance at 442 aligned sites. Numbers on the branches represent percentages of 1000 bootstrap samples supporting each branch; only values ≥ 50 % are shown.
Figure 5.

Figure 3. NJ tree of vertebrate VEGFA and PDGF and homologs from poxviruses of mammals. The tree was based on the Poisson-corrected amino acid distance at 116 aligned sites. Numbers on the branches represent percentages of 1000 bootstrap samples supporting each branch; only values ≥ 50 % are shown.
The tree showed two major clusters, separated by a strongly supported (99% bootstrap support) internal branch: (1) IL-10 from both tetrapods (including birds and mammals) and bony fishes; and (2) tetrapod IL-19, IL-20, and IL-24 together with bony fish homologs (Figure 3). In the latter cluster, the fish sequences clustered outside terrapod IL-19, IL-20, and IL-24, though with low bootstrap support (Figure 3). This topology suggests that IL-19, IL-20, and IL-24 arose by gene duplication in the tetrapod lineage after its separation from the bony fishes. Because each of the two clusters included sequences from both tetrapods (birds and mammals) and bony fishes (Figure 3), the tree supports the hypothesis that these two major clusters originated prior to the divergence of tetrapods and bony fishes.
Poxviruses homologs of vertebrate members of this family of cytokines fell in both of the two major clusters (Figure 3). Sequences from Yaba-like disease virus (YLDV) and Tanapox virus (TANV) belonged to the cluster including IL-19, IL-20, and IL-24 (Figure 3). YLDV and TANV sequences clustered together and both clustered with mammalian IL-24, although the bootstrap support was not strong (53%; Figure 3). It would be interesting to know whether the function of the latter molecules resembles that of host IL-24, which activates the Stat-1 and Stat-3 transcription factors, thereby contributing to the survival and proliferation of cells in processes such as wound healing (Wang and Liang 2005). There is experimental evidence that, in the case of YLDV, its interleukin-like molecule serves to reduce the virulence of viral infection (Bartlett et al. 2004).
In the phylogenetic tree, bovine papular stomatitis virus (BPSV) and Orf virus (ORFV) sequences belonged to the IL-10 cluster (Figure 3). In fact, ORFV IL-10 clustered with that of its host, the sheep, with 99% bootstrap support (Figure 3). BPSV IL-10 also clustered with that of its bovine host with more modest (77%) bootstrap support (Figure 3). In a recent paper, Odom and colleagues (2009) presented a tree based on DNA sequences in which ORFV and BPSV IL-10 sequences clustered together, although with only modest bootstrap support (78%). However, a tree based on DNA sequences is likely to be unreliable in this case because synonymous sites are saturated with changes in a number of comparisons.
As noted by Bratke and McLysaght (2008), the genes encoding IL-10 in BPSV and ORFV occur at corresponding map locations, which would seem to argue for a single event of horizontal gene transfer into the common ancestor of ORFV and BPSV. If so, the clustering pattern observed in our phylogeny (Figure 3) might be explained by convergent or parallel evolution at the amino acid sequence level, whereby the viral IL-10 has come to resemble that of its host. However, an examination of the sequences suggests that the hypothesis of convergent or parallel evolution at the sequence level is unlikely in this case.
The conserved portion of the IL-10 sequence, which includes the receptor binding sites, corresponds to residues 20–151 of the human mature IL-10 chain (Josephson et al. 2001). In this region of IL-10 of bovine, sheep, BPSV, and ORFV, there are six variable amino acid sites that are informative for the phylogeny of the four taxa: residues 44, 46, 123, 129, 130, and 152 of the mature mammalian IL-10 chain. In all six sites, BPSV has the same residue as bovine and ORFV the same residue as sheep. Such extensive convergent/parallel evolution has not, to our knowledge, been documented elsewhere (Hughes 1999). Moreover, only two of these sites (positions 44 and 46) are involved in the interface of IL-10 with its receptor (Josephson et al. 2001). Convergent or parallel evolution might be plausibly explained in the case of these two contact residues, but it is harder to account for in the case of the other four residues. Rather than convergent or parallel substitutions occurring at a total of six amino acid sites, it seems more parsimonious to conclude that incorporation of a host IL-10 gene in a corresponding map location has occurred independently in the BPSV and ORFV lineages.
