

Abstract
Endothelial barrier dysfunction contributes to morbidity in sepsis. We tested the hypothesis that raising the intracellular ascorbate concentration protects the endothelial barrier from septic insult by inhibiting protein phosphatase type 2A. Monolayer cultures of microvascular endothelial cells were incubated with ascorbate, dehydroascorbic acid (DHAA), NADPH oxidase inhibitors apocynin and diphenyliodonium, or PP2A inhibitor okadaic acid, and then were exposed to septic insult (lipopolysaccharide and interferon-γ). Under standard culture conditions that depleted intracellular ascorbate, septic insult stimulated oxidant production and PP2A activity, dephosphorylated phosphoserine and phosphothreonine residues in the tight junction-associated protein occludin, decreased the abundance of occludin at cell borders, and increased monolayer permeability to albumin. NADPH oxidase inhibitors prevented PP2A activation and monolayer leak, showing that these changes required reactive oxygen species. Okadaic acid, at a concentration that inhibited PP2A activity and monolayer leak, prevented occludin dephosphorylation and redistribution, implicating PP2A in the responses of occludin to septic insult. Incubation with ascorbate or DHAA raised intracellular ascorbate concentrations and mitigated the effects of septic insult. In conclusion, ascorbate acts within microvascular endothelial cells to inhibit septic stimulation of oxidant production by NADPH oxidase and thereby prevents PP2A activation, PP2A-dependent dephosphorylation and redistribution of occludin, and disruption of the endothelial barrier.
Keywords: Ascorbate, Endothelial cells, Interferon-γ, Lipopolysaccharide, NADPH oxidase, Occludin, Permeability, Protein phosphatase type 2A, Sepsis
Introduction
Rising rates of hospitalization and death due to sepsis show that this disease is a worsening health care problem [1]. Endothelial barrier dysfunction occurs in systemic capillaries and venules of patients with sepsis. This dysfunction increases the microvessels’ permeability to macromolecules, such as albumin, and frequently leads to plasma extravasation, edema, organ failure and septic shock [2–5]. Since no effective pharmacological therapy is available to prevent the increase in permeability, there is a pressing need to identify the molecular mechanisms of this pathology and potential treatments.
Endothelial barrier dysfunction is part of the systemic inflammatory response commonly initiated by pathogenic bacteria, their components (e.g. lipopolysaccharide, LPS), and inflammatory cytokines [e.g. interferon (IFN)-γ]. Studies of barrier function in endothelial and epithelial cell cultures have shown that these septic insults decrease phosphorylation of threonine residues in occludin, stimulate redistribution of occludin and other tight junction proteins away from cell borders, and increase paracellular permeability to macromolecules [6–8]. It has also been observed, in epithelial cell cultures, that serine/threonine protein phosphatase 2A (PP2A) dephosphorylates threonine residues in occludin and this change is associated with disassembly of tight junctions and increased paracellular permeability [9–11]. Septic insult (LPS + IFN-γ) in microvascular endothelial cell cultures stimulates production of NADPH oxidase-derived superoxide, which forms peroxynitrite that nitrates tyrosine residues in the PP2A catalytic subunit (PP2Ac). This nitration in PP2Ac is associated with PP2A activation and endothelial barrier dysfunction [12].
Pretreatment with the antioxidant ascorbate attenuates the increase in monolayer permeability caused by LPS in aortic endothelial cell cultures [13]. Parenteral administration of ascorbate also decreases edema formation in LPS-injected or burn-injured animals, as well as in patients with severe burn injury [14–19]. The present study was designed to identify the mechanism of action of ascorbate. We tested the hypothesis that intracellular ascorbate stabilizes the endothelial barrier during septic insult by inhibiting PP2A.
Materials and Methods
Cell cultures
The University at Buffalo Institution Animal Care and Use Committee approved the procedures. Microvascular endothelial cells were isolated from murine skeletal muscles and grown in DMEM/F12 medium containing 10% fetal bovine serum, 2 mM L-glutamine, 5 U/ml heparin, 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 μg/ml endothelial cell growth supplement, as described previously [20].
Chemicals
L-Ascorbic acid was purchased from Sigma-Aldrich. This molecule becomes ascorbate when put into solution. Dehydro-L-(+)-ascorbic acid dimer (DHAA) was purchased as a dry powder from Fluka Chemical (a division of Sigma-Aldrich) and stored at −20°C. The Certificate of Analysis indicated that the purity of DHAA was 98.3%. The identity of the product was confirmed to be DHAA by spectroscopy in the vendor’s lab, as well as by HPLC-based electrochemical assay and quantitative NMR in our labs. Fetal bovine serum was purchased from Hyclone. Catalase and p-nitrophenylphosphate eiwo (p-NPP) were from Calbiochem. Anti-occludin antibody(catalog number 71–1500), L-glutamine, penicillin, phosphate buffered saline (PBS) and streptomycin were from Invitrogen. Anti-phosphoserine and anti-threonine antibodies (catalog numbers sc-81514 and sc-5267, respectively) were from Santa Cruz. Anti-PP2Ac antibody(catalog number 610556), endothelial cell growth supplement and protein-A-agarose beads were from BD Biosciences. Anti-β-actin antibody (lot number 21107) was from Rockland. CellTiter-Fluor™ cell viability assay kit was from Promega. Competitive ELISA assay kit for murine type IV collagen (1014 Strip Plate) was purchased from Exocell. Protease inhibitor cocktail was from Roche. Radioimmunoprecipitation assay buffer was from Cell Signaling Technology. Bicinchoninic acid protein assay kit was from Pierce. All other chemicals were obtained from Sigma-Aldrich.
Experimental procedures
Microvascular endothelial cells were maintained without endothelial cell growth supplement for 7 days, and the serum concentration was decreased to 2% for the final 2 days, before being used for experiments. The cells were incubated with ascorbate, DHAA or other drugs for the indicated periods before exposure to either septic insult [25 ng/ml LPS (Escherichia coli 055:B5) and 100 U/ml IFN-γ dissolved in bovine serum albumin (BSA) solution] or control (BSA only).
