

Abstract
We present a refined alkane charge equilibration (CHEQ) force field, improving our previously reported CHEQ alkane force field[1] to better reproduce experimental hydration free energies. Experimental hydration free energies of ethane, propane, butane, pentane, hexane, and heptane are reproduced to within 3.6% on average. We demonstrate that explicit polarization results in a shift in molecular dipole moment for water molecules associated with the alkane molecule. We also show that our new parameters do not have a significant effect on the alkane-water interactions as measured by the radial distribution function (RDF).
Keywords: alkanes, hydration free energy, thermodynamic integration, force field, charge equilibration
1. Introduction
Alkanes are an important class of compounds with a variety of commercial and industrial uses, such as in petroleum refining, as industrial solvents, and in a wide range of products including oils and cosmetics[2, 3]. Aliphatic groups are also widely present in organic and biological systems; therefore, accurate representations of alkanes and aliphatic groups are desirable towards the goal of efficient molecular modeling of such systems. Another consideration is the treatment of electrostatic interactions in molecular simulations; traditionally, atoms are assigned fixed charges which interact via a Coulomb potential. Recent efforts have led to the development of polarizable force fields that explicitly model non-additive electrostatic effects arising from polarization. In the coarsest sense, these models attempt to capture changes in molecular electrostatic properties with changing local (electro)chemical environment; moreover, these are implemented within a classical, deterministic formalism. One such model is the charge equilibration (CHEQ) model, which is described in detail elsewhere[4, 5, 6, 7, 8]. Our previous studies have focused on refining the CHEQ hexane force field of Patel and Brooks[9] for more accurate bulk property predictions of larger n-alkanes[1] and extending the CHEQ model for more accurate modeling of phospholipid bilayers[10]. In this work, we present a revision to the alkane force field of Davis et al [1] to obtain more accurate hydration free energies.
We consider polarizable models for alkanes (as has been done in the past by several groups) as it has been shown that incorporating such effects improves the prediction of alkane dielectric constants. Accurately capturing the dielectric environment in alkyl environments has implications in modeling processes involving strongly charged systems within phospholipid bilayer systems (see for instance, Allen et al [11], Roux et al [12], and Bucher et al [13]).
2. Hydration Free Energy Calculations
The free energies of hydration are computed by decoupling a single alkane molecule from bulk TIP4P-FQ[14] water by thermodynamic integration (TI) [15]. A more detailed explanation can be found elsewhere[1]. Briefly, we followed a two-step path in which the electrostatic terms were first decoupled before decoupling the Lennard-Jones (LJ) interactions. This two-step scheme prevents the polarization catastrophe that could result from close range charge-charge interactions in the absence of LJ repulsion. Using the TI approach, the total hydration free energy can be expressed as
| (1) |
where λ1 and λ2 are path variables representing the electrostatic and LJ decoupling processes, respectively, and H(λ) is the λ-parameterized Hamiltonian of the system. Each path was divided into windows corresponding to a specific value of λ over which ensemble averages were sampled. Along the λ1 path, the solute-water electrostatic interactions were decoupled by linearly scaling them to zero. For the LJ terms, a soft-core potential was used since linear scaling of the LJ terms along the λ2 path leads to a rapidly changing Hamiltonian near λ2 = 0 that is difficult to sample. Therefore, we use the separation-shifted scaling method of Zacharias et al [16], in which the λ-dependent LJ terms are expressed as
| (2) |
where the summation is over all solute-water interactions. Ai and Bi are the atom-specific LJ parameters, r is the interaction distance, and the shift parameter δ attenuates the potential for a given solute model. As in our previous study[1], we found that shift parameters in the range of 7–8 Å2 were suitable for the alkanes presented here.
3. Simulation Methods
Free energy calculations were carried out in the NPT ensemble using the CHARMM molecular modeling package[17]. The simulation system consisted of a single alkane molecule and 256 TIP4P-FQ water molecules with cubic periodic boundary conditions. The alkanes investigated were ethane, propane, butane, pentane, hexane, and heptane. Temperature was maintained at 298 K using the Nosé-Hoover method[18, 19] with a thermal piston mass of 500 kcal mol−1 ps−2. Pressure was kept constant at 1 atm using the Langevin piston method[18] with a piston mass of 300 amu. Long-range electrostatics were accounted for by the Particle Mesh Ewald method[20, 21] with a screening parameter κ = 0.320, a 40 × 40 × 40 Fast Fourier Transform grid, and fourth-order B-spline interpolation. The LJ interactions were smoothly tapered over a 1 Å region to a cutoff of 12 Å. The leapfrog Verlet integrator[18] with a time step of 0.5 fs was used to propagate the dynamics. The fluctuating charges were thermostatted using a 1 K Nosé-Hoover bath, with a charge mass of 8.9 × 10−4 ps2 e−2 for the alkane charges and a charge mass of 3.9 × 10−5 ps 2 e−2 for the TIP4P-FQ charges; we find no difference using these slightly smaller charge massess for water relative to the original model of Rick et al [14].