In both BPSV and ORFV, the IL-10 gene is located between two genes encoding ankyrin repeat-containing proteins. Genes encoding ankyrin repeat-containing proteins are found in two separate locations in both the BPSV and ORFV genomes. In BPSV, these groups of genes include the following predicted open reading frames (orfs): (1) orfs 3, 4, and 8; and (2) orfs 123, 126, 128, and 129. In ORFV, the former group is represented by just orf8, while the latter group includes orfs 123, 126, 128, and 129. In both viruses, the IL-10 gene is located between orfs 126 and 128. A phylogenetic tree of the ankyrin repeat-containing proteins of BPSV and ORFV supports the hypothesis that orf 126 of BPSV is orthologous to orf 126 of ORFV and that orf 128 of BPSV is orthologous to ORF 128 of ORFV (Figure 4). The phylogeny of ankyrin repeat-containg proteins thus supports the hypothesis that the IL-10 genes of BPSV and ORFV are inserted in precisely syntenic locations.
4.3. VEGFA
It is possible that the clusters of genes encoding ankyrin repeat-containg proteins are particularly prone to recombinational events, including horizontal gene transfer. Both BPSV and ORFV encode homologs of vertebrate vascular endothelial growth factor A (VEGFA). In BPSV, the VEGFA gene (orf 6) is located in within the first group of linked ankyrin repeat-containing proteins, whereas in ORFV the VEGFA gene (orf 132) is located close to the second group of ankyrin repeat-containing proteins.
The VEGFA genes of BPSV and ORFV provide a good example of the complexities one encounters in attempting to unravel the evolutionary history of horizontal gene transfer events in the poxviruses. Figure 5 shows a phylogenetic tree of VEGFA from these viruses and selected vertebrates, rooted by sequences of the related protein PDGF. In the phylogenetic tree, VEGFA homologs from BPSV and ORFV clustered together and outside mammalian and chicken VEGFA sequences; and this topology was supported by a significant (98% bootstrap support) branch (Figure 5). Taken at face value, the tree supports the hypothesis that there was a single ancient event of horizontal transfer of a VEGFA to the ancestor of BPSV and ORFV.
It might be argued that the clustering of the two viral VEGFA sequences represents long-branch attraction. The maximum likelihood (ML) method of phylogenetic reconstruction may be less prone to long-branch than NJ, (Anderson and Swofford 2004; Hughes and Friedman 2007). However, the present case ML produced the same topology as NJ (not shown). Whether or not any phylogenetic method can reconstruct the true tree in this case, it is nonetheless noteworthy that the two viral VEGFA sequences (Figure 5) do not show any hint of the close similarity to the corresponding host sequences that was seen in the case of IL-10 (Figure 3). Thus, the pattern of similarity to the host genes differs dramatically between VEGFA and IL-10.
These examples suggest that it may be misleading to use either phylogenetic analysis alone or map position alone as a criterion for determining the number of horizontal transfer events. In the case of IL-10, the most reasonable hypothesis seems to be that two independent events of insertion of a host-derived IL-10 gene in the same genomic location occurred independently in BPSV and ORFV. In the case of VEGFA, there may have been a single horizontal transfer event, but there have been subsequent genomic rearrangements that have led to the VEGFA genes occupying different map positions in the two viruses. The latter events may also have been involved in the reduction of the number of genes encoding ankyrin repeat-containing proteins in ORFV compared to BPSV (Figure 4).