Intracellular ascorbate concentrations were determined by HPLC with electrochemical detection using a previously described method [20]. Cells in 35 mm dishes were washed twice with 2.5 ml of ice-cold PBS and then scrape-harvested into 500 μl of cold water. Aliquots were combined with metaphosphoric acid (final concentration, 0.85%) for subsequent ascorbate assay and the remainder of the cell harvest was analyzed for total cell protein content.
Oxidant production was measured using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF diacetate). This molecule diffuses passively into cells, is de-esterified of diacetate by intracellular esterases, and then is oxidized to fluorescent dichlorofluorescein by oxidants such as peroxynitrite and hydroxyl radical [21,22]. Confluent microvascular endothelial cells, in 96-well plates, were washed with PBS and incubated 30 min with H2DCF diacetate (10 μM) in the dark. Subsequently the cells were washed twice with PBS and their fluorescence was measured at excitation and emission wavelengths of 485/20 nm and 528/20 nm, respectively.
The permeability of endothelial monolayers to Evans blue-coupled BSA was determined as described previously [12]. In brief, the microvascular endothelial cells were grown on gelatin-coated inserts (3 μm pore size) in 12-well plates (BD Biosciences). Evans blue-coupled BSA and uncoupled BSA were added to the upper chamber and lower chamber, respectively, and incubated 1 h with cells. Finally, the Evans blue-coupled BSA in the lower chamber was measured at 595 nm.
Cell viability was measured by Promega CellTiter-Fluor™ cell viability assay according to the manufacturer’s protocol. Briefly, endothelial cells in 96-well plates were incubated with 100 μl of CellTiter-Fluor™ reagent for 30 min at 37°C and then fluorescence was determined at 400 nm/505 nm. Type IV collagen was measured by Exocell 1014 Strip Plate competitive ELISA assay according to the manufacturer’s instructions.
PP2A activity was measured as okadaic acid-inhibitable phosphatase activity by the method described previously [12]. A 100 μl aliquot of cell harvest (containing protein concentration of 500 μg/ml) was mixed with 100 μl of assay buffer [5 mM p-NPP, 3 mM MnCl2, 0.1 mM EDTA, 50 mM Tris-Cl, pH 7.0] with or without 50 nM okadaic acid, and then it was incubated 10 min at 30°C. The hydrolysis of p-NPP was determined at 405 nm and the PP2A activity was calculated as the difference between total phosphatase activity and okadaic acid-insensitive phosphatase activity.
Western blot analysis of proteins was performed as follows. Cells were rinsed twice with PBS and scrape-harvested in radioimmunoprecipitation assay buffer containing protease inhibitor cocktail. The cell harvests were sonicated on ice and then centrifuged for 10 min at 14,000 g at 4°C. Next, the supernatants were collected and protein concentration was determined by bicinchoninic acid protein assay. Cell proteins were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane. Then the blocked membranes were incubated with anti-PP2Ac antibody or anti-β-actin antibody for 2 h, followed by incubation with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Protein bands were detected by ECL chemiluminescence and quantified with Quantity One (Bio-Rad) software.
Immunoprecipitation of proteins was performed using aliquots of cell harvests containing 800 μg of total protein each. The cell harvests were incubated overnight at 4°C with anti-occludin antibody and then incubated for 3 h at 4°C with protein-A-agarose beads. The immunoprecipitates were washed four times with ice-cold radioimmunoprecipitation assay buffer and then boiled in Laemmli’s sample buffer and separated by electrophoresis. Subsequently the proteins were transferred to polyvinylidene difluoride membrane. Finally, specific antibodies were used to detect phosphoserine, phosphothreonine and occludin.
Immunofluorescence microscopy was performed using cells grown on glass coverslips. The cells were fixed by incubation with 3% paraformaldehyde for 20 min, permeabilized by incubation with 0.2% Triton X-100 for 20 min, and blocked by incubation with 5% BSA for 30 min at room temperature. Next, the cells were incubated sequentially with anti-occludin antibody for 2 h and goat AlexaFluor 488-conjugated anti-rabbit IgG antibody for 1 h. Finally, the cells’ labeled occludin was visualized by fluorescence microscopy.
Statistics
Data are presented as mean ± SD values and were analyzed with the Prism statistical program. Comparisons between mean values were performed with one-way ANOVA followed by the Tukey multiple comparison test. P<0.05 was considered significant.
Results
Intracellular ascorbate and oxidant production
Microvascular endothelial cells were incubated for 12 h with ascorbate, DHAA or DMEM vehicle and then were incubated for 24 h with LPS+IFNγ (septic insult) or BSA (control). The concentration of ascorbate and DHAA added to the cell cultures was 500 μM, because this was the peak ascorbate concentration achieved in plasma by an ascorbate infusion protocol that inhibited edema formation in patients [19]. Ascorbate was not detectable in the cells or medium of cell cultures that were treated with vehicle instead of ascorbate or DHAA (Fig. 1A). Intracellular ascorbate concentrations were elevated to similar levels by incubation with either ascorbate or DHAA (Fig. 1A). Delayed exposure to LPS+IFNγ did not alter the intracellular ascorbate concentration derived from exogenous ascorbate or DHAA (Fig. 1A).
Fig. 1.

Incubations with ascorbate and DHAA increase intracellular ascorbate concentration and prevent LPS+IFNγ-induced oxidant production in microvascular endothelial cells. The cells were incubated with ascorbate (500 μM), DHAA (500 μM) or DMEM vehicle (Vehicle) for 12 h. Subsequently they were incubated with LPS (25 ng/ml) + IFNγ (100 U/ml) or vehicle (BSA, Control) for 24 h. (A) Intracellular ascorbate concentration was determined by HPLC-based electrochemical assay and expressed as nmol ascorbate/mg cell protein. (B) Oxidant production was assessed by dichlorodihydrofluorescein oxidation assay and expressed as a percentage of the value for vehicle control. Plotted are mean ± SD values for 3 experiments. *P<0.05 compared to vehicle control. #P<0.05 compared to the combination of vehicle (DMEM) and LPS+IFNγ.