We calculate the long-range correction (LRC) to the LJ potential using the approach previously described by Anisimov et al[22]. Briefly, a normal (without TI modifications) 500 ps simulation was performed for each system (single alkane molecule in 256 waters). For a given trajectory snapshot, the LRC was calculated as the difference in the LJ energy using nonbonded cutoffs of 12 Å and 30 Å. The LRC was then averaged over the entire trajectory.
Since adequate convergence of free energy calculations is a common problem [23] and since a single trajectory is not statistically meaningful[24], uncertainties were estimated from the standard deviation of multiple independent trajectories using the approach previously described by Warren et al[25]. Five replicate trajectories, each approximately 20 ns in length, were used to determine the total hydration free energy for each alkane. Along the electrostatic decoupling path, 10 linearly spaced windows were used, each consisting of 25 ps of equilibration followed by 100 ps of sampling time. For the LJ decoupling path, much more sampling was required for the desired convergence. Therefore, 15 windows were used, each consisting of 200 ps of equilibration and 1 ns of sampling time. The LJ decoupling windows were spaced nonlinearly such that the window density increased quadratically near full decoupling, allowing more complete sampling of the regions of greatest curvature in λ. Statistical uncertainties were calculated by adding the variances from the two paths.
4. Force Field Parameterization
The parameterization of the CHEQ force field for alkanes is discussed in detail in our previous study[1]. Carrying over the alkane electrostatic and polarization model from that work [1], we here present a modification to the water-alkane Lennard-Jones interaction parameters that results in more accurate alkane hydration free energies in water. The functional form of the potential between two sites, i and j separated by a distance rij, is given by,
| (3) |
where ∈ij and are the interaction potential well depth and the distance at which the potential is at a minimum, respectively. Since traditionally the cross-interaction parameters are derived from the values for the individual interacting atoms, and , the adjustable parameters are ∈i, ∈j, , and .
For the present work, in order to retain the previously refined alkane atomic Lennard-Jones parameters (thus retaining the alkane-alkane interaction) [1], we consider specific Lennard-Jones interaction parameters (NBFIX) between the alkane carbons and the water oxygens. This need for unique interaction parameters between certain atom types to achieve better agreement with target data has been demonstrated and validated by Vorobyov et al[26].
We consider several reasons for adopting such an approach, again emphasizing the earlier use of such a protocol by Vorobyov et al[26]. First, as suggested by Equation 3, the Lennard-Jones interaction depends on atom-pair parameters, ∈ij and . These cross interaction parameters between atoms of different types have traditinally been determined from the individual atom-type parameters using empirical combining rules for purposes of convenience (thus obviating the need for explicitly determining individual atom-pair interaction parameters); however, there is no a priori requirement for using a particular combining rule. Thus, the current approach merely derives Lennard-Jones interaction parameters between heterogeneous atom-type pairs explicitly without relying on restrictive empirical combining rules. Second, when implementing polarizable force fields within a point-charge formalism (as considered presently with the charge equilibration model), molecules naturally encounter changes in charge distribution (coarsely represented as changes in partial charges at atomic sites) when placed in different media (solvents). Since the Lennard-Jones and electrostatic interactions are coupled, it is clear that for a particular atomic site within a molecule, changing the partial charge of the site requires attention to the attendant change in the Lennard-Jones parameters for the site. Thus, using a single Lennard-Jones parameter set for a given atom type and invoking a (restrictive) empricial combining rule may not afford the flexibility to accurately represent relevant interactions. Thus, we adopt an approach that allows greater flexibility to this end. We note that in the ideal case, one would use a more flexible interaction model that couples the electrostatic and Lennard-Jones parameters (see for instance, Chen et al [27]; Bauer et al [28]). However, such models may become prohibitively costly and cumbersome; the current approach is a lower-level approximation to such a force field. We assert that further studies along these lines are needed and would prove insightful.