5. Population Biology
Because of the comparatively large genome sizes of poxviruses, relatively few studies so far have addressed the population genetics of poxviruses at the whole-genome level, although recent technological advances should soon make such studies affordable. To date, most population surveys have focused on individual genes or gene regions. Hughes and Hughes (2007) compared the sequences obtained by a large number of such studies of both RNA viruses and DNA viruses, including a number of poxviruses. These broad-scale comparisons showed that purifying selection is more efficient overall in RNA viruses than DNA viruses (Hughes and Hughes 2007).
To provide a further test of this hypothesis, we estimated dS and dN (Nei and Gojobori 1986) pairwise between 132 aligned orthologous genes of 46 VARV genomes (for accession numbers see Figure 2). These VARV genomes were sequenced by Esposito et al. (2006), who noted a relatively low level of sequence polymorphism in comparison to many other viral sequence data sets. In order to compare VARV with an RNA virus with similarly low polymorphism, we estimated dS and dN pairwise among the polyprotein genes of 53 West Nile virus (WNV) isolates from the North American clade (Hughes et al. 2007). WNV is a flavivirus that has spread rapidly in the Americas since it was first observed in the Western Hemisphere in 1999 (Nash et al. 2001).
When dN was plotted against dS for pairwise comparisons among VARV genomes, the points formed two distinct clusters (Figure 6). These two clusters correspond to comparisons within and between the two major VARV clades (P-I and P-II; Figure 2). When dN was plotted against dS for pairwise comparisons among WNV genomes, the points were in general shifted to the right relative to VARV, indicating that dN was generally lower relative to dS in WNV than in VARV (Figure 6). Mean dN/dS was 0.230 in the case of VARV and 0.066 in the case WNV. Because of the non-independence of pairwise comparisons, we used a randomization test to test the equality of these means by creating pseudo-datasets by sampling (with replacement) from the data 1000 times. By this test the difference in means was highly significant (P < 0.001). Values of dS for the most distant comparisons among WNV isolates were higher than any observed in VARV (Figure 6A). However, even when we excluded at WNV comparisons for which dS was greater than the highest value observed in VARV (0.0086; Figure 6), mean dN/dS in the case of WNV (0.098) was still less than half that of VARV; and the difference was highly significant (P < 0.001; randomization test).
Figure 6.
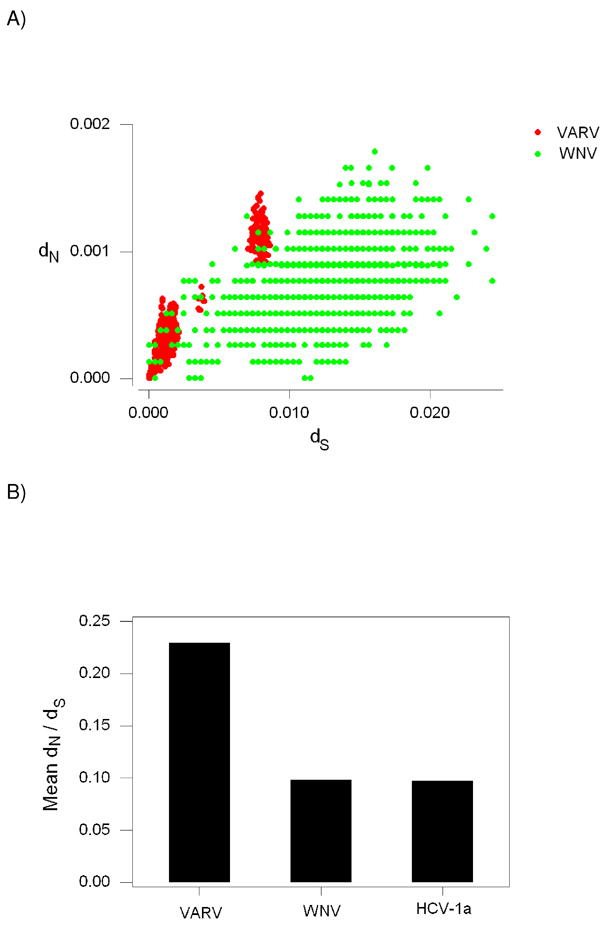
(A) Plot of the number of nonsynonymous substitutions per nonsynonymous site (dN) against the number of synonymous substitutions per synonymous site (dS) for pairwise comparisons among: (1) 132 orthologous protein-coding genes of Variola virus (VARV; red); and (2) complete polyprotein genes of North American West Nile virus (WNV; green). (B) Mean dN/dS in pairwise comparisons of VARV, WNV, and Hepatitis C virus 1a (HCV-1a).