LPS+IFNγ increased oxidant production in cells that had not received ascorbate or DHAA (Fig. 1B). This effect of LPS+IFNγ was abolished in cells previously incubated with ascorbate or DHAA(Fig. 1B). Although donation of reducing equivalents from ascorbate to transition metals may increase the concentrations of ascorbate radical and hydrogen peroxide [23], this mechanism did not account for the effect of ascorbate on the oxidant production we measured, because DHAA had an equally large effect (Fig. 1B). Taken together, the results of Fig. 1A and B indicate that intracellular ascorbate inhibits the oxidant production induced by septic insult.
Endothelial barrier dysfunction, collagen production and PP2A activity
LPS+IFNγ increased microvascular endothelial cell monolayer permeability to Evans blue-coupled BSA markedly (Fig. 2A). This effect was prevented by pretreating the cells with ascorbate or DHAA(Figure 2A). Although reactions of ascorbate with transition metals may increase hydrogen peroxide in cell culture media, addition of a hydrogen peroxide scavenger (catalase, 500 U/ml) to the medium did not alter the protective effects of ascorbate and DHAA on monolayer permeability (data not shown). Cell viability was not altered by these treatments (Figure 2B). The similar viabilities of control and LPS+IFNγ-stimulated cells suggest that cell death was not the cause of the change in monolayer permeability induced by septic insult.
Fig. 2.

Ascorbate and DHAA prevent LPS+IFNγ-induced barrier dysfunction. Microvascular endothelial cells were treated as described in Fig. 1. (A) Permeability of endothelial monolayers was measured with Evans blue-coupled albumin and expressed as a percentage of the value for vehicle control. (B) Cell viability was measured by CellTiter-Fluor™ cell viability assay and expressed as a percentage of the value for vehicle control. Plotted are mean ± SD values for 4 experiments. *P<0.05 compared to vehicle control. #P<0.05 compared to the combination of vehicle (DMEM) and LPS+IFNγ.
To elucidate the mechanism by which intracellular ascorbate stabilizes the endothelial barrier, type IV collagen production, PP2Ac protein expression and PP2A activity were measured. Incubations of cell cultures for 36 h with ascorbate or DHAA increased the production of type IV collagen and these effects were not altered by addition of LPS+IFNγ during the final 24 h (Fig. 3). LPS+IFNγ did not change PP2A protein expression (Fig. 4A) but did increase PP2A activity (Fig. 4B). Further, both ascorbate and DHAA inhibited the PP2Aactivation induced by LPS+IFNγ (Fig. 4B).
Fig. 3.

Incubations with ascorbate and DHAA increase type IV collagen production in the presence or absence of delayed LPS+IFNγ. Microvascular endothelial cells were treated as described in Fig. 1 and then aliquots of media were collected for competitive ELISA assay of type IV collagen. *P<0.05 compared to vehicle control. #P<0.05 compared to the combination of vehicle (DMEM) and LPS+IFNγ.
Fig. 4.

Ascorbate and DHAA prevent LPS+IFNγ-induced PP2A activation. Microvascular endothelial cells were treated as described in Fig. 1. (A) PP2Ac and β-actin (loading control) protein expression was measured by western blot analysis and the cell treatments (LPS+IFNγ, ascorbate and DHAA) were found to have no detectable effect on the PP2Ac:β-actin ratio. A representative western blot is shown. (B) PP2A activity was measured as okadaic acid-inhibitable phosphatase activity and expressed as a percentage of the value for vehicle control. Plotted are mean ± SD values for 9 experiments. *P<0.05 compared to vehicle control. #P<0.05 compared to the combination of vehicle (DMEM) and LPS+IFNγ.
NADPH oxidase is the principal source of superoxide in LPS+IFNγ treated microvascular endothelial cells and inhibition of NADPH oxidase by apocynin or diphenyliodonium (DPI) prevents stimulation by LPS+IFNγ of superoxide production [12, 24]. Apocynin and DPI had no significant effects on the basal level of endothelial cell monolayer permeability or PP2A activity (i.e., absent LPS+IFNγ; Figs. 5 and 6). The increase in monolayer permeability induced by LPS+IFNγ was prevented by apocynin and DPI (Fig. 5). The induction of PP2A activity by LPS+IFNγ was inhibited by apocynin and DPI, while PP2Ac protein expression was not changed (Fig. 6). These results implicated NADPH oxidase in the endothelial barrier dysfunction and PP2A activation following exposure to LPS+IFNγ.
Fig. 5.

NADPH oxidase inhibitors prevent LPS+IFNγ-induced barrier dysfunction in microvascular endothelial cell monolayers. The cells were incubated with apocynin (250 μM), DPI (5 μM) or DMEM vehicle (Vehicle) for 1 h. Subsequently they were incubated with LPS (25 ng/ml) + IFNγ (100 U/ml) or vehicle (BSA, Control) for 24 h and then monolayer permeability was measured. Plotted are mean ± SD values for 3 experiments. *P<0.05 compared to vehicle control. #P<0.05 compared to the combination of vehicle (DMEM) and LPS+IFNγ.
Fig. 6.

NADPH oxidase inhibitors prevent LPS+IFNγ-induced PP2A activation. Microvascular endothelial cells were treated as described in Fig. 4. (A) PP2Ac and β-actin protein expression was measured by western blot analysis and the cell treatments (LPS+IFNγ, apocynin and DPI) were found to have no detectable effect on the PP2Ac:β-actin ratio. A representative western blot is shown. (B) PP2A activity. Plotted are mean ± SD values for 6 experiments. *P<0.05 compared to vehicle control. #P<0.05 compared to the combination of vehicle (DMEM) and LPS+IFNγ.
Occludin phosphorylation and localization
LPS+IFNγ treatment of cells decreased the abundance of phosphorylated serine and threonine residues in occludin immunoprecipitates (Fig. 7). The decreased phosphorylation of occludin was associated with loss of the protein from regions of cell-cell contact (Fig. 7). Pretreatment of the cells with ascorbate or DHAA prevented these effects of LPS+IFNγ (Fig. 7). To explore if PP2A mediated the dephosphorylation of occludin, okadaic acid was used to inhibit PP2A specifically. The concentration of okadaic acid we used has been shown to inhibit PP2A activity and septic induction of endothelial monolayer leak [12]. Okadaic acid prevented the LPS+IFNγ-induced decrease in phosphoserine and phosphothreonine levels and maintained the normal localization of occludin at cell borders (Fig. 8). These results suggested that intracellular ascorbate protects the endothelial barrier from septic insult by preventing PP2A-mediated dephosphorylation and redistribution of occludin.