Returning briefly to the parameterization strategy, the adjustable parameters are ∈CT2−OT, ∈CT3−OT, , and . CT2 and CT3 represent the methylene and methyl carbons, respectively, while OT represents the water oxygens. Taking ∈CT2−OT and ∈CT3−OT to be equivalent results in a total of three adjustable paramters. Briefly, ∈ was first adjusted to make the hydration free energy more favorable overall. Then, the CT2−OT rmin was adjusted to attain a more accurate hydration free energy for ethane. Finally, various values of the CT3−OT rmin were tested for propane and hexane; short simulations were used to obtain several values of the hydration free energy as a function of rmin. Linear regression of this data allowed extrapolation to a set of parameters that minimized the error in the hydration free energy for both propane and hexane. The final parameter set is shown in Table 1.
Table 1.
NBFIX terms applied to the LJ interactions between alkane and water. CT2 and CT3 represent methylene and methyl carbons, respectively, while OT represents the water oxygens.
| ∈ (kcal mol−1) | rmin (Å) | |
|---|---|---|
| CT2−OT | -0.14525 | 3.5347 |
| CT3−OT | -0.14525 | 3.6294 |
5. Results
5.1. Hydration Free Energies
The hydration free energies computed using the refined parameters are shown in Table 2. The values are in very good agreement with experiment, with an RMS error of 3.6% over the series. This is a significant improvement over our previous study[1] in which we calculated hydration free energies for pentane, hexane, and heptane using our alkane force field without the modification described in this work. For example, we previously obtained a hydration free energy of 1.80 ± 0.5 kcal mol−1 for hexane, compared to the value of 2.507 ± 0.200 kcal mol−1 from Table 2. Even taking into consideration the fact that our previous analysis used less sampling resulting in a larger error, it is clear that the current force field gives much more accurate results.
Table 2.
Components of the free energy calculations in kcal mol−1 for the alkanes. The LJ decoupling portion (ΔGLJ), electrostatic decoupling portion (ΔGelec), long-range correction (LRC), total hydration free energy (ΔGtotal) with error, and the experimental free energy[29] (ΔGexpt) are shown.
| ΔGLJ | ΔGelec | LRC | ΔGtotal | ΔGexpt | |
|---|---|---|---|---|---|
| ethane | 2.065 | -0.058 | -0.301 | 1.706 ± 0.147 | 1.768 |
| propane | 2.480 | -0.039 | -0.432 | 2.010 ± 0.253 | 1.975 |
| butane | 2.783 | -0.108 | -0.562 | 2.113 ± 0.219 | 2.148 |
| pentane | 3.117 | -0.172 | -0.693 | 2.252 ± 0.109 | 2.338 |
| hexane | 3.558 | -0.223 | -0.827 | 2.507 ± 0.200 | 2.550 |
| heptane | 4.002 | -0.280 | -0.954 | 2.768 ± 0.329 | 2.627 |
5.2. Radial Distribution Functions
Next, we consider the water-alkane interactions as measured by the radial distribution function (RDF). For all systems, we have calculated the RDFs for water oxygen to alkane carbons using both the original model and our revised model. The system of a single alkane molecule with 256 TIP4P-FQ waters was simulated for ~ 5 ns for each alkane to obtain these results, shown in Figure 1. It is clear from these profiles that the changes made to the model have virtually no effect on the solvation structure of the alkanes in water. This is important, since in parameterizing a model it is possible to reproduce some properties very accurately while others very poorly. We wish to ensure that increasing the accuracy of the free energies does not affect how accurately the alkane-water interactions are captured. Therefore it is encouraging that the radial distribution functions are virtually unchanged compared to the original model.
Figure 1.