It might be argued that the higher dN/dS in VARV reflects positive selection, but there was no evidence to support such a hypothesis. In the VARV data overall mean dS for all pairwise comparisons (typically designated πS) was 0.00289 ± 0.00013, while overall mean dN (designated πN) was 0.00050 ± 0.00003. The difference between πS and πN for VARV was highly significant (P < 0.001; Z-test). Similarly, when πS and πN were computed for individual genes, the former was invariably larger, and in most cases significantly so (not shown). In the case of WNV, πS (0.01224 ± 0.00098) was also significantly greater than πN (0.00073 ±0.00010; P < 0.001; Z-test).
Previous studies have suggested that purifying selection is generally more efficient in RNA viruses with arthropod vectors than in other RNA viruses (Woelk and Holmes 2002; Hughes and Hughes 2007). The causes of this difference are not obvious. It is possible that exposure to both arthropod and vertebrate hosts increases the intensity of purifying selection. Another possible explanation is that the effective population sizes of RNA viruses with arthropod vectors are in general larger than of those transmitted by other means. In order to test whether the lower dN relative to dS in WNV than in VARV was due only to the fact that WNV is insect-borne, we also included a comparison with another flavivirus, Hepatitis C virus 1a (HCV-1a), which is not arthropod-transmitted (Figure 6B). Mean dN/dS was significantly greater in VARV than in either WNV or HCV-1a (P < 0.001 in both cases; randomization test; Figure 6B). On the other hand, mean dN/dS did not differ significantly between the two flaviviruses (Figure 6B).
These results support the hypothesis that purifying selection is more efficient at removing deleterious mutations in flaviviruses than in VARV, consistent with other results (Hughes and Hughes 2007). One possible explanation for this difference is that the rate of recombination per nucleotide is higher in RNA viruses than in large DNA viruses like poxviruses (Hughes and Hughes 2007). Such a conclusion is consistent with the prediction that genomes or genomic regions with low recombination rates will in general show elevated accumulation of slightly deleterious mutations, which in coding regions will be mainly nonsynonymous. This prediction is supported by data from sex chromosomes (Berlin and Ellegren 2006; Wykoff et al. 2002), mitochondrial genomes (Nachman et al. 1994, 1996; Rand and Kann 1996) and Drosophila genomic regions with different recombination rates (Haddrill et al. 2007).
An alternative explanation relates to the fact that many dsDNA viruses have large genomes encoding multiple genes. The presence of multiple genes may confer a certain degree of functional redundancy, allowing for a relaxation of some degree of functional constraints. However, the DNA virus datasets analyzed by Hughes and Hughes (2007) included many from small DNA viruses, such as Papillomaviridae and Circoviridae. The latter showed patterns of polymorphism similar to those seen in large DNA viruses such as poxviruses and herpesviruses. Thus, the comparative inefficiency of purifying selection in DNA viruses does not appear to be an effect of genome size alone.
6. Conclusion
This brief review has shown that the study of poxviruses raises a number of intriguing evolutionary questions, which are far from being revolved. These questions include the following: the origin of poxviruses and dsDNA viruses in general; the origin of specific poxviruses such as VARV; the role of horizontal gene transfer in poxvirus evolution; and the population processes that explain patterns of nucleotide sequence polymorphism. Modern evolutionary genetics, thanks to an increased understanding of the stochastic nature of the evolutionary process (Kimura 1983; Nei 1987), provides a powerful conceptual arsenal for understanding population processes. These concepts are particularly useful when applied with an appreciation for the realities of each species natural history, including (in the case of viruses infecting humans) time of origin as human pathogens, existence of non-human reservoirs, modes of transmission, and so forth. Unfortunately, progress in evolutionary biology has been stalled by the widespread use of invalid statistical methods and by a mentality inherited from the early days of Neo-Darwinism that sees natural selection as an all-powerful force fine-tuning every aspect of the phenotype (Hughes 2007b). This review is written in the hope that virologists will take a lead in combating such outworn attitudes and in advancing a genuine understanding of the process that shape viral evolution.