Fig. 7.

Ascorbate and DHAA prevent LPS+IFNγ-induced dephosphorylation and redistribution of occludin. Microvascular endothelial cells were treated as described in Fig. 1. (A) p-Serine and p-threonine levels were measured in occludin immunoprecipitates by western blot analysis. A representative western blot is shown. (B) Ratios of phosphoserine:occludin and phosphothreonine:occludin in occludin immunoprecipitates. Plotted are mean ± SD values for 3 experiments. *P<0.05 compared to vehicle control. #P<0.05 compared to the combination of vehicle (DMEM) and LPS+IFNγ. (C) Cellular distribution of immunoreactive occludin was visualized by immunofluorescence microscopy. Representative micrographs are shown.
Fig. 8.

LPS+IFNγ-induced dephosphorylation and redistribution of occludin are prevented by the PP2A inhibitor, okadaic acid. Microvascular endothelial cells were incubated with okadaic acid (OA, 0.5 nM) or DMEM vehicle (Vehicle) for 1 h and then incubated with LPS+IFNγ or vehicle (BSA, Control) for 24 h. (A) p-Serine and p-threonine levels were measured in occludin immunoprecipitates by western blot analysis. A representative western blot is shown. (B) Ratios of phosphoserine:occludin and phosphothreonine:occludin in occludin immunoprecipitates. Plotted are mean ± SD values for 3 experiments. *P<0.05 compared to vehicle control. #P<0.05 compared to the combination of vehicle (DMEM) and LPS+IFNγ. (C) Immunofluorescence micrographs of occludin in microvascular endothelial cells.
Discussion
Septic insults induce leaks in endothelial and epithelial barriers by disrupting intercellular tight junctions [6–8]. This disruption is mediated by activation of PP2A [12]. Because endothelial barrier dysfunction contributes to morbidity in sepsis [2–5], there is an urgent need to identify the molecular mechanisms of this pathology and potential treatments. The present study discovered that intracellular ascorbate protects endothelial barrier function during septic insult and that this protection is associated with inhibition of oxidant production, PP2A activation, and occludin dephosphorylation and redistribution.
There is abundant evidence that ascorbate influences endothelial barrier function under certain conditions. Observations in patients with scurvy and in scorbutic animals show that severe ascorbate deficiency can increase capillary permeability and edema formation, even in the absence of sepsis [25–27]. In contrast, administration of ascorbate does not affect acutely the microvascular permeability to protein in normal, non-septic animals. For example, bolus intravenous injection of ascorbate (100 mg/kg) does not change microvascular permeability in normal rats, even though this dose is large enough to prevent the microvascular leak induced by intravenous injection of arachidonate [28].
Why endothelial barrier dysfunction occurs under non-septic, scorbutic conditions in situ is not known. In vitro approaches to solving this problem have yielded conflicting results. Addition of ascorbate to the medium of non-septic endothelial cell cultures has been reported to decrease monolayer permeabilityto macromolecules in some studies [29,30]. For instance, incubation of EA.hy926 endothelial cells with extracellular ascorbate increases intracellular ascorbate concentration and inhibits the diffusion of inulin (molecular weight range 5,000–5,500) across the endothelial monolayer [30]. Also, daily addition of ascorbate for 5 days to artery endothelial cell cultures inhibits the trans-monolayer diffusion of small and large fluorescein dextrans (molecular weights 4,000 and 70,000, respectively) [29]. In the present experiments, microvascular endothelial cell monolayer cultures that were grown under standard conditions did not contain detectable levels of ascorbate in either the cells or medium. Compared to these ascorbate-deficient cultures, elevation of intracellular ascorbate concentration by incubations with ascorbate or DHAA did not change monolayer permeability to Evans blue-coupled BSA (molecular weight 70,000) in the absence of septic insult. This failure of ascorbate to alter monolayer permeability under non-septic conditions may be clinically relevant because it was observed in experiments with endothelial cells derived from exchange vessels (capillaries and venules) and with a macromolecule (albumin) that is abundant in plasma.
Ascorbate clearly has an important role in sepsis. Critically ill patients have decreased plasma levels of ascorbate [31–37]. Further, in septic patients the plasma ascorbate concentration correlates inversely with multiple organ failure incidence and directly with survival [31,33]. Experiments with animal models have shown that ascorbate increases survival after septic insults [38–41]. There is also clinical evidence that parenteral administration of ascorbate may improve outcome. In a randomized, prospective, double-blind, placebo-controlled trial with 226 critically ill patients, 28-day mortality was decreased in the patients who received combined ascorbate and vitamin E by intravenous infusion compared to those who did not [42]. Another randomized, prospective trial found a decreased incidence of organ failure and shortened ICU stay for critically ill patients who began receiving intravenous injections of combined ascorbate (3 g/day for up to 28 days) and vitamin E within 24 h of traumatic injury or major surgery [37]. Finally, a study of patients who received high-dose intravenous ascorbate (1584 mg/kg/24 h) in the first 24 h after burn injury observed that ascorbate treatment was associated with lower resuscitation fluid volume requirements, less weight gain, and less wound edema [19]. It may be inferred that ascorbate prevents endothelial barrier dysfunction, because the latter is a major contributor to fluid requirements and edema formation [2]. Parenteral administration of ascorbate also decreases edema formation in LPS-injected or burn-injured animal models of critical illness [14–18].
Ascorbate pretreatment attenuates the increase in monolayer permeability to albumin caused by LPS+IFN-γ (25 ng/ml E. coli LPS and 100 U/ml IFN-γ) in microvascular endothelial cell cultures (present experiments) and by LPS (1 μg/ml E. coli LPS) in aortic endothelial cell cultures [13]. The septic insults that increased permeability did not change cell viability in our experiments or those of Dimmeler [13]. Therefore the barrier dysfunction and protection caused by septic insult and ascorbate, respectively, do not depend on changes in endothelial cell viability.