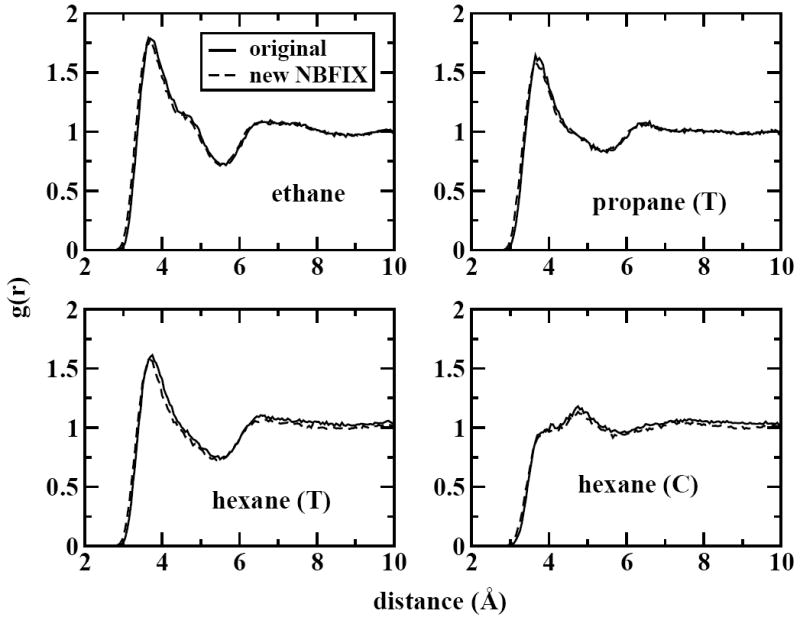
Radial distribution functions for water oxygen to alkane carbons; T denotes the average of terminal carbons (i.e. C1 and C6 for hexane) while C denotes the average of center carbons (i.e. C3 and C4 for hexane).
5.3. Enthalpy of Solvation
We also measured the change in enthalpy upon solvation, ΔHsolv of the alkanes in water. This data was obtained from 5 ns simulations of a single alkane molecule in water (without TI modifications) as well as pure water as described in Section 3. ΔHsolv was calculated as
| (4) |
where 〈U + PV〉a+w is the average system enthalpy of the simulation of a single alkane molecule in water, 〈U + PV〉w is the average system enthalpy of the pure water simulation, and 〈U + RT〉a is the average gas phase enthalpy of the single alkane molecule (where RT = 0.58 kcal mol−1 at 298 K). The calculated values of Hsolv are shown in Table 3. For comparison, we also show the values computed using our original alkane model. Experimentally derived values[30], also shown in Tble 3, exhibit a more negative trend as alkane size increases. Our results show more fluctuation as a function of alkane size and do not quite follow this trend. However, comparing to the values of ΔHsolv calculated from our original model, we see a significant improvement. Neither model is able to accurately capture the experimental trend, but the magnitudes predicted by our revised model are more in line with experiment.
Table 3.
Change in enthalpy (in kcal mol−1) upon solvation (ΔHsolv) for the series of alkanes. Results from both the original and revised models are shown, as well as experimental data[30] for comparison.
| ΔHsolv | ΔHsolv (orig) | ΔHsolv (expt) | |
|---|---|---|---|
| ethane | -2.82 | -4.13 | -1.14 |
| propane | -4.33 | -6.17 | -1.92 |
| butane | -3.61 | -5.84 | -2.69 |
| pentane | -4.02 | -7.43 | -3.47 |
| hexane | -3.72 | -7.58 | -4.24 |
| heptane | -4.32 | -7.24 | -5.01 |
5.4. Polarization Effects
Since the CHEQ model is a polarizable model, we wish to probe the effects of explicit polarization on the alkane-water systems. Specifically, we have investigated the shift in the molecular dipole moment of water when associated with an alkane. This is shown by the water molecular dipole moment distributions in Figure 2. It is clear that there is a nontrivial shift in the molecular dipole moment (~ 0.06 D) for water molecules associated with the alkane. This is because water molecules surrounded by bulk water experience stronger polarization effects than those associated with the nonpolar alkane molecule. It is encouraging that the CHEQ model can capture even the subtle polarization effects caused by a single alkane molecule in bulk water.
Figure 2.