Supplementary Material
Acknowledgments
This research was supported by grant GM43940 from the National Institutes of Health to A.L.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson FE, Swofford DL. Should we be worried about long-branch attraction in real data sets? : Investigations using metazoan 18S rRNA. Mol Phyl Evol. 2004;33:440–451. doi: 10.1016/j.ympev.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Artenstein AW, Grabenstein JD. Smallpox vaccines for biodefense: need and feasibility. Expert Rev Vaccines. 2008;7:1225–1237. doi: 10.1586/14760584.7.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babkin IV, Shchelkunov SN. [The time scale in poxvirus evolution] Mol Biol (Mosk) 2006;40:20–24. doi: 10.1134/s0026893306010043. [in Russian]. [DOI] [PubMed] [Google Scholar]
- Bartlett NW, Dumoutier L, Renauld JC, Kotenko SV, McVey CE, Lee HJ, Smith GL. A new member of the interleukin 10-related cytokine family encoded by a poxvirus. J Gen Virol. 2004;85:1401–1412. doi: 10.1099/vir.0.79980-0. [DOI] [PubMed] [Google Scholar]
- Baxby D. Jenner’s Smallpox Vaccine: the Riddle of the Origin of Vaccinia Virus. Heinemann; London: 1981. [Google Scholar]
- Berlin S, Ellegren H. Fast accumulation of nonsynonymous mutations on the female-specific W chromosome in birds. J Mol Evol. 2006;62:66–72. doi: 10.1007/s00239-005-0067-6. [DOI] [PubMed] [Google Scholar]
- Bratke KA, McLysaght A. Identification of multiple independent horizontal gene transfers into poxviruses using a comparartive genomics approach. BMC Evolutionary Biology. 2008;8:67. doi: 10.1186/1471-2148-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugert JJ, Darai G. Poxvirus homologies of cellular genes. Virus Genes. 2000;21:111–133. [PubMed] [Google Scholar]
- Commons S, Steinke JW, Borish L. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J Allergy Clin Immunol. 2008;121:1108–1111. doi: 10.1016/j.jaci.2008.02.026. [DOI] [PubMed] [Google Scholar]
- Diamond J. Guns, Germs, and Steel. Norton; New York: 1997. [Google Scholar]
- Dixon CW. Smallpox. Churchill; London: 1962. [Google Scholar]
- Drake JW, Holland JJ. Mutation rates among RNA viruses. Proc Natl acad Sci USA. 1999;96:13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop LR, Oehlberg KA, Reid JJ, Avci D, Rosengard AM. Variola virus immune evasion proteins. Microbes Infect. 2003;5:1049–1056. doi: 10.1016/s1286-4579(03)00194-1. [DOI] [PubMed] [Google Scholar]
- Esposito JJ, Sammons SA, Frace AM, Osborne JD, Olsen-Rasmussen M, Zhang M, Govril D, Damon IK, Kline R, Lakes M, Li Y, Smith GL, Meyer H, LeDur JW, Wohlhueter RM. Genome sequence diversity and clues to the evolution of variola (smallpox) virus. Science. 2006;313:807–812. doi: 10.1126/science.1125134. [DOI] [PubMed] [Google Scholar]
- Everett H, McFadden G. Poxviruses and apoptosis: a time to die. Curr Opin Microbiol. 2002;5:395–402. doi: 10.1016/s1369-5274(02)00340-5. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Fenner F, Kerr PJ. Evolution of the poxviruses, including the coevolution of virus and host in myxomatosis. In: Morse SS, editor. The Evolutionary Biology of Viruses. Raven Press; New York: 1994. pp. 273–292. [Google Scholar]
- Gubser C, Hué S, Kellam P, Smith GL. Poxvirus genomes: a phylogenetic analysis. J Gen Virol. 2004;85:105–117. doi: 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- Haddrill PR, Halligan DL, Tomaras D, Charlesworth B. Reduced efficacy of selection in regions of the Drosophila genome that lack crossing over. Genome Biology. 2007;8:R18. doi: 10.1186/gb-2007-8-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Adaptive Evolution of Genes and Genomes. Oxford University Press; New York: 1999. [Google Scholar]