Reaction of ascorbate with transition metals can produce hydrogen peroxide, which is a precursor of the strongly oxidizing hydroxyl radical [23]. However, the similar degree of barrier protection conferred by ascorbate and DHAA indicates that hydrogen peroxide production in the medium is not required. Instead, the stabilization of the barrier is likely due to the intracellular ascorbate that endothelial cells derive from extracellular ascorbate and DHAA.
Ascorbate and DHAA decreased monolayer permeability in LPS+IFNγ-stimulated (septic) cells but not in unstimulated (non-septic) control cells, despite similar increases in collagen production in the septic and non-septic groups. We infer that the increases in collagen production induced by ascorbate and DHAA were not sufficient to decrease monolayer permeability. This inference is supported by the observation of May et al. [30] that incubation for 18 h with 300 μM ascorbate did not change the extracellular barrier to macromolecular diffusion through endothelial cell cultures. These observations must be compared to those of Utoguchi et al. [29], who reported that inhibitors of collagen synthesis prevented the decrease in monolayer permeability caused by daily additions of ascorbate (10–100 μM) to bovine artery endothelial cells that were growing to confluence on porous membrane filters [29]. However, as remarked by May et al. [30], the ascorbate-induced decrease in permeability of the bovine artery endothelial cells alone was relatively greater than that of the filters and extracellular matrix [29], indicating that ascorbate had an effect on the cells beyond that due to collagen deposition.
LPS lowers intracellular cAMP concentration as it increases monolayer permeability in endothelial cell cultures [43]. Moreover, the monolayer leak can be blocked with drugs that elevate intracellular cAMP [43]. This mechanism is unlikely to account for the protective effect of intracellular ascorbate that we observed, however, because ascorbate is a competitive inhibitor of adenylate cyclase and decreases intracellular cAMP levels [44]. A more probable explanation of how intracellular ascorbate stabilizes the endothelial barrier during septic insult involves NADPH oxidase, superoxide, peroxynitrite and PP2A (Fig. 9), and these mediators will be discussed next.
Fig. 9.
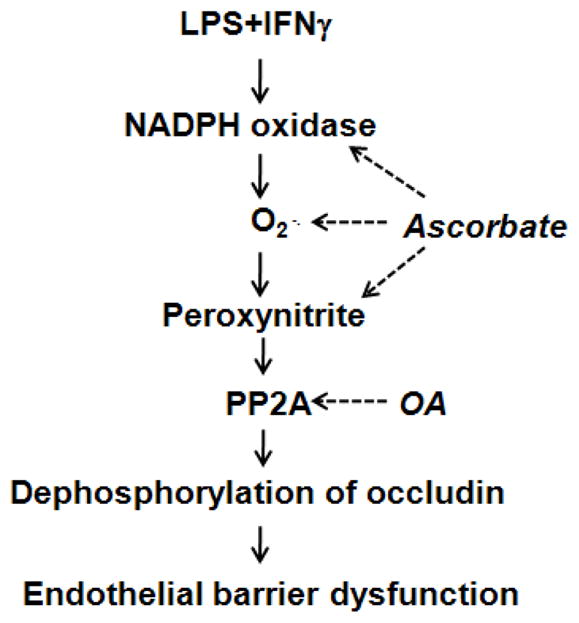
Diagram of the mechanism by which intracellular ascorbate and okadaic acid (OA) prevent the induction of endothelial barrier dysfunction by LPS+IFNγ. The solid arrows indicate stimulation and the dotted arrows indicate inhibition.
In microvascular endothelial cells, LPS+IFNγ increases protein expression of the NOX1 and p47phox subunits of NADPH oxidase, resulting in increased NADPH oxidase activity that becomes the principle source of superoxide in these cells [24]. Superoxide is a precursor of peroxynitrite and the latter increases paracellular permeability in endothelial cell monolayers [45,46]. Apocynin decreased the basal level of endothelial cell monolayer permeability (i.e., absent LPS+IFNγ) in a previous experiment [Fig. 6 of reference 12], but neither apocynin nor DPI had this effect in the present study and the reason for this discrepancy is not known. However, the present study observed a protective effect of NADPH oxidase inhibitors (apocynin and DPI) on cell monolayer leak, which shows that the products of this enzyme’s activity contribute to septic endothelial barrier dysfunction. Two previous studies also support this inference [47,48], although it should be noted that knockout of p47phox or gp91phox does not confer protection against LPS-induced death [49]. Intracellular ascorbate prevents the induction of endothelial barrier dysfunction by inhibiting the septic activation of NADPH oxidase and by scavenging superoxide and peroxynitrite (Fig. 9). Intracellular ascorbate abrogates NADPH oxidase activation by preventing septic stimulation of the Jak2/Stat1/IRF1 signaling pathway that leads to p47phox expression [24].
LPS+IFNγ increases the serine/threonine protein phosphatase activity of PP2A in microvascular endothelial cells. The septic insult does not alter PP2Ac protein expression. Instead it stimulates production of NADPH oxidase-derived superoxide, which then reacts with nitric oxide to form peroxynitrite that activates PP2A [12]. Recent experiments with enterocytes and myocardial cells have confirmed that PP2A activity is increased by peroxynitrite [50,51]. Peroxynitrite nitrates the tyrosine residues of PP2Ac, thereby preventing their phosphorylation and consequent inactivation of PP2A [12]. Additionally, PP2Ac nitration may also restrict the access of methylesterase enzymes to PP2Ac and thereby prevent demethylation of the catalytic subunit and consequent inactivation of the phosphatase, since PP2Ac methylation facilitates assembly of the PP2A holoenzyme [52]. Lastly, nitration of PP2Ac at the cell membrane may prevent translocation of the enzyme to the cytosol and thus accelerate dephosphorylation of occludin at the membrane [53].