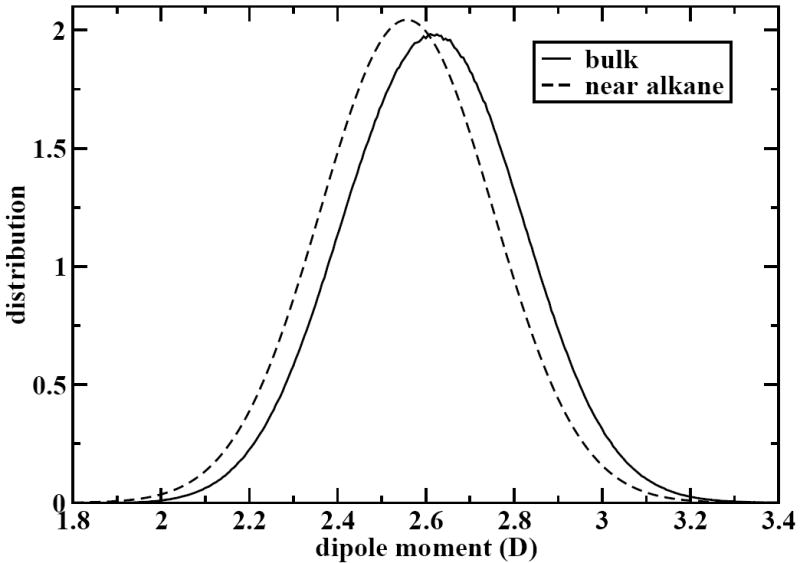
Water molecular dipole moment distributions averaged over all alkane-water systems. We show the distribution over all water molecules (“bulk”) as well as only those waters within 3 Å of an alkane terminal carbon (“near alkane”). The “near alkane” distribution has been smoothed by fitting to a gaussian function.
6. Conclusion
We have presented a revised set of nonbonded parameters for our CHEQ alkane force field. The CHEQ force field can capture subtle polarization effects of a single alkane molecule on nearby waters as shown by water dipole moment distributions. Furthermore, we have shown that our force field modfications have not affected the alkane-water interactions as measured by the RDF. Finally, our revised force field gives more accurate hydration free energies. The hydration free energy is important for modeling the interactions of water with aliphatic groups. Specifically, our previous work has shown that inclusion of polarization enhances the amount of water penetration into phospholipid bilayers compared to traditional fixed-charge force fields[10]. While it is difficult to ascertain whether this is a favorable result, we believe that the current force fields are deficient in their treatment of the interactions between water and the nonpolar interior of the bilayer. More rigorous treatment of the hydration free energy, and therefore the solvation process in general, is an important step towards a better representation of the interaction of water with the bilayer interior. Future work will explore the application of the refined parameters to bilayer simulations.
Acknowledgments
The authors gratefully acknowledge support from the National Institute of Health sponsored COBRE (Center of Biomedical Research) Grant Numbers P20-RR017716 at the University of Delaware (Department of Chemistry and Biochemistry) and P20-RR015588 (Department of Chemical Engineering).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis JE, Warren GL, Patel S. A revised charge equilibration potential for liquid alkanes. J Phys Chem B. 2008;112:8298–8310. doi: 10.1021/jp8003129. [DOI] [PubMed] [Google Scholar]
- 2.Dikshith TSS, Diwan PV. Industrial Guide to Chemical and Drug Safety. John Wiley & Sons; Hyderabad, India: 2003. [Google Scholar]
- 3.Borbon A, Fontaine H, Locoge N, Veillerot M, Galloo JC. Developing receptor-oriented methods for non-methane hydrocarbon characterization in urban air - Part 1: source identification. Atmospheric Environment. 2003;37:4051–4064. [Google Scholar]
- 4.Rick SW, Berne BJ. Dynamical Fluctuating Charge Force Fields: The Aqueous Solvation of Amides. J Am Chem Soc. 1996;118:672–679. [Google Scholar]
- 5.Mortier WJ, Ghosh SK, Shankar S. Electronegativity Equalization Method for the Calculation of Atomic Charges in Molecules. J Am Chem Soc. 1986;108:4315–4320. [Google Scholar]
- 6.Nalewajski RF, Korchowiec J, Zhou Z. Molecular Hardness and Softness Parameters and Their Use in Chemistry. International Journal of Quantum Chemistry: Quantum Chemistry Symposium 22. 1988;22:349–366. [Google Scholar]
- 7.Patel S, Brooks I, Charles L. CHARMM Fluctuating Charge Force Field for Proteins: I. Parameterization and Application to Bulk Organic Liquid Simulations. J Comp Chem. 2004;25:1–15. doi: 10.1002/jcc.10355. [DOI] [PubMed] [Google Scholar]
- 8.Rappe A, Goddard WA., I Charge Equilibration for Molecular Dynamics Simulations. J Phys Chem. 1991;95:3358–3363. [Google Scholar]
- 9.Patel S, Brooks CL. Revisiting the Hexane-Water Interface via Molecular Dynamics Simulations Using Non-Additive Alkane-Water Potentials. J Chem Phys. 2006;124:204706. doi: 10.1063/1.2198538. [DOI] [PubMed] [Google Scholar]