- Hughes AL. Origin and evolution of viral interleukin-10 and other DNA virus genes with vertebrate homologues. J Mol Evol. 2002;54:90–101. doi: 10.1007/s00239-001-0021-1. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Evidence for abundant slightly deleterious polymorphisms in bacterial populations. Genetics. 2005;169:553–558. doi: 10.1534/genetics.104.036939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Micro-scale signature of purifying selection in Marburg virus genomes. Gene. 2007a;392:266–272. doi: 10.1016/j.gene.2006.12.038. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level. Heredity. 2007b;99:364–373. doi: 10.1038/sj.hdy.6801031. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Near neutrality: leading edge of the neutral theory of molecular evolution. Ann NY Acad Sci. 2008;1133:162–179. doi: 10.1196/annals.1438.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Relaxation of purifying selection on live attenuated vaccine strains of the family Paxamyxoviridae. Vaccine. 2009;27:1685–1690. doi: 10.1016/j.vaccine.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Variation in the pattern of synonymous and nonsynonymous difference between two fungal genomes. Mol Biol Evol. 2005a;22:1320–1324. doi: 10.1093/molbev/msi120. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Poxvirus genome evolution by gene gain and loss. Mol Phyl Evol. 2005b;35:186–195. doi: 10.1016/j.ympev.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. The effect of branch lengths on phylogeny: an empirical study using highly conserved orthologs from mammalian genomes. Mol Phyl Evol. 2007;45:81–88. doi: 10.1016/j.ympev.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Codon-based tests of positive selection, branch lengths, and the evolution of mammalian immune system genes. Immunogenetics. 2008;60:495–506. doi: 10.1007/s00251-008-0304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Hughes MA. More effective purifying selection in RNA viruses than in DNA viruses. Gene. 2007;404:117–125. doi: 10.1016/j.gene.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Piontkivska H. Nucleotide sequence polymorphism in circoviruses. Infection, Genetics, and Evolution. 2008;8:130–138. doi: 10.1016/j.meegid.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Verra F. Extensive polymorphism and ancient origin of Plasmodium falciparum. Trends Parasitol. 2002;18:348–351. doi: 10.1016/s1471-4922(02)02290-0. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Friedman R, Murray M. Genomewide pattern of synonymous nucleotide substitution in two complete genomes of Mycobacterium tuberculosis. Emerg Infect Dis. 2002;8:1342–1346. doi: 10.3201/eid0811.020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Packer B, Welsch R, Bergen AW, Chanock SJ, Yeager M. Widespread purifying selection at polymorphic sites in human protein-coding loci. Proc Natl Acad Sci USA. 2003;100:15754–15757. doi: 10.1073/pnas.2536718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Piontkivska H, Krebs KC, O’Connor DH, Watkins DI. Within-host evolution of CD8+-TL epitopes encoded by overlapping and non-overlapping reading frames of simian immunodeficiency virus. Bioinformatics. 2005;21(Suppl 3):iii39–iii44. doi: 10.1093/bioinformatics/bti1203. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Piontkivska H, Foppa I. Rapid fixation of a distinctive sequence motif in the 3′ noncoding region of the clade of West Nile virus invading North America. Gene. 2007;399:152–161. doi: 10.1016/j.gene.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irausquin SJ, Hughes AL. Distinctive pattern of sequence polymorphism in the NS3 protein of hepatitis C virus type 1b reflects conflicting evolutionary pressures. J Gen Virol. 