The endothelial barrier dysfunction induced by septic insult evidently depends on PP2A activation, because inhibition of PP2A activity by okadaic acid or siRNA knockdown of PP2Ac stabilizes the barrier [12]. The present study found that NADPH oxidase inhibitors (apocynin and DPI) and intracellular ascorbate prevent LPS+IFNγ-induced increases in PP2A activity. Since ascorbate blocks septic activation of NADPH oxidase, the latter results support the hypothesis that intracellular ascorbate abrogates septic activation of PP2A by decreasing the production of superoxide and its product peroxynitrite. An alternative hypothesis is that ascorbate-derived hydrogen peroxide inhibits PP2A in endothelial cells, because hydrogen peroxide has been shown to inhibit phosphatase activity in cell-free preparations of PP2A [54]. However, the effect on PP2Athat we observed in endothelial cells after incubation with ascorbate was also evident after incubation with DHAA, and therefore it is not attributable to generation of hydrogen peroxide by reactions of ascorbate with transition metals in the culture medium.
Inflammatory signals stimulate myosin light chain kinase-dependent contractions of the actin-myosin cytoskeleton, which widen the intercellular space for transendothelial diffusion of macromolecules, and these signals dissociate intercellular junctions [55]. Phosphorylation of occludin on tyrosine, serine and threonine residues, which is controlled by protein kinases and protein phosphatases, normally regulates tight junction permeability[56]. It has been observed in epithelial cell cultures that PP2A dephosphorylates threonine residues in occludin and this change is associated with disassembly of tight junctions and increased paracellular permeability [9–11]. The present study found that serine and threonine residues in occludin become dephosphorylated, and occludin becomes less abundant at cell borders, when endothelial cell monolayer permeability is increased by septic insult. Ascorbate, DHAA and okadaic acid prevent septic stimulation of PP2A activity and this effect is associated with preservation of occludin phosphorylation, occludin localization at cell borders, and low permeability of the endothelial monolayer to albumin (Fig. 9). The modulations of PP2A activity and monolayer permeability by intracellular ascorbate are consequences of the latter preventing activation of NADPH oxidase. Taken together, these results support the conclusion that intracellular ascorbate prevents microvascular endothelial barrier dysfunction after septic insult by inhibiting NADPH oxidase-dependent activation of PP2A, PP2A-induced dephosphorylation of occludin on serine and threonine residues, and loss of occludin from tight junctions.
Acknowledgments
The excellent technical assistance of Dr. David P. Schuster and George Kamenos is gratefully acknowledged. The project described was supported by Award Number R01AT003643 from the National Center for Complementary and Alternative Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Complementary and Alternative Medicine or the National Institutes of Health.
Abbreviations
- BSA
bovine serum albumin
- DHAA
dehydroascorbic acid
- eNOS
endothelial nitric oxide synthase
- H2DCF
2′,7′-dichlorodihydrofluorescein
- IFNγ
interferon-γ
- LPS
lipopolysaccharide
- OA
okadaic acid
- PBS
phosphate buffered saline
- p-NPP
p-nitrophenylphosphate eiwo
- PP2A
protein phosphatase type 2A
- PP2Ac
PP2A catalytic subunit
References
- 1.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med. 2007;35:1244–1250. doi: 10.1097/01.CCM.0000261890.41311.E9. [DOI] [PubMed] [Google Scholar]
- 2.Christ F, Gamble J, Gartside IB, Kox WJ. Increased microvascular water permeability in patients with septic shock, assessed with venous congestion plethysmography (VCP) Intensive Care Med. 1998;24:18–27. doi: 10.1007/s001340050509. [DOI] [PubMed] [Google Scholar]
- 3.Dhillon SS, Mahadevan K, Bandi V, Zheng Z, Smith CW, Rumbaut RE. Neutrophils, nitric oxide, and microvascular permeability in severe sepsis. Chest. 2005;128:1706–1712. doi: 10.1378/chest.128.3.1706. [DOI] [PubMed] [Google Scholar]
- 4.Hauptmann S, Klosterhalfen B, Weis J, Mittermayer C, Kirkpatrick CJ. Skeletal muscle oedema and muscle fibre necrosis during septic shock. Observations with a porcine septic shock model. Virchows Arch. 1994;424:653–659. doi: 10.1007/BF00195781. [DOI] [PubMed] [Google Scholar]
- 5.Holbeck S, Grande PO. Endotoxin increases both protein and fluid microvascular permeability in cat skeletal muscle. Crit Care Med. 2003;31:560–565. doi: 10.1097/01.CCM.0000048620.88344.70. [DOI] [PubMed] [Google Scholar]
- 6.Oshima T, Laroux FS, Coe LL, Morise Z, Kawachi S, Bauer P, Grisham MB, Specian RD, Carter P, Jennings S, Granger DN, Joh T, Alexander JS. Interferon-gamma and interleukin-10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc Res. 2001;61:130–143. doi: 10.1006/mvre.2000.2288. [DOI] [PubMed] [Google Scholar]
- 7.Sheth P, Delos Santos N, Seth A, LaRusso NF, Rao RK. Lipopolysaccharide disrupts tight junctions in cholangiocyte monolayers by a c-Src-, TLR4-, and LBP-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2007;293:G308–G318. doi: 10.1152/ajpgi.00582.2006. [DOI] [PubMed] [Google Scholar]