- 10.Davis JE, Rahaman O, Patel S. Molecular Dynamics Simulations of a DMPC Bilayer Using Nonadditive Interaction Models. Biophys J. 2009;96:385–402. doi: 10.1016/j.bpj.2008.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen TW, Andersen OS, Roux B. Energetics of Ion conduction through the gramicidin channel. PNAS. 2004;101(1):117–122. doi: 10.1073/pnas.2635314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roux B, Allen T, Berneche S, Im W. Theoretical and Computational Models of Ion Channels. Quarterly Reviews of Biophysics. 2004;37:15–103. doi: 10.1017/s0033583504003968. [DOI] [PubMed] [Google Scholar]
- 13.Bucher D, Kuyucak S. Importance of water polarization for ion permeation in narrow pores. Chem Phys Lett. 2009;447:207–210. [Google Scholar]
- 14.Rick SW, Stuart SJ, Berne BJ. Dynamical fluctuating charge force fields: Application to liquid water. J Chem Phys. 1994;101(7):6141–6156. [Google Scholar]
- 15.Straatsma TP, McCammon JA. Multiconfiguration thermodynamic integration. J Chem Phys. 1991;95:1175–1188. [Google Scholar]
- 16.Zacharias M, Straatsma TP, McCammon JA. Separation-shifted scaling, a new scaling method for Lennard-Jones interactions in thermodynamic integration. J Chem Phys. 1994;100:9025–9031. [Google Scholar]
- 17.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 18.Allen MP, Tildesley DJ. Computer Simulation of Liquids. Clarendon Press; Oxford: 1987. [Google Scholar]
- 19.Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol Phys. 1984;52:255–268. [Google Scholar]
- 20.Darden T, York D, Pederson L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 21.Essmann U, Darden T, Lee H, Perera L, Berkowitz ML, Pederson L. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 22.Anisimov VM, Vorobyov IV, Roux B, Alexander J, MacKerell D. Polarizable Empirical Force Field for the Primary and Secondary Alcohol Series Based on the Classical Drude Model. J Chem Theory Comput. 2007;3:1927–1946. doi: 10.1021/ct700100a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vorobyov IV, Anisimov VM, Alexander J, MacKerell D. Polarizable Empirical Force Field for Alkanes Based on the Classical Drude Oscillator Model. J Phys Chem B. 2005;109:18988–18999. doi: 10.1021/jp053182y. [DOI] [PubMed] [Google Scholar]
- 24.Shirts MR, Pitera JW, Swope WC, Pande VS. Extremely precise free energy calculations of amino acid side chain analogs: Comparison of common molecular mechanics force fields for proteins. J Chem Phys. 2003;119:5740–5761. [Google Scholar]
- 25.Warren GL, Patel S. Hydration free energies of monovalent ions in transferable intermolecular potential four point fluctuating charge water: An assessment of simulation methodology and force field performance and transferability. J Chem Phys. 2007;127:064509. doi: 10.1063/1.2771550. [DOI] [PubMed] [Google Scholar]
- 26.Vorobyov IV, Anisimov VM, Greene S, Venable RM, Moser A, Pastor RW, MacKerell AD. Additive and classical drude polarizable force fields for linear and cyclic ethers. J Chem Theory Comput. 2007;3:1120–1133. doi: 10.1021/ct600350s. [DOI] [PubMed] [Google Scholar]
- 27.Chen B, Xing J, Siepmann JI. Development of Polarizable Water Force Fields for Phase Equilibrium Calculations. J Phys Chem B. 2000;104:2391–2401. [Google Scholar]
- 28.Bauer BA, Patel S. Properties of water along the liquid-vapor coexistence curve via molecular dynamics simulations using the polarizable TIP4P-QDP-LJ model. Journal of Chemical Physics. 2009 doi: 10.1063/1.3200869. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ben-Naim A, Marcus Y. Solvation thermodynamics of nonionic solutes. J Chem Phys. 1984;81:2016–2027. [Google Scholar]
- 30.Cabani S, Gianni P, Mollica V, Lepori L. Group contributions to the thermodynamic properties of non-ionic organic solutes in dilute aqueous solution. J Solution Chem. 1981;10:563–595. [Google Scholar]