2008;89:1921–1929. doi: 10.1099/vir.0.2008/000992-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Aravind L, Koonin EV. Common origin of four diverse families of large eukaryotic DNA viruses. J Virol. 2001;75:11720–11734. doi: 10.1128/JVI.75.23.11720-11734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Balaji S, Koonin EV, Aravind L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Research. 2006;117:156–184. doi: 10.1016/j.virusres.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Josephson K, Logsdon NJ, Walter MR. Crystal structure of the IL-10/IL-10R1 complex reveals a shared receptor binding site. Immunity. 2001;14:35–46. doi: 10.1016/s1074-7613(01)00169-8. [DOI] [PubMed] [Google Scholar]
- Kanai K, Satoh Y, Yamanaka H, Kawaguchi A, Horie K, Sugata K, Hoshikawa Y, Sata T, Sairenji T. The vIL-10 gene of Epstein-Barr virus (EBV) is conserved in a stable manner except for a few point mutations in various EBV isolates. Virus Genes. 2007;35:563–569. doi: 10.1007/s11262-007-0153-5. [DOI] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution. Cambridge University Press; Cambridge: 1983. [Google Scholar]
- Koonin EV, Senkevich TG, Dolja VV. The ancient virus world and evolution of cells. Biol Direct. 2006;1:9. doi: 10.1186/1745-6150-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz EJ, Wang C, Upton C. Poxviruses: past, present, and future. Virus Research. 2006;117:105–118. doi: 10.1016/j.virusres.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Li WH. Unbiased estimates of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- Li Y, Carroll DS, Gardner SN, Walsh MW, Vitalis EA, Damon IK. On the origin of smallpox: Correlating variola phylogenetics with historical smallpox records. Proc Natl Acad Sci USA. 2007;40:15787–15792. doi: 10.1073/pnas.0609268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourie B, Nakano JH, Kemp GE, Setzer HW. Isolation of poxvirus from an African rodent. J Infect Dis. 1975;132:677–681. doi: 10.1093/infdis/132.6.677. [DOI] [PubMed] [Google Scholar]
- McFadden G. Viroceptors, Virokines, and Related Immune Modulators Encoded by DNA Viruses. Landes; Austin TX: 1995. [Google Scholar]
- McLysaght A, Baldi PF, Gaut BS. Extensive gene gain associated with adaptive evolution of poxviruses. Proc Natl Acad Sci USA. 2003;26:15655–15660. doi: 10.1073/pnas.2136653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse S. Toward an evolutionary biology of viruses. In: Morse SS, editor. The Evolutionary Biology of Viruses. Raven Press; New York: 1994. pp. 1–28. [Google Scholar]
- Moss B. Poxvirus entry and membrane fusion. Virology. 2006;344:48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Moss B, Shisler JL. Immunology 101 at poxvirus U: immune evasion genes. Semin Immunol. 2001;13:59–66. doi: 10.1006/smim.2000.0296. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Boyer SN, Aquadro CF. Nonneutral evolution at the mitochondrial NADH dehydrogenase subunit 3 gene in mice. Proc Natl Acad Sci USA. 1994;91:6364–6368. doi: 10.1073/pnas.91.14.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Brown WM, Stoneking M, Aquadro CF. Nonneutral mitochondrial DNA variation in humans and chimpanzees. Genetics. 1996;142:953–963. doi: 10.1093/genetics/142.3.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash D, Mostashari F, Fine A, Miller J, O’Leary D, Murray K, Huang A, Rosenberg A, Greenberg A, Sherman M, Wong S, Layton M. The outbreak of West Nile virus infection in the New York City area in 1999. New England J Med. 2001;344:1807–1814. doi: 10.1056/NEJM200106143442401. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press; New York: 1987. [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nowak MA, May RM. Virus Dynamics. Oxford University Press; Oxford: 2000. [Google Scholar]