- 8.Simonovic I, Rosenberg J, Koutsouris A, Hecht G. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol. 2000;2:305–315. doi: 10.1046/j.1462-5822.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- 9.González-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778:729–756. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Nunbhakdi-Craig V, Machleidt T, Ogris E, Bellotto D, White CL, 3rd, Sontag E. Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction complex. J Cell Biol. 2002;158:967–978. doi: 10.1083/jcb.200206114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheth P, Samak G, Shull JA, Seth A, Rao R. Protein phosphatase 2A plays a role in hydrogen peroxide-induced disruption of tight junctions in Caco-2 cell monolayers. Biochem J. 2009;421:59–70. doi: 10.1042/BJ20081951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu F, Wilson JX. Peroxynitrite-dependent activation of protein phosphatase type 2A mediates microvascular endothelial barrier dysfunction. Cardiovasc Res. 2009;81:38–45. doi: 10.1093/cvr/cvn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimmeler S, Brinkmann S, Neugebauer E. Endotoxin-induced changes of endothelial cell viability and permeability: protective effect of a 21-aminosteroid. Eur J Pharmacol. 1995;287:257–61. doi: 10.1016/0014-2999(95)00499-8. [DOI] [PubMed] [Google Scholar]
- 14.Dubick MA, Williams C, Elgjo GI, Kramer GC. High-dose vitamin C infusion reduces fluid requirements in the resuscitation of burn-injured sheep. Shock. 2005;24:139–44. doi: 10.1097/01.shk.0000170355.26060.e6. [DOI] [PubMed] [Google Scholar]
- 15.Dwenger A, Pape HC, Bantel C, Schweitzer G, Krumm K, Grotz M, Lueken B, Funck M, Regel G. Ascorbic acid reduces the endotoxin-induced lung injury in awake sheep. Eur J Clin Invest. 1994;24:229–35. doi: 10.1111/j.1365-2362.1994.tb01079.x. [DOI] [PubMed] [Google Scholar]
- 16.Feng NH, Chu SJ, Wang D, Hsu K, Lin CH, Lin HI. Effects of various antioxidants on endotoxin-induced lung injury and gene expression: mRNA expressions of MnSOD, interleukin-1beta and iNOS. Chin J Physiol. 2004;47:111–120. [PubMed] [Google Scholar]
- 17.Sakurai M, Tanaka H, Matsuda T, Goya T, Shimazaki S, Matsuda H. Reduced resuscitation fluid volume for second-degree experimental burns with delayed initiation of vitamin C therapy (beginning 6 h after injury) J Surg Res. 1997;73:24–27. doi: 10.1006/jsre.1997.5203. [DOI] [PubMed] [Google Scholar]
- 18.Shen KP, Lo YC, Yang RC, Liu HW, Chen IJ, Wu BN. Antioxidant eugenosedin-A protects against lipopolysaccharide-induced hypotension, hyperglycaemia and cytokine immunoreactivity in rats and mice. J Pharm Pharmacol. 2005;57:117–125. doi: 10.1211/0022357055137. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka H, Matsuda T, Miyagantani Y, Yukioka T, Matsuda H, Shimazaki S. Reduction of resuscitation fluid volumes in severely burned patients using ascorbic acid administration: a randomized, prospective study. Arch Surg. 2000;135:326–331. doi: 10.1001/archsurg.135.3.326. [DOI] [PubMed] [Google Scholar]
- 20.Wilson JX, Dixon SJ, Yu J, Nees S, Tyml K. Ascorbate uptake by microvascular endothelial cells of rat skeletal muscle. Microcirculation. 1996;3:211–221. doi: 10.3109/10739689609148290. [DOI] [PubMed] [Google Scholar]
- 21.Crow JP. Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide. 1997;1:145–157. doi: 10.1006/niox.1996.0113. [DOI] [PubMed] [Google Scholar]
- 22.Wrona M, Patel K, Wardman P. Reactivity of 2′,7′-dichlorodihydrofluorescein and dihydrorhodamine 123 and their oxidized forms toward carbonate, nitrogen dioxide, and hydroxyl radicals. Free Radic Biol Med. 2005;38:262–270. doi: 10.1016/j.freeradbiomed.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 23.Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, Khosh DB, Drisko J, Levine M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci USA. 2008;105:11105–11109. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu F, Schuster DP, Tyml K, Wilson JX. Ascorbate inhibits NADPH oxidase subunit p47phox expression in microvascular endothelial cells. Free Radic Biol Med. 2007;42:124–131. doi: 10.1016/j.freeradbiomed.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 25.Friederici HH, Taylor H, Rose R, Pirani CL. The fine structure of capillaries in experimental scurvy. Lab Invest. 1966;15:1442–1458. [PubMed] [Google Scholar]
- 26.Gore I, Wada M, Goodman ML. Capillary hemorrhage in ascorbic-acid-deficient guinea pigs. Ultrastructural basis. Arch Pathol. 1968;85:493–502. [PubMed] [Google Scholar]
- 27.Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895–906. doi: 10.1016/s0190-9622(99)70244-6. [DOI] [PubMed] [Google Scholar]
- 28.Alvarez-Guerra M, Hannaert P, Hider H, Chiavaroli C, Garay RP. Vascular permeabilization by intravenous arachidonate in the rat peritoneal cavity: antagonism by antioxidants. Eur J Pharmacol. 2003;466:199–205. doi: 10.1016/s0014-2999(03)01544-9. [DOI] [PubMed] [Google Scholar]
- 29.Utoguchi N, Ikeda K, Saeki K, Oka N, Mizuguchi H, Kubo K, Nakagawa S, Mayumi T. Ascorbic acid stimulates barrier function of cultured endothelial cell monolayer. J Cell Physiol. 1995;163:393–399. doi: 10.1002/jcp.1041630219. [DOI] [PubMed] [Google Scholar]
- 30.May JM, Qu Z, Qiao H. Transfer of ascorbic acid across the vascular endothelium: mechanism and self-regulation. Am J Physiol Cell Physiol. 2009;297:C169–C178. doi: 10.1152/ajpcell.00674.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borrelli E, Roux-Lombard P, Grau GE, Girardin E, Ricou B, Dayer J, Suter PM. Plasma concentrations of cytokines, their soluble receptors, and antioxidant vitamins can predict the development of multiple organ failure in patients at risk. Crit Care Med. 1996;24:392–397. doi: 10.1097/00003246-199603000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Doise JM, Aho LS, Quenot JP, Guilland JC, Zeller M, Vergely C, Aube H, Blettery B, Rochette L. Plasma antioxidant status in septic critically ill patients: a decrease over time. Fundam Clin Pharmacol. 2008;22:203–209. doi: 10.1111/j.1472-8206.2008.00573.x. [DOI] [PubMed] [Google Scholar]
- 33.Galley HF, Davies MJ, Webster NR. Ascorbyl radical formation in patients with sepsis: effect of ascorbate loading. Free Radic Biol Med. 1996;20:139–143. doi: 10.1016/0891-5849(95)02022-5. [DOI] [PubMed] [Google Scholar]
- 34.Long CL, Maull KI, Krishnan RS, Laws HL, Geiger JW, Borghesi L, Franks W, Lawson TC, Sauberlich HE. Ascorbic acid dynamics in the seriously ill and injured. J Surg Res. 2003;109:144–148. doi: 10.1016/s0022-4804(02)00083-5. [DOI] [PubMed] [Google Scholar]