- O’Connor DH, McDermott AB, Krebs AC, Dodds EJ, Miller JE, Gonzalez EJ, Jacoby TJ, Yant L, Piontkivska H, Pantophlet R, Burton DR, Rehrauer WR, Wilson N, Hughes AL, Watkins DI. A dominant role for CD8+-T-lymphocyte selection in Simian Immunodeficiency Virus sequence variation. J Virol. 2004;78:14012–14022. doi: 10.1128/JVI.78.24.14012-14022.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom MR, Hendrickson RC, Lefkowitz EJ. Poxvirus protein evolution: family wide assessment of possible horizontal gene transfer events. Virus Research. 2009;144:233–249. doi: 10.1016/j.virusres.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sakaoka H, Kurita K, Iida Y, Takada S, Umene K, Kim YT, Ren CS, Nahmias AJ. Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. J Gen Virol. 1994;75:513–527. doi: 10.1099/0022-1317-75-3-513. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Schneider A, Souvorov A, Sabath N, Landan G, Gonnet GH, Graur D. Estimates of positive Darwinian selection are inflated by errors in sequencing, annotation, and alignment. Genome Biol Evol. 2009;1:114–118. doi: 10.1093/gbe/evp012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seet BT, Johnston JB, Brunetti CR, Barrett JW, Everett H, Cameron C, Sypula J, Nazarian SH, Lucas A, McFadden G. Poxviruses and immune evasion. Annu Rev Immunol. 2003;21:377–423. doi: 10.1146/annurev.immunol.21.120601.141049. [DOI] [PubMed] [Google Scholar]
- Shisler JL, Moss B. Immunology at poxvirus U: avoiding apoptosis. Semin Immunol. 2001;13:67–72. doi: 10.1006/smim.2000.0297. [DOI] [PubMed] [Google Scholar]
- Smith GL. Vaccinia: virus, vector and vaccine. In: Dimock NJ, Griffiths PD, Madeley CR, editors. Control of Virus Diseases. Cambridge University Press; Cambridge: 1990. pp. 77–121. [Google Scholar]
- Smith SA, Kotwal GJ. Immune response to poxvirus infections in various animals. Crit Rev Microbiol. 2002;28:149–185. doi: 10.1080/1040-840291046722. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Van Regenmortel MH, Fauquest CM, Bishop DH, Carstens EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, McGoech DJ, Pringle CR, Wickner RB. Virus Taxonomy. Academic Press; San Diego: 2000. [Google Scholar]
- Wang M, Liang P. Interlekin-24 and its receptors. Immunology. 2005;114:166–170. doi: 10.1111/j.1365-2567.2005.02094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. Adaptation and Natural Selection. Princeton: Princeton University Press; 1966. [Google Scholar]
- Woelk CH, Holmes EC. Reduced positive selection in vector-borne RNA viruses. Mol Biol Evol. 2002;19:2333–2336. doi: 10.1093/oxfordjournals.molbev.a004059. [DOI] [PubMed] [Google Scholar]
- Wyckoff GJ, Joyce L, Wu CI. Molecular evolution of functional genes on the mammalian Y chromosome. Mol Biol Evol. 2002;19:1633–1636. doi: 10.1093/oxfordjournals.molbev.a004226. [DOI] [PubMed] [Google Scholar]
- Xing K, Deng R, Wang J, Feng J, Huang M, Wang X. Genome-based phylogeny of poxviruses. Intervirology. 2006;49:207–214. doi: 10.1159/000090790. [DOI] [PubMed] [Google Scholar]
- Zhang J. Frequent false detection of positive selection by the likelihood method with branch-site models. Mol Biol Evol. 2004;21:1332–1339. doi: 10.1093/molbev/msh117. [DOI] [PubMed] [Google Scholar]
- Zhang L, Villa NY, McFadden G. Interplay between poxviruses and the cellular ubiquitin/ubiquitin-like pathways. FEBS Lett. 2009;583:607–614. doi: 10.1016/j.febslet.2009.01.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.