- 35.Luo M, Fernandez-Estivariz C, Jones DP, Accardi CR, Alteheld B, Bazargan N, Hao L, Griffith DP, Blumberg JB, Galloway JR, Ziegler TR. Depletion of plasma antioxidants in surgical intensive care unit patients requiring parenteral feeding: effects of parenteral nutrition with or without alanyl-glutamine dipeptide supplementation. Nutrition. 2007;24:37–44. doi: 10.1016/j.nut.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metnitz PG, Bartens C, Fischer M, Fridrich P, Steltzer H, Druml W. Antioxidant status in patients with acute respiratory distress syndrome. Intensive Care Med. 1999;25:180–185. doi: 10.1007/s001340050813. [DOI] [PubMed] [Google Scholar]
- 37.Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT, Radella F, Garcia I, Maier RV. Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg. 2002;236:814–822. doi: 10.1097/00000658-200212000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armour J, Tyml K, Lidington D, Wilson JX. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. J Appl Physiol. 2001;90:795–803. doi: 10.1152/jappl.2001.90.3.795. [DOI] [PubMed] [Google Scholar]
- 39.Gaut JP, Belaaouaj A, Byun J, Roberts LJ, 2nd, Maeda N, Frei B, Heinecke JW. Vitamin C fails to protect amino acids and lipids from oxidation during acute inflammation. Free Radic Biol Med. 2006;30:1494–1501. doi: 10.1016/j.freeradbiomed.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 40.Tyml K, Li F, Wilson JX. Septic impairment of capillary blood flow requires nicotinamide adenine dinucleotide phosphate oxidase but not nitric oxide synthase and is rapidly reversed by ascorbate through an endothelial nitric oxide synthase-dependent mechanism. Crit Care Med. 2008;36:2355–2362. doi: 10.1097/CCM.0b013e31818024f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu F, Wilson JX, Tyml K. Ascorbate protects against impaired arteriolar constriction in sepsis by inhibiting inducible nitric oxide synthase expression. Free Radic Biol Med. 2004;37:1282–1289. doi: 10.1016/j.freeradbiomed.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 42.Crimi E, Liguori A, Condorelli M, Cioffi M, Astuto M, Bontempo P, Pignalosa O, Vietri MT, Molinari AM, Sica V, Della Corte F, Napoli C. The beneficial effects of antioxidant supplementation in enteral feeding in critically ill patients: a prospective, randomized, double-blind, placebo-controlled trial. Anesth Analg. 2004;99:857–863. doi: 10.1213/01.ANE.0000133144.60584.F6. [DOI] [PubMed] [Google Scholar]
- 43.Schlegel N, Baumer Y, Drenckhahn D, Waschke J. Lipopolysaccharide-induced endothelial barrier breakdown is cyclic adenosine monophosphate dependent in vivo and in vitro. Crit Care Med. 2009;37:1735–1743. doi: 10.1097/CCM.0b013e31819deb6a. [DOI] [PubMed] [Google Scholar]
- 44.Kaya F, Belin S, Diamantidis G, Fontes M. Ascorbic acid is a regulator of the intracellular cAMP concentration: old molecule, new functions? FEBS Lett. 2008;582:3614–3618. doi: 10.1016/j.febslet.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Zhao S, Gu Y, Lewis DF, Alexander JS, Wang Y. Effects of peroxynitrite and superoxide radicals on endothelial monolayer permeability: potential role of peroxynitrite in preeclampsia. J Soc Gynecol Investig. 2005;12:586–592. doi: 10.1016/j.jsgi.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 46.Neumann P, Gertzberg N, Vaughan E, Weisbrot J, Woodburn R, Lambert W, Johnson A. Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am J Physiol Lung Cell Mol Physiol. 2006;290:L674–L684. doi: 10.1152/ajplung.00391.2005. [DOI] [PubMed] [Google Scholar]
- 47.Gertzberg N, Neumann P, Rizzo V, Johnson A. NAD(P)H oxidase mediates the endothelial barrier dysfunction induced by TNF-alpha. Am J Physiol Lung Cell Mol Physiol. 2004;286:L37–L48. doi: 10.1152/ajplung.00116.2003. [DOI] [PubMed] [Google Scholar]
- 48.Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, Liu QH, Malik AB. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox−/− and gp91phox−/− mice. J Immunol. 2002;168:3974–3982. doi: 10.4049/jimmunol.168.8.3974. [DOI] [PubMed] [Google Scholar]
- 49.Zhang WJ, Wei H, Frei B. Genetic deficiency of NADPH oxidase does not diminish, but rather enhances, LPS-induced acute inflammatory responses in vivo. Free Radic Biol Med. 2009;46:791–798. doi: 10.1016/j.freeradbiomed.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guner YS, Ochoa CJ, Wang J, Zhang X, Steinhauser S, Stephenson L, Grishin A, Upperman JS. Peroxynitrite-induced p38 MAPK pro-apoptotic signaling in enterocytes. Biochem Biophys Res Commun. 2009;384:221–225. doi: 10.1016/j.bbrc.2009.04.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohr MJ, Davis JP, Ziolo MT. Peroxynitrite increases protein phosphatase activity and promotes the interaction of phospholamban with protein phosphatase 2A in the myocardium. Nitric Oxide. 2009;20:217–221. doi: 10.1016/j.niox.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tolstykh T, Lee J, Vafai S, Stock JB. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000;19:5682–5691. doi: 10.1093/emboj/19.21.5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sim AT, Ludowyke RI. The complex nature of protein phosphatases. IUBMB Life. 2002;53:283–286. doi: 10.1080/15216540213462. [DOI] [PubMed] [Google Scholar]
- 54.Yu JS. Activation of protein phosphatase 2A by the Fe2+/ascorbate system. J Biochem. 1998;124:225–230. doi: 10.1093/oxfordjournals.jbchem.a022084. [DOI] [PubMed] [Google Scholar]
- 55.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med. 2009;11:e19. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rao R. Occludin phosphorylation in regulation of epithelial tight junctions. Ann N Y Acad Sci. 2009;1165:62–68. doi: 10.1111/j.1749-6632.2009.04